
Abstract
Background
Energy metabolism disorder, especially lipid metabolism disorder, is an important biological characteristic of colon cancer. This research sought to examine the association between lipid metabolism-related long non-coding RNAs (lncRNAs) and prognoses among colon cancer patients.
Methods
The transcriptome profile and clinical data of patients with colon cancer were retrieved from The Cancer Genome Atlas database. Using consensus clustering, cases were divided into two clusters and Kaplan–Meier analysis was executed to analyze differences in their prognoses. The gene set enrichment analysis (GSEA) was used to discover biological processes and signaling pathways. A lipid metabolism-related lncRNA prognostic model (lipid metabolism-LncRM) was created utilizing the least absolute shrinkage and selection operator (LASSO) regression. The tumor microenvironment was evaluated on the basis of the composition of immune and stromal cells.
Results
The patients in Cluster 2 were found to have a better prognosis and higher expression of programmed cell death 1 (PD-1) and programmed cell death ligand 1 (PD-L1) relative to Cluster 1. The results of GSEA showed the enrichment of energy metabolism pathways in Cluster 2. LASSO regression was used to identify the five LncRNAs that were shown to be most substantially linked to patient prognosis. These were NSMCE1-DT, LINC02084, MYOSLID, LINC02428, and MRPS9-AS1. Receiver operating characteristic (ROC) curves and survival analysis illustrated that the lipid metabolism-LncRM had a significant prognostic value. Further analysis showed that high- and low-risk groups were significantly different in terms of clinical characteristics and immune cells infiltration.
Conclusions
Lipid metabolism-related lncRNAs could predict the prognoses and tumor microenvironment of colon cancer and might be important biomarkers relevant to immunotherapy.
Similar content being viewed by others


Introduction
Colon cancer is the most prevalent malignant illness of the gastrointestinal tract [1], whereby the incidence and mortality rates account for 6.0% and 5.8% of all cancers [2, 3]. At present, the treatment modalities include chemotherapy, radiotherapy, endoscopic and surgical local excision, targeted therapy, and immunotherapy. These treatment regimens can improve the patients' 5-year overall survival (OS) rate is 60–70% [4]. Tumors are often heterogeneous. Characterizing tumors into different subtypes and designing individualized treatment strategies according to their biological characteristics can improve cancer control rates. Hence, it is necessary to recognize new markers for anticipating the prognoses of colon cancer patients and screening those who may stand to substantially benefit from immunotherapy.
Metabolic reprogramming has emerged as a prominent feature of cancer; it promotes tumor cell proliferation and survival [5], whereby lipid metabolism disorder is a prominent metabolic change in tumors. Active fat metabolism often occurs in patients with aggressive metastatic colon cancer [6]. Lipid metabolism disorder can lead to the alteration of the expression and activity of lipid metabolic enzymes due to abnormally activated carcinogenic signal pathway, redistribution of lipid metabolism in cancer cells, along with enhanced rapid proliferation, migration, and invasion of cancer. In addition, Gharib et al. [7] report that microRNA-497-5p facilitates colon cancer cell death induced by starvation via targeting acyl-CoA synthase-5 and regulating lipid metabolism. Nevertheless, as far as we know, there are no studies on the association of lipid metabolism, long non-coding RNAs (lncRNAs) with colon cancer advancement.
lncRNAs are a widely present type of RNA molecule with a length exceeding 200 nucleotides but no ability for protein-coding [8]. According to recent research, lncRNAs may influence tumor growth by modifying fatty acid metabolism [9,10,11]. Because it binds to the arginine residues of the hnRNPA1-RGG motif, the lncRNA HOXB-AS3 has the potential to significantly suppress glucose metabolic reprogramming in colon cancer cells [12]. This study aimed at evaluating whether targeting both lipid metabolism-related genes and lncRNAs may be utilized as novel therapeutic targets against colon cancer.
Herein, the data on colon cancer were extracted from the cancer genome atlas (TCGA) database and we investigated the association of lipid metabolism-related lncRNAs with clinical characteristics and immunotherapy. Based on lipid metabolism-related lncRNAs, the Least Absolute Shrinkage Selection Operator (LASSO) was utilized to construct a prognostic risk stratification model. In addition, we also evaluated the immune statuses of patients in different risk groups.
Methods
Data collection
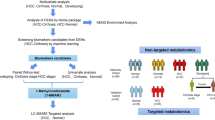
The study flow is illustrated in Additional file 1: Figure S1. We acquired the RNA-seq and clinical data of patients with colon cancer from the TCGA database (https://portal.gdc.cancer.gov/); a total of 4,234 lncRNAs were obtained. We extracted the data of 514 samples, of which 473 were from tumor tissues and the remaining were from adjacent normal tissues. The clinical information was available for 446 cases which also had complete RNA-seq data. Using the Molecular Signatures Database v7.0, 189 genes related to lipid metabolism were identified (Additional file 2: Table S1) [13, 14]. Ethics approval and informed consent were not required in this study since the data were obtained from public databases. The study was conducted in accordance with the declaration of Helsinki.
Identification of lipid metabolism-related lncRNAs
The differential expressions of lncRNAs between cancer and normal specimens were compared utilizing the R package, “limma”. Spearman correlation analysis was carried out to screen lipid metabolism-related lncRNAs correlated with at least one lipid metabolism gene (| Pearson R |> 0.6 and p < 0.001). The co-expression network of lipid metabolism-related genes and lncRNAs was created utilizing the R package, “igraph”. For the purpose of screening lncRNAs that were correlated with OS, a univariable Cox regression was utilized.
Analysis of functional enrichment
On the basis of the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways [15,16,17], the Gene Set Enrichment Analysis (GSEA) (https://www.gsea-msigdb.org/gsea/index.jsp) was employed to identify biological functions and signaling pathways in different clusters. Enriched genes were defined according to the threshold values of p < 0.05 and false discovery rate < 25%.
Evaluation of cell compositions in tumor microenvironment
Using ESTIMATE, we assessed the immune and stromal cells in malignant tissues, computed stromal and immune scores, and predicted the cellular infiltration in the tumor microenvironment (TME) [18]. StromalScore refers to the infiltration level of stromal cells in the tumor tissue; ImmunoScore represents the infiltration level of immune cells; and ESTIMATEScore is an indicator that combines StromalScore with ImmunoScore to infer tumor purity. By employing CIBERSORT (https://cibersort.stanford.edu/), we computed the proportion of 22 distinct immune cell infiltrates. Based on the R package “ImmuneSubtypeClassifier” and “TCGAbiolinks”, samples were divided into different immune subtypes and molecular subtypes [19, 20].
Construction and verification of the risk signature
The LASSO regression analysis was carried out with the help of the R package “glmnet” to create a prognostic model based on lipid metabolism-related lncRNAs (lipid metabolism-LncRM). The calculation for the model was as follows:
We calculated the risk scores according to the level of each lncRNA expression and the matching regression coefficient. The estimation of the risk score was achieved according to the equation below: Risk score = (0.7641 × NSMCE1-DT) + (0.1906 × LINC02084) + (1.2843 × MYOSLID) + (0.2206 × LINC02428) + (0.1157 × MRPS9-AS1). Patients were categorized at random into training (n = 224) and testing subsets (n = 222) with the help of the R package, “caret”, and assigned to high- and low-risk groups on the basis of the mean risk score obtained in the training set. Univariate and multivariate Cox regression analyses were employed to examine the independent prognostic values of risk scores.
Statistical analysis
The R statistical program version 3.6.1. was utilized to execute all analyses of statistical data. The survival in subgroups was computed utilizing the Kaplan–Meier curves. We used the “survivalROC” package for receiver operating characteristic (ROC) curve analysis to examine the prediction accuracy. The differential expressions of lncRNAs between cancer and normal samples were compared utilizing the Wilcoxon signed-rank test. Differences in qualitative variables were examined using the χ2 test. The relationship among the quantitative variables was assessed utilizing Spearman's correlation analysis. P < 0.05 was established as a determinant of statistical significance.
Results
Selection of lipid metabolism-related lncRNAs
The clinical information of 446 colon cancer patients in TCGA is shown in Table 1. In total, 27 genes and 349 lncRNAs related to lipid metabolism were screened using the Spearman correlation analysis (Fig. 1A). Next, based on the clinical data, we filtered out 18 lipid metabolism-related lncRNAs significantly related to prognosis. The forest plot shows the hazards ratio and 95% confidence intervals (CIs) of the 18 lipid metabolism-related lncRNAs (all p < 0.05, Fig. 1B). Figures 1C and 1D demonstrate that in tumor tissues, the expressions of 8 lipid metabolism-related prognostic lncRNAs (NSMCE1 − DT, STAG3L5P − PVRIG2P − PILRB, MYOSLID, LINC02428, NCBP2 − AS1, WARS2 − AS1, MRPS9 − AS1, and LENG8 − AS1) were upregulated, whereas that of the remaining 10 lncRNAs (SNHG26, LINC02084, PCED1B − AS1, ATP2B1 − AS1, LINC01857, LINC00861, LINC02381, PRKAR1B − AS2, MIR600HG, and LINC01679) were markedly downregulated (all p < 0.05).

Identification of lipid metabolism-related lncRNAs in colon cancer. A The co-expression network of lipid metabolism-related genes and lncRNAs. B Forest plot based on the univariate Cox regression analysis for OS. C Heatmap and D box plot for the differential expressions of 18 lipid metabolism-related lncRNAs between tumor and normal tissues. lncRNAs, long non-coding RNAs; OS, overall survival. *p < 0.05, **p < 0.01, and ***p < 0.001
Definition of colon cancer subtypes according to lipid metabolism-related lncRNAs
The consensus clustering analysis based on the lipid metabolism-related lncRNAs was used to examine the molecular subtypes of colon cancer. At k = 2, the consensus matrix heatmap showed sharp and crisp boundaries (Fig. 2A; Additional file 3: Figure S2), which indicated that the samples could be divided into stable and robust clusters. Therefore, all data were assigned to two clusters. Patients in Cluster 2 showed significantly longer OS relative to those in Cluster 1 (p = 0.011, Fig. 2B). Association analysis of subgroups and clinical features presented that only the distribution of the metastatic status was different between clusters (p < 0.05, Fig. 2C). This suggested that the difference in survival between the two clusters might be related to their different risk of metastasis.

Identification of tumor subtypes based on the prognostic lipid metabolism-related lncRNAs. A Consensus clustering matrix at k = 2. B Kaplan–Meier plot for patients in Clusters 1 and 2. C Heatmap of 18 prognostic lipid metabolism-related lncRNAs and clinicopathological features in Clusters 1 and 2. *p < 0.05
Next, the relationship among lncRNAs related to lipid metabolism and immune characteristics in colon cancer was investigated. First, the levels of expression of immune checkpoints (programmed cell death 1 [PD-1], programmed cell death ligand 1 [PD-L1], cytotoxic T lymphocyte antigen-4 [CTLA-4], indoleamine 2,3-dioxygenase 1 [IDO1], indoleamine 2,3-dioxygenase 2 [IDO2], hepatitis A virus cellular receptor 2 [HAVCR2], and lymphocyte activating gene 3 [LAG3]) in different subgroups were estimated and these results are shown in Fig. 3A-E and Additional file 4: Figure S3A-G. Relative to Cluster 1, the PD-1, PD-L1, IDO1, HAVCR2, and LAG3 expression levels were all significantly higher in Cluster 2 (all p < 0.05). This suggested that Cluster 2 patients might benefit more from treatment with immune checkpoint inhibitors.

Relationship between the Clusters and immune characteristics. The expression of immune checkpoints A PD-1, B PD-L1, C IDO1, D HAVCR2, and E LAG3 in Clusters 1 and 2. F-G Correlations among the 18 lipid metabolism-related lncRNAs and immune checkpoints. Differences in H immune, I stromal, and J estimate scores in Clusters 1 and 2. K Differences in the abundances of 22 immune cell types between Clusters 1 and 2. PD-1, programmed cell death 1; PD-L1, programmed cell death ligand 1; IDO1, indoleamine 2,3-dioxygenase 1; HAVCR2, hepatitis A virus cellular receptor 2; LAG3, lymphocyte activating gene 3. *p < 0.05, **p < 0.01, and ***p < 0.001
Further analysis indicated that LINC02084, PCED1B-AS1, LINC01857, LINC00861, LINC02381, and LINC01679 were positively correlated with the expression of PD-1, while STAG3L5P-PVRIG2P-PILRB, NCBP2-AS1, WARS2-AS1, and LENG8-AS1 were negatively correlated with the expressions of PD-1 (Fig. 3F). SNHG26, NSMCE1 − DT, LINC02084, PCED1B-AS1, MYOSLID, LINC02428, ATP2B1 − AS1, LINC01857, LINC00861, LINC02381, PRKAR1B − AS2, MRPS9 − AS1, and LINC01679 were positively correlated with the expression of PD-L1, while STAG3L5P-PVRIG2P-PILRB and LENG8-AS1 were negatively correlated with the expressions of PD-L1 (Fig. 3G). Moreover, the findings from the ESTIMATE analysis suggested that the immune (p < 0.001), stromal (p < 0.05), and estimate (p < 0.01) scores in Cluster 2 were markedly higher than those in Cluster 1 (Fig. 3H-J). Finally, we evaluated the abundances of 22 distinct kinds of immune cells between the two subtypes and found no differences (all p > 0.05, Fig. 3K). This suggested that the composition of immune cells in Cluster 2 was not significantly different from that in Cluster 1; however, the number of infiltrating immune cells and stromal cells was higher, and the tumor purity was lower in Cluster 2 than those in Cluster 1.
Gene set enrichment analysis for clusters 1 and 2
To understand the mechanisms underlying prognostic and immunophenotypic differences between Clusters 1 and 2, we performed GSEA. The pathways related to citrate cycle and tricarboxylic acid cycle (TCA) cycle (p = 0.010), galactose metabolism (p = 0.002), proline and arginine metabolism (p < 0.001), glutathione metabolism (p = 0.002), proteasome (p = 0.010), and oxidative phosphorylation (p < 0.001) were considerably enriched in Cluster 2 (Fig. 4A–F; Additional file 5: Table S2). Previous results showed that Cluster 2 patients had better survival, indicating that the signaling pathway enriched in Cluster 2 might be involved in the inhibition of the occurrence and development of colon cancer.

Gene set enrichment analysis in Clusters 1 and 2 on the basis of the KEGG pathways. The pathways related to A citrate cycle and TCA cycle, B galactose metabolism, C arginine and proline metabolism, D glutathione metabolism, E proteasome metabolism, and F oxidative phosphorylation are enriched in Cluster 2. KEGG, Kyoto Encyclopedia of Genes and Genomes; TCA, tricarboxylic acid cycle
Construction of a lipid metabolism-LncRM for predicting survival
For an accurate prediction of colon cancer prognoses, five lncRNAs were filtered out by LASSO regression from among the 18 lipid metabolism-related prognostic lncRNAs (Fig. 5A, 5B). The patients were categorized into low- and high-risk groups predicated on the mean risk score (Fig. 5C, 5D). In both training (p = 0.001) and testing cohorts (p = 0.012), high-risk patients exhibited considerably reduced OS duration in contrast with that in the low-risk group (Fig. 5E, 5F). In addition, the ROC curves for OS at 1, 3, and 5 years in the training and testing cohorts are illustrated in Figs. 5G and 5H, respectively. As depicted in Fig. 5I, the high-risk patients showed elevated levels of NSMCE1-DT, LINC02084, MYOSLID, LINC02428, and MRPS9-AS1 expressions (all p < 0.05). This is consistent with the results shown in Fig. 1B, indicating that these five lncRNAs were all related to poor prognosis.

Construction and evaluation of the lipid metabolism-lncRNA prognostic model for colon cancer. A, B LASSO regression analysis of lipid metabolism-related lncRNAs that are most significantly correlated with OS. C Risk score and D survival status of each patient. OS of patients in high- and low-risk groups in E training and F testing cohorts. ROC analysis for risk scores in G training and H testing cohorts. I Expressions of five prognostic lipid metabolism-related lncRNAs in different risk groups. LASSO, least absolute shrinkage and selection operator; OS, overall survival; ROC, receiver operating characteristic. *p < 0.05, **p < 0.01, and ***p < 0.001
According to the R package “ImmuneSubtypeClassifier,” tumor samples were divided into five immune subtypes: C1 (wound healing), C2 (IFN-gamma dominant), C3 (inflammatory), C4 (lymphocyte depleted), and C6 (TGF-b dominant). However, C5 (immunologically quiet) subtype was not present. The relationships between the five lncRNAs in the prognostic model and immune subtypes are shown in Additional file 6: Figure S4. Only LINC02084 was differentially expressed among the different immune subtypes (p < 0.001), indicating that the expression level of LINC02084 was related to immune subtypes. In addition, the relationships between lncRNAs and the four molecular subtypes of colon cancer are shown in Additional file 7: Figure S5. In addition to MRPS9-AS1, the expression levels of the remaining four lncRNAs were associated with the molecular subtypes (all p < 0.01).
Association between clinical characteristics and risk score
Figures 6A and 6B indicated that risk score independently served as a predictor of OS (both p < 0.001). In addition, high-risk scores had a high likelihood of being associated with distant metastases, higher N staging, total staging, and immunity scores, and a high likelihood of belonging to the Cluster 1 subtype (all p < 0.05, Fig. 6C-G and Additional file 8: Figure S6). This may be part of the reason for the worse prognosis of high-risk patients.

Relationship among risk scores and clinical characteristics. Forest plots based on A univariate and B multivariate Cox regression analysis for OS. Correlations of risk scores and C N stage, D metastatic status, E total stage, F immune score, and G Clusters. OS, overall survival. *p < 0.05, **p < 0.01, and ***p < 0.001
Correlation between lipid metabolism-LncRM and tumor microenvironment
As shown in Fig. 7A-C and Additional file 4: Figure S3H-N, as opposed to the low-risk colon cancer patients, the high-risk cases showed higher expressions of CTLA-4 (p < 0.001), PD-1 (p < 0.01), PD-L1 (p < 0.001), IDO1 (p < 0.05), IDO2 (p < 0.05), HAVCR2 (p < 0.001), and LAG3 (p < 0.05). The estimate, stromal, and immune scores were elevated in the high-risk group in comparison to the low-risk subgroup (all p < 0.001, Fig. 7D). The abundances of immune cells estimated by CIBERSORT analysis are depicted in Fig. 8A–H. Mast cells activated, CD4 + T cells naive, NK cells resting, and Neutrophils activated were positively correlated with risk scores (all p < 0.05), while CD8 + T cells, T regulatory cells, mast cells resting, and dendritic cells resting were found to have a negative link to risk scores (all p < 0.05). This phenomenon shows that the higher the risk score, the worse the anti-tumor immune ability.

Correlation of risk score and tumor microenvironment. The expressions of A PD-L1, B PD-1, and C CTLA-4 in different risk groups. D Tumor microenvironment scores for different risk groups. PD-1, programmed cell death 1; PD-L1, programmed cell death ligand 1; CTLA-4, cytotoxic T lymphocyte antigen-4. **p < 0.01, and ***p < 0.001

Relationship among risk scores and immune cell infiltrates. Correlations of risk scores and A mast cells activated, B neutrophils, C NK cells resting, D T cells CD4 naive, E dendritic cells resting, F mast cells resting, G T cells CD8, and H T cells regulatory
Discussion
In the present study, 18 lipid metabolism-related prognostic lncRNAs were analyzed. By consensus clustering, colon cancer patients were classified into two distinct categories. In Cluster 2, the survival time of patients was prolonged, and the expressions of PD-1, PD-L1, IDO1, HAVCR2, and LAG3 were elevated. In addition, metabolic-related pathways were highly enriched in Cluster 2. Five lipid metabolism-related lncRNAs most significantly correlated with prognosis were selected using LASSO analysis to construct the lipid metabolism-LncRM. Using multivariate analysis, we identified the risk score as an independent and adverse predictor for OS. Moreover, patients with distinct risk scores exhibited varying survival status, clinical characteristics, and immune cell infiltration. We confirmed that lipid metabolism-related lncRNAs may serve as a signature for predicting survival and is a potential immunotherapeutic target for patients with colon cancer.
Energy metabolism reprogramming is an important feature of tumors [21, 22]. The imbalance among oncogenic pathways interferes with the synthesis and metabolism of various substances, including glucose, glutamine, and fatty acids, thereby promoting the growth of tumor cells [23,24,25]. To date, studies on the association of lncRNA expression with lipid metabolism in colon cancer are scarcely reported. Therefore, it is necessary to investigate lipid metabolism-related lncRNAs using “big data”. Christensen et al. [26] show that the lncRNA SNHG16 performs an important function in the transcription of genes related to lipid metabolism and can target up to 26 microRNA families. Yurui et al. [27] have constructed a competing endogenous RNA (ceRNA) network related to fatty acid metabolism. They report that lipid metabolism-related lncRNAs can potentially affect the prognoses of colon cancer patients. Metabolic reprogramming varies between different cell subtypes. In this study, the population was divided into two subgroups based on lipid metabolism-related lncRNAs Cluster recognition. Different subgroups showed varying survival times and risks of distant metastases. Further, significant differences were found in glucose metabolism and lipid metabolism pathways. Therefore, the study of lipid metabolism-related lncRNAs may help to further classify patients and provide individualized treatment options.
To better predict the prognoses of patients, we finally identified five prognostic-associated lipid metabolism-related lncRNAs, namely NSMCE1-DT, LINC02084, MYOSLID, LINC02428, and MRPS9-AS1. LINC02084 has been previously linked to the prognoses of patients with hepatocellular carcinoma [28]. MYOSLID is involved in the advancement of head and neck squamous cell carcinomas (HNSCC) and osteosarcoma, and its mechanism of action is by promoting RAB13 expression by sponging microRNA-1286 or promoting invasion and metastases by regulating a part of the epithelial-mesenchymal transformation [29, 30]. As far as we know, this is the first research on the relationship of NSMCE1-DT, LINC02428, and MRPS9-AS1 with tumor prognosis. The underlying mechanisms that affect patient survival warrant further investigations.
In addition, based on the lipid metabolism-LncRM, we categorized the patients into low- and high-risk groups and compared the survival, clinicopathological features, immune cell infiltrates, and the expressions of immune checkpoints between the two groups. The high-risk group was found to show higher staging and poorer survival. Further analysis demonstrated that low infiltration levels of CD8 + T cells and dendritic cells resting, and elevated expression levels of immune checkpoints in these patients, thereby suggesting that immunotherapeutic intervention for patients in the high-risk group might improve tumor control.
Currently, immunotherapy is an emerging and dynamic area in cancer treatment, including, but not limited to, cancer vaccines, oncolytic viruses, and immune checkpoint inhibitors [31,32,33]. CTLA-4, PD-1, and PD-L1 are responsible for the immunotherapeutic responses in patients with colon cancer. Fiegle et al. [34] demonstrate the association of PD-1 and CTLA-4 overexpression with a worse OS and disease-free survival. In the CheckMate-142 (NCT02060188) study, the effects of nivolumab monotherapy on 74 patients with microsatellite instability-high (MSI-H) or mismatch repair deficiency (dMMR) metastatic colorectal carcinoma (mCRC) were reported, wherein the estimated rates at 12 months were 50% for progression-free survival and 73% for OS [35]. In addition, nivolumab plus ipilimumab in the treatment of MSI-H MCRC showed a disease control rate of 80%, and the objective remission rate was 55% (95% CI 45.2–63.8) [36]. Therefore, although high-risk patients identified through the prognostic model established in this study showed poor survival, they may potentially benefit more from therapy using immune checkpoint inhibitors.
In theory, PD-L1 is a viable marker of anti-PD-1 immunotherapy. It is reported that mCRC patients who responded to PD-1 inhibitors show dMMR status or PD-L1 up-regulation in tumor cells [37, 38]. Further studies show that PD-L1 overexpression is related to better clinical prognoses. This may be due to compensatory regulation of this marker, thereby threatening the tumor with an aggressive immune response [39]. Therefore, PD-L1 expression and dMMR genotype are considered possible markers of responses to PD-1 inhibitors. However, some studies report that the level of PD-L1 expression is not linked to the treatment efficacy in CRC patients [40, 41]. In addition to PD-L1, earlier studies have also illustrated that the existence of tumor-infiltrating lymphocytes in the TME is closely related to a better response rate toward anti-tumor immunotherapy [42,43,44]. In our study, the expression level of PD-L1 was discovered to be higher in the high-risk subgroup, accompanied by significant infiltrations of immune cells in the TME, indicating that the high-risk group could be highly suited for immunotherapeutic treatments.
Using lipid metabolism-related lncRNAs, herein, we have provided a new perspective for prognostic prediction and treatment formulation for patients with colon cancer. Nevertheless, there are several drawbacks to the research. Our results were based entirely on bioinformatic analyses, and more trials are required to confirm the mechanism behind lncRNA action. Further, prospective clinical data are needed to verify the prognostic and therapeutic values of lipid metabolism-LncRM, accounting for the inherent deficiencies of this retrospective analysis.
Conclusions
In conclusion, we confirmed the association of lipid metabolism-related lncRNAs in colon cancer with the prognoses of patients, the expressions of immune checkpoints, and the infiltrations of different immune cells in the TME. These lncRNAs could function as new potential targets for treating colon cancer.
Availability of data and materials
Publicly available datasets were analyzed in this study. This data can be found here: http://cancergenome.nih.gov/.
Abbreviations
- OS:
-
Overall survival
- lncRNAs:
-
Long non-coding RNAs
- TCGA:
-
The cancer genome atlas
- LASSO:
-
The least absolute shrinkage selection operator
- KEGG:
-
The kyoto encyclopedia of genes and genomes
- GSEA:
-
Gene set enrichment analysis
- TME:
-
Tumor microenvironment
- lipid metabolism-LncRM:
-
Prognostic model based on lipid metabolism-related lncRNAs
- ROC:
-
Receiver operating characteristic
- CI:
-
Confidence interval
- PD-1:
-
Programmed cell death 1
- PD-L1:
-
Programmed cell death ligand 1
- CTLA-4:
-
Cytotoxic T lymphocyte antigen-4
- IDO1:
-
Indoleamine 2,3-dioxygenase 1
- IDO2:
-
Indoleamine 2,3-dioxygenase 2
- HAVCR2:
-
Hepatitis A virus cellular receptor 2
- LAG3:
-
Lymphocyte activating gene 3
- TCA:
-
Tricarboxylic acid cycle
- ceRNA:
-
Competing endogenous RNA
- HNSCC:
-
Head and neck squamous cell carcinomas
- MSI-H:
-
Microsatellite instability-high
- dMMR:
-
Mismatch repair deficiency
- mCRC:
-
Metastatic colorectal carcinoma
References
Kanth P, Inadomi JM. Screening and prevention of colorectal cancer. BMJ (Clinic Res Ed). 2021;374: n1855.
Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin. 2022;72:7–33.
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49.
RL Siegel, KD Miller, A Goding Sauer, SA Fedewa, LF Butterly, JC Anderson, et al. Colorectal cancer statistics 2020. CA: Cancer J Clinic 2020;70:145–64.
Xia L, Oyang L, Lin J, Tan S, Han Y, Wu N, et al. The cancer metabolic reprogramming and immune response. Mol Cancer. 2021;20:28.
Luo X, Cheng C, Tan Z, Li N, Tang M, Yang L, et al. Emerging roles of lipid metabolism in cancer metastasis. Mol Cancer. 2017;16:76.
E Gharib, P Nasri Nasrabadi, M Reza Zali. miR-497-5p mediates starvation-induced death in colon cancer cells by targeting acyl-CoA synthetase-5 and modulation of lipid metabolism. J Cellular Physiol. 2020;235(7):5570–5589 https://doi.org/10.1002/jcp.29488.
Evans JR, Feng FY, Chinnaiyan AM. The bright side of dark matter: lncRNAs in cancer. J Clin Investig. 2016;126:2775–82.
Wu H, Liu B, Chen Z, Li G, Zhang Z. MSC-induced lncRNA HCP5 drove fatty acid oxidation through miR-3619-5p/AMPK/PGC1α/CEBPB axis to promote stemness and chemo-resistance of gastric cancer. Cell Death Dis. 2020;11:233.
Shang C, Wang W, Liao Y, Chen Y, Liu T, Du Q, et al. LNMICC promotes nodal metastasis of cervical cancer by reprogramming fatty acid metabolism. Can Res. 2018;78:877–90.
He W, Liang B, Wang C, Li S, Zhao Y, Huang Q, et al. MSC-regulated lncRNA MACC1-AS1 promotes stemness and chemoresistance through fatty acid oxidation in gastric cancer. Oncogene. 2019;38:4637–54.
Huang JZ, Chen M, Chen D, Gao XC, Zhu S, Huang H, et al. A peptide encoded by a putative lncRNA HOXB-AS3 suppresses colon cancer growth. Mol Cell. 2017;68:171-84.e6.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–50.
Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–73.
Kanehisa M, Furumichi M, Sato Y, Ishiguro-Watanabe M, Tanabe M. KEGG: integrating viruses and cellular organisms. Nucleic Acids Res. 2021;49:D545–51.
Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30.
Kanehisa M. Toward understanding the origin and evolution of cellular organisms. Protein Sci Publ Protein Soc. 2019;28:1947–51.
Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4:2612.
Thorsson V, Gibbs DL, Brown SD, Wolf D, DS Bortone,TH Ou Yang, et al. The immune landscape of cancer. Immunity. 2018;48:812-30.e14.
Mounir M, Lucchetta M, Silva TC, Olsen C, Bontempi G, Chen X, et al. New functionalities in the TCGAbiolinks package for the study and integration of cancer data from GDC and GTEx. PLoS Comput Biol. 2019;15: e1006701.
Cao Y. Adipocyte and lipid metabolism in cancer drug resistance. J Clin Investig. 2019;129:3006–17.
Biswas SK. Metabolic reprogramming of immune cells in cancer progression. Immunity. 2015;43:435–49.
Ma Y, Temkin SM, Hawkridge AM, Guo C, Wang W, Wang XY, et al. Fatty acid oxidation: an emerging facet of metabolic transformation in cancer. Cancer Lett. 2018;435:92–100.
Jiang P, Du W, Wu M. Regulation of the pentose phosphate pathway in cancer. Protein Cell. 2014;5:592–602.
Z Ghasemishahrestani, LM Melo Mattos, TM Tilli, A Santos, MD Pereira. Pieces of the complex puzzle of cancer cell energy metabolism: an overview of energy metabolism and alternatives for targeted cancer therapy. Curr Med Chem. 2021;28:3514–3534.
Christensen LL, True K, Hamilton MP, Nielsen MM, Damas ND, Damgaard CK, et al. SNHG16 is regulated by the Wnt pathway in colorectal cancer and affects genes involved in lipid metabolism. Mol Oncol. 2016;10:1266–82.
Peng Y, Xu C, Wen J, Zhang Y, Wang M, Liu X, et al. Fatty acid metabolism-related lncRNAs are potential biomarkers for predicting the overall survival of patients with colorectal cancer. Front Oncol. 2021;11: 704038.
Lv Y, Wei W, Huang Z, Chen Z, Fang Y, Pan L, et al. Long non-coding RNA expression profile can predict early recurrence in hepatocellular carcinoma after curative resection. Hepatol Res: Off J Japan Soc Hepatol. 2018;48:1140–8.
Xiong HG, Li H, Xiao Y, Yang QC, Yang LL, Chen L, et al. Long noncoding RNA MYOSLID promotes invasion and metastasis by modulating the partial epithelial-mesenchymal transition program in head and neck squamous cell carcinoma. J Experiment Clinic Cancer Res CR. 2019;38:278.
Yang S, Chen M, Lin C. A novel lncRNA MYOSLID/miR-1286/RAB13 axis plays a critical role in osteosarcoma progression. Cancer Manage Res. 2019;11:10345–51.
Igarashi Y, Sasada T. Cancer vaccines: toward the next breakthrough in cancer immunotherapy. J Immunol Res. 2020;2020:5825401.
RL Siegel, KD Miller, A Jemal. Cancer statistics 2020. CA: Cancer J Clinic. 2020;70(1):7–30.
Zimmermannova O, Caiado I, Ferreira AG, Pereira CF. Cell fate reprogramming in the Era of cancer immunotherapy. Front Immunol. 2021;12: 714822.
Fiegle E, Doleschel D, Koletnik S, Rix A, Weiskirchen R, Borkham-Kamphorst E, et al. Dual CTLA-4 and PD-L1 blockade inhibits tumor growth and liver metastasis in a highly aggressive orthotopic mouse model of colon cancer. Neoplasia. 2019;21:932–44.
HJ Lenz, E Van Cutsem, M Luisa Limon, KYM Wong, A Hendlisz, M Aglietta, et al. First-line nivolumab plus low-dose ipilimumab for microsatellite instability-high/mismatch repair-deficient metastatic colorectal cancer: the phase II checkmate 142 study. J Clinic Oncol Off J Am Soc Clinic Oncol. 2022;40:161–70.
Overman MJ, Lonardi S, Wong KYM, Lenz HJ, Gelsomino F, Aglietta M, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J Clin Oncol. 2018;36:773–9.
Lizardo DY, Kuang C, Hao S, Yu J, Huang Y, Zhang L. Immunotherapy efficacy on mismatch repair-deficient colorectal cancer: From bench to bedside. Biochim Biophys Acta. 2020;1874: 188447.
Noh BJ, Kwak JY, Eom DW. Immune classification for the PD-L1 expression and tumour-infiltrating lymphocytes in colorectal adenocarcinoma. BMC Cancer. 2020;20:58.
Li Y, Liang L, Dai W, Cai G, Xu Y, Li X, et al. Prognostic impact of programed cell death-1 (PD-1) and PD-ligand 1 (PD-L1) expression in cancer cells and tumor infiltrating lymphocytes in colorectal cancer. Mol Cancer. 2016;15:55.
Gutting T, Burgermeister E, Härtel N, Ebert MP. Checkpoints and beyond - Immunotherapy in colorectal cancer. Semin Cancer Biol. 2019;55:78–89.
Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017;18:1182–91.
Mei Z, Liu Y, Liu C, Cui A, Liang Z, Wang G, et al. Tumour-infiltrating inflammation and prognosis in colorectal cancer: systematic review and meta-analysis. Br J Cancer. 2014;110:1595–605.
Li M, Wang H, Li W, Peng Y, Xu F, Shang J, et al. Identification and validation of an immune prognostic signature in colorectal cancer. Int Immunopharmacol. 2020;88: 106868.
Kitsou M, Ayiomamitis GD, Zaravinos A. High expression of immune checkpoints is associated with the TIL load, mutation rate and patient survival in colorectal cancer. Int J Oncol. 2020;57:237–48.
Acknowledgements
We acknowledge TCGA and Molecular Signatures database for providing their platforms and contributors for uploading their meaningful datasets. We thank Bullet Edits Limited for the linguistic editing and proofreading of the manuscript.
Funding
This research was funded by the Fujian Province Natural Science Foundation [grant number 2021J01438] and the National Clinical Key Specialty Construction Program [grant number not applicable]. The funders had no role in the study design, data collection and analysis, data interpretation, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
YL and YX conceived and designed the study. YL and YX collected and analyzed the data. YL, SL, and LH wrote the manuscript. YL and YX prepared Figs. 1, 2, 3, 4, 5, 6, 7 and 8 and Additional file Figs. 1, 2, 3, 4, 5 and 6. YL, LS, and JW revised the manuscript. JW supervised the study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethics approval and informed consent of patients were not required in this study since TCGA and Molecular Signatures Database belong to public databases. The study was conducted in accordance with the declaration of Helsinki.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
Flow chart of the search protocol and study design. TCGA, The Cancer Genome Atlas; lncRNAs, long non-coding RNAs; LASSO, Least Absolute Shrinkage Selection Operator; GSEA, gene set enrichment analysis.
Additional file 2: Table S1.
Genes related to lipid metabolism.
Additional file 3: Figure S2.
Consensus clustering based on prognostic lipid metabolism-related lncRNAs. A Consensus clustering cumulative distribution function (CDF) for k = 2 to 9. B Relative changes in the areas under the CDF curve for k = 2 to 9. Consensus matrix similarity for C 2, D 3, E 4, and F 5 clusters.
Additional file 4: Figure S3.
Association of immune checkpoints with Clusters and prognostic risk groups. The expression of immune checkpoints A PD-1, B PD-L1, C CTLA-4, D IDO1, E IDO2, F HAVCR2, and G LAG3 in Clusters 1 and 2. The expressions of H PD-L1, I PD-1, J CTLA-4, K IDO1, L IDO2, M HAVCR2, and N LAG3 in different risk groups. PD-1, programmed cell death 1; PD-L1, programmed cell death ligand 1; CTLA-4, cytotoxic T lymphocyte antigen-4; IDO1, indoleamine 2,3-dioxygenase 1; IDO2, indoleamine 2,3-dioxygenase 2; HAVCR2, hepatitis A virus cellular receptor 2; LAG3, lymphocyte activating gene 3. ns, not significant; *p < 0.05, **p < 0.01, and ***p < 0.001.
Additional file 5: Table S2.
The enriched pathways of the Cluster 1 and 2.
Additional file 6: Figure S4.
Association of each lncRNA in the identified signature with the immune subtypes. The expression of lncRNA A NSMCE1-DT, B LINC02084, C MYOSLID, D LINC02428, and E MRPS9-AS1 in different immune subtypes. -, not significant, *p < 0.05, and ***p < 0.001.
Additional file 7: Figure S5.
Association of each lncRNA in the identified signature with the molecular subtypes. The expression of lncRNA A NSMCE1-DT, B LINC02084, C MYOSLID, D LINC02428, and E MRPS9-AS1 in different molecular subtypes. MSI, microsatellite instability; CIN, chromosomal instability; GS, genome-stable; HM-SNV, hypermutated single nucleotide variants. -, not significant; *p < 0.05, **p < 0.01, and ***p < 0.001.
Additional file 8: Figure S6.
Relationship among risk scores and clinical characteristics. Correlations of risk scores and A age, B sex, C T stage, D N stage, E metastatic status, F total stage, G immune score, and H Clusters. ns, not significant; *p < 0.05, **p < 0.01, and ***p < 0.001.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lin, Y., Xiao, Y., Liu, S. et al. Role of a lipid metabolism-related lncRNA signature in risk stratification and immune microenvironment for colon cancer. BMC Med Genomics 15, 221 (2022). https://doi.org/10.1186/s12920-022-01369-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-022-01369-8