
Abstract
Background
Hypophosphatemic rickets (HR) is a rare genetic disorder associated with renal phosphate wasting and characterized by bone defects. Inactivating mutations in the phosphate regulating endopeptidase homolog X‑linked gene (PHEX) account for most cases of HR. The aim of this study was to identify causative variants in nine unrelated Chinese families associated with HR, and to determine potential pathogenicity of the identified variants.
Methods
Genomic DNA was isolated from the peripheral blood of HR patients and their healthy relatives, followed by next-generation sequencing and/or Sanger sequencing. In silico prediction combined with conservation analysis was performed to assess the effects of the variants, and 3D protein modeling was conducted to predict the functional effects on the encoded protein.
Results
All HR patients recruited in this study displayed bone deformities and tooth agenesis, as well as reduced serum phosphate levels and elevated urine phosphate levels. Nine PHEX variants were identified in eight families, including four novel variants (c.1661_1726del, c.980A > G, c.1078A > T, and c.1017_1051dup). Of the nine identified PHEX variants, five caused a truncated protein, two caused an altered amino acid, and the other two were the canonical splicing variants. Novel variants c.1336G > A and c.1364 T > C in SLC34A3 were also found in one family. Conservation analysis showed that all the amino acids corresponding to the missense variants were highly conserved. In silico analysis and 3D protein structure modeling confirmed the pathogenicity of these variants.
Conclusions
This study identified four novel variants in PHEX and two novel variants in SLC34A3 in a Chinese cohort with HR. Our findings highlight the dominant role of PHEX in HR, and expand the genotypic and phenotypic spectra of this disorder.
Similar content being viewed by others

Background
Hypophosphatemic rickets (HR) is a group of metabolic bone diseases characterized by growth retardation, progressive bowing, abnormal bone and dentin formation, and short stature [1]. HR was first described as vitamin D-resistant rickets by Albright et al. in 1937, which differentiated it from general rickets caused by nutritional deficiency [2]. Although HR is not affected by the level of vitamin D, an excess of the hormone fibroblast growth factor 23 (FGF23) in circulation is believed to play a critical role in many HR cases. Current biochemical indicators of HR include elevated levels of FGF23 and serum alkaline phosphatase, as well as urinary phosphate excretion, and reduced levels of serum phosphate, and low to normal levels of serum calcium and serum 1,25(OH)2D [3, 4].
Up to now, there are 10 candidate genes contribute to congenital HR [3]. Most HR types are FGF23-dependent, including X-linked dominant HR (XLHR) (MIM 307800, caused by mutations in PHEX), autosomal dominant HR (ADHR) (MIM 193100, caused by mutations in FGF23), autosomal recessive HR (ARHR) type 1 (MIM 241520, caused by mutations in DMP1), and autosomal recessive HR (ARHR) type 2 (MIM 613312, caused by mutations in ENPP1). Meanwhile, FGF23-independent types of HR include HR with hypercalciuria (HHRH) (MIM 241530, caused by mutations in SLC34A3), HR with nephrolithiasis and osteoporosis type 1 (MIM 612286, caused by mutations in SLC34A1), HR with nephrolithiasis and osteoporosis type 2 (MIM 612287, caused by mutations in SLC9A3R1), HR with hyperparathyroidism (MIM 612089, caused by mutations in KLOTHO), X-linked HR (Dent syndrome) (MIM 300554, caused by mutations in CLCN5), and X-linked HR (Lowe syndrome) (MIM 309000, caused by mutations in OCRL1) [3, 5].
XLHR is the most common inherited HR type [6, 7] and accounts for more than 80% of all documented HR cases, with an incidence of 1 in 20,000 live births [8]. This disorder is caused by inactivating mutation in the PHEX gene. The PHEX gene, involved in the etiology of XLHR, encodes a membrane endopeptidase that is abundantly expressed in osteoblasts, odontoblasts, and chondrocytes [9, 10]. Levels of FGF23 is well known to be elevated in XLHR patients [3], as well as in a HR mouse model [11], and plays a key role in HR progression [12]. A study of Hyp mice, an animal model of XLHR caused by mutations in the PHEX gene [13], showed that FGF23 acted downstream of PHEX and that a PHEX mutation directly enhanced the secretion of FGF23 by osteoblasts, leading to reduced renal phosphate resorption and disrupted bone phenotypes [14]. Elevated FGF23 levels have been shown to inhibit the transcription of the genes SLC34A1 and SLC34A3, encoding NaPi-IIa and NaPi-IIc, respectively [15]. In addition, suppression of NaPi-IIa and NaPi-IIc results in impaired renal phosphate reabsorption, and thus an accelerated manifestation of the HR phenotype [16].
Although different types of HR share common clinical manifestations, they have distinct causative factors and follow different patterns of inheritance. Therefore, genetic analysis of HR can aid precise diagnosis and direct therapeutic interventions. Prompted by the availability of next-generation sequencing, molecular diagnosis of HR has been performed in many populations including Europeans [17], Turkish [18], Americans [19], Chinese [20,21,22], and Japanese [23]. To date, 623 mutations in PHEX have been found to contribute to the development of XLHR (HGMD Professional, release 2020.04). In the present study, we recruited 17 HR patients from nine Chinese families to further investigate the pathogenic genes. Nine pathogenic variants in PHEX, and two variants in SLC34A3 were identified, which included four novel variants in PHEX and two in SLC34A3. These findings expand the mutational spectra of PHEX and SLC34A3, and provide evidence of the dominant role of PHEX in HR development.
Materials and methods
Subjects
Nine unrelated Chinese families with HR were recruited in this study. In total, 56 individuals (27 male and 29 female) were included, 17 of whom had been diagnosed with HR (5 male and 12 female). Data on variables such as sex, age, height, weight, bone phenotypes, dental phenotypes, and available biochemical indicators were collected at their first visit. Height was converted to age- and sex-specific SDS Z-score based on the standard of the Chinese population [24]. Peripheral blood was collected from all probands and their available family members.
DNA/RNA isolation
Genomic DNA was isolated from peripheral blood using the conventional proteinase K‐phenol‐chloroform method [25]. Total RNA was obtained using Trizol reagent (Invitrogen, CA, USA) from peripheral blood, in accordance with the manufacturer’s instructions. Subsequently, cDNA was prepared from 1 µg of RNA using PrimeScript RT reagent kit with gDNA eraser (TaKaRa).
Sanger sequencing and genomic panel sequencing
Sanger sequencing was employed to screen the variants in PHEX. Primers designed for each exon are listed in Additional file 1: Table S1. DNA was amplified using a PCR system (TaKaRa). Sequencing was performed using Applied Biosystems 3739xl DNA analyser (Thermo Fisher Scientific, Waltham, MA, USA). For the probands with no variant identified in PHEX, customized panel sequencing including 184 skeleton-related genes (Additional file 1: Table S2) was conducted as previously described [25]. A total of 1–3 μg genomic DNA was used for panel sequencing. Briefly, DNA samples were sheared into 200 bp fragments using a Bioruptor NGS sonication device (Diagenode, Seraing, Belgium). After purification and library construction, hybridization reactions were performed, followed by sequencing performed on a HiSeq 2500 system (Illumina, San Diego, CA, USA). Sanger sequencing was used to verify the variants in the candidate genes. The sequencing results were analyzed using CodonCode Aligner (version 6.0.2.6; CodonCode, Centerville, MA, USA).
Reverse-transcription PCR (RT-PCR) and amplicon sequencing
RT-PCR was carried out to determine the effects of the variants at the mRNA level. Primers were designed (Additional file 1: Table S1) via the online tool Primer 3 (http://primer3.ut.ee/), followed by examination using in silico PCR (http://genome.ucsc.edu/cgi-bin/hgPcr). The PCR products were separated on 2% agarose gel, and separated bands were isolated and processed for amplicon sequencing.
Bioinformatic analysis
Conservation analysis was conducted via multiple alignments using Molecular Evolutionary Genetics Analysis version X (MEGA-X, https://www.megasoftware.net/). To predict the likelihood of mutation pathogenicity, Protein Variation Effect Analyzer version 1.1 (PROVEAN, http://provean.jcvi.org/seq_submit.php) and Polymorphism Phenotyping version 2 (PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/) were employed. Human Splicing Finder version 3.1 (http://www.umd.be/HSF3/) was used to predict the effects of intronic variants on splicing. Protein modeling of PHEX and the protein encoded by the PHEX variant c.1661_1726del (p.Glu554_Gly575del) was performed using the online tool Phyre2 (http://www.sbg.bio.ic.ac.uk) [26]. The 3D structure of the PHEX protein was visualized by EZMOL (http://www.sbg.bio.ic.ac.uk/ezmol/). 3D models of wild-type proteins and the mutant proteins c.980A > G(p.Tyr327Cys) and c.1735G > A(p.Gly579Arg) were constructed using Swiss-Model (http://swissmodel.expasy.org) and residue analysis was conducted by PyMOL (http://www.pymol.org). The template used for PHEX protein modeling was based on Neprilysin (NEP, PBD: 5JMY).
Results
Clinical manifestations
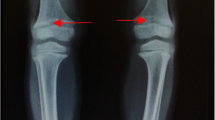
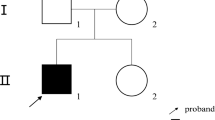
In this study, nine Chinese HR families were recruited, and the pedigrees are presented in Fig. 1A. The clinical symptoms are briefly summarized in Table 1. In general, poor dental development (Fig. 1C, E, F, I, M, P, T) and bone deformities (Fig. 1B, D, G, J, N, O, Q–S, U–X) were present in all HR patients within the recruited families. Notably, most HR patients were experiencing short stature (II-1 (family 1), I-2/II-2 (family 3), II-3 (family 5), III-9 (family 6), as well as II-1 (family 7)) and frequent fractures (family 2, 4 (Fig. 1K, L), 5, 6, 7 and 9). HR patients in family 1, 2, 3 (Fig. 1H), and 8 also displayed impaired physical mobility, and bone pain was reported in family 2, 3, and 4. In addition to these common HR characteristics, HR patients in family 5 also displayed knee valgus and scoliosis. The proband II-1 in family 9 showed kyphosis (Fig. 1Y, Z), as well as insensitivity to pain and temperature in the left arm.

Clinical information and genetic background of the nine HR families. A Pedigrees of nine Chinese families with HR. Black symbols indicate individuals with HR, while open symbols represent unaffected individuals. Arrows indicate probands and slashes indicate deceased individuals. Squares represent males and circles represent females. In Family 6, gray symbols indicate affected individuals with hereditary spastic paraplegia and black symbols indicate affected individuals with HR. B–Z Clinical manifestations of HR patients
Assessment of biochemical indicators in HR patients
Biochemical data were collected and are summarized in Table 2. It was found that the serum phosphate and serum creatinine levels were reduced in all HR patients, while β-CTX and urine phosphate levels were elevated, compared to healthy controls. Normal to high alkaline phosphatase (ALP) levels were found in patients’ serum, and very high ALP levels were detected in proband 6 (575.00 U/L), proband 8 (757.90 U/L), and proband 9 (555.00 U/L). Levels of parathyroid hormone (PTH), an important hormone that regulates calcium and phosphorus metabolism, varied between individuals, with elevated levels detected in the majority of the HR patients.
Identification of variants in PHEX and SLC34A3
Sanger sequencing of the exons and associated splicing sites, as well as both UTR regions of the PHEX gene revealed nine PHEX variants in family 1–8 (Table 1, Fig. 2). Among these nine PHEX variants, four were novel (c.1661_1726del, c.980A > G, c.1078A > T, and c.1017_1051dup), and five were previously reported in HGMD (c.591A > G, c.1735G > A, c.1079 + 1G > A, c.1965 + 1G > A, and c.1699C > T). All families carrying PHEX variants exhibited an X-linked dominant inheritance, with the exceptions of family 2, 4, 7, and 8, in which de novo variants were detected. In family 1, although variant c.591A > G (p.Gln197Gln) was supposed to be a synonymous variant, sequencing at the mRNA level identified the deletion of 77 bp at the 3′ region of exon 5 (Fig. 2A, B). mRNA sequencing was also conducted in family 2 (Fig. 2C). This confirmed that the variant c.1661_1726del did not lead to alternative splicing, but rather to the deletion of 66 bp spanning from 3′ of exon 16 to 5′ of exon 17 (Fig. 2C). A cis double variant c.[980A > G; 1078A > T] was found in family 3 (Fig. 2D), with both variants being inherited from the mother. In family 6, the proband (III-9) with HR (c.1017_1051dup in PHEX) married III-8 with hereditary spastic paraplegia harboring the variant c.715C > T(p.Arg239Cys) in ALT1. Their daughter (IV-1) inherited both variants and exhibited the phenotypes of both diseases.

Variant analysis of Families 1–9. A In proband 1, a synonymous variant c.591A > G(p.Gln197Gln) was identified in genomic DNA. B Sanger sequencing at the mRNA level indicated that part of exon 5 was deleted, caused by variant c.591A > G, leading to a frameshift inducing a premature stop codon (p.Gly196Alafs*16). C In proband 2, Sanger sequencing confirmed that c.1661_1726del did not induce alternative splicing. D–J Sanger sequencing results of probands 3–9
The pathogenic gene of family 9 was SLC34A3, and proband 9 was a heterozygote of variants c.1336G > A(p.Val446Ile) and c.1364T > C(p.Leu455Pro) (Table 1). The variant c.1336G > A was inherited from the father, while c.1364 T > C was inherited from the mother. The younger brother of the proband (II-2) inherited neither of these variants from the parents (Fig. 2J, Additional file 1: Fig. S2).
Bioinformatic analysis of the variants
It was confirmed that all point mutation sites, p.Y327C, p.K360*, and p.G579R in PHEX, and p.V446I and p.L455P in SLC34A3, were highly evolutionarily conserved (Fig. 3A). To predict the likelihood of pathogenicity, two online tools (PROVEAN and PolyPhen-2) were used and the predicted scores are summarized in Additional file 1: Table S3. In addition, the splice site variants were predicted as “most probably affecting splicing” by Human Splicing Finder (version 3.1) (Additional file 1: Table S3). The 3D structure of the PHEX protein was modeled by Phyre2 for the deletion c.1661_1726del in PHEX. In comparison to the wild-type PHEX protein, the mutant PHEX protein lacked an α-helix (5′ of exon 17) and a β-sheet (3′ of exon 16) (Fig. 3B), which altered its 3D conformation. The 3D structure of the PHEX protein was also modeled by Swiss-Model and the functional consequences of the missense variants c.980A > G(p.Tyr327Cys) and c.1735G > A(p.Gly579Arg) were simulated by Pymol (Fig. 3B). This analysis indicated altered interactions of the substituted amino acids with their neighboring residues.

Bioinformatic analysis of the variants. A Evolutionary conservation of amino acids in the protein products of PHEX and SLC34A3. Variant sites are indicated by black arrows. B Comparison of the 3D structures of mutant proteins with the wild type. The 3D structures of the protein products of c.1661_1726del(p.Glu554_Gly575del) (left), missense variant c.980A > G(p.Tyr327Cys) (middle), and missense variant c.1735G > A(p.Gly579Arg) (right) are compared with the corresponding wild type
Discussion
To date, 623 variants have been identified in PHEX, including 215 missense/nonsense mutations (34%), 104 splicing substitutions (17%), 4 regulatory substitutions (1%), 128 small deletions (20%), 76 small insertions/duplications (12%), 13 small indels (2%), 66 gross deletions (11%), 12 gross insertions/duplications (2%), and 5 complex rearrangements (1%) (Fig. 4B). Point mutations were predominant, constituting 51% of all mutations, followed by small copy number variations (34%). Here, the pathogenic mutations in each exon across the PHEX gene identified to date have been summarized (Fig. 4A, C), and it was confirmed that the mutation spectrum spanned the whole gene without any hotspot regions [17, 27, 28].

Genotypic characterization of PHEX mutations. A Mutation spectrum of PHEX. Each numbered box represents a corresponding exon in PHEX. Numbers of missense/nonsense mutations (blue), splicing substitutions (yellow), small deletions (red), and small insertions (green) are presented above the 22 exons. The novel variants found in this study are listed below in pink, while previously reported variants found in this study are listed below in brown. B Distribution of mutations in PHEX (HGMD Professional, release 2020.04). C Distribution of mutations in each exon of PHEX (HGMD Professional, release 2020.04)
Common consequences of gene mutations are dysfunction of protein, frameshift, truncated protein, protein redundancy, and lack of protein product resulting from mRNA degradation. It has been reported that most cases of XLHR induced by PHEX mutations are caused by the generation of a truncated protein or by the translation of a dysfunctional PHEX product [29], and this was further confirmed in the current study. (i) It has previously been reported that about 85% of PHEX mutations result in a truncated PHEX protein [22]. Three novel variants identified in this study, c.1661_1726del(p.Glu554_Gly575del), c.1078A > T(p.Lys360*), and c.1017_1051dup(p.Phe351Trpfs*16), as well as two previously reported variants c.591A > G(p.Gly196Alafs*16) and c.1699C > T(p.Arg567*), resulted in a truncated PHEX protein and the characteristic HR phenotype. The synonymous variant c.591A > G(p.Gln197Gln) resulted in a frameshift-induced premature termination codon (p.Gly196Alafs*16) (Fig. 2A, B), which was caused by the recognition of an alternative splice donor site “GT” located 3 bp upstream of the mutant site in exon 5. This is in line with findings reported by Liao et al. [30]. (ii) Some PHEX mutations, such as missense mutations and a few splicing mutations, resulted in dysfunction of the PHEX protein. Most of the missense mutations found in PHEX, although located at different sites, affect post-translational modification of the protein, thus leading to loss of protein function [27]. For example, the reported missense mutation c.1735G > A(p.Gly579Arg) identified in family 4 [31, 32] was located at the extracellular C-terminal region, which could affect the secondary structure of the PHEX protein by disrupting disulfide bonds [31]. The predicted 3D structure of the PHEX protein (Fig. 3B) also indicated that Gly579Arg, a change involving substitution of the short-side-chain glycine for the long-side-chain arginase, would possibly push away the interacting α-helix, having a negative effect on protein function. Compared with the severely impacted physical mobility reported in a Turkish family with the same variant [31], the clinical symptoms of the HR patients in this study were limited to mild bone deformities.
The proband in family 3 carried a missense variant c.980A > G(p.Tyr327Cys) and a nonsense variant c.1078A > T(p.Lys360*) (Fig. 2D), both located on the same allele and inherited from his mother (Additional file 1: Fig. S1C). It has previously been shown that a truncated protein generated from a nonsense mutation can cause abnormal bone mineralization and hypophosphatemia [33]. The above-mentioned nonsense variant was therefore predicted to be pathogenic, which was supported by in silico tools (Additional file 1: Table S3). Meanwhile, for the novel missense variant c.980A > G also located in exon 9, there were conflicting results on its pathogenicity (Additional file 1: Table S3). According to Gaucher et al. [17], missense mutations at highly conserved sites encoding residues in the interior of the PHEX protein could alter protein folding and trafficking. The missense variant c.980A > G(p.Tyr327Cys) resulted in an exchanged of tyrosine with cysteine, suggesting a potential role in disulfide bond formation and protein folding [31, 34]. In line with this, the predicted 3D protein structure and residue interactions (Fig. 3B) suggested that the wild-type tyrosine closely interacted with glutamine 515 at the neighboring α-helix via a hydrogen bond. However, the mutant cysteine could not form this hydrogen bond and did not interact with the α-helix, potentially changing the stability of the protein. The above findings show that the nonsense variant in family 3 contribute to the development of HR, and indicate a potential role also for the missense variant.
No genotypic and phenotypic correlation was found in this study, which is in line with previous findings [22, 35]. Nevertheless, the reported variants were concentrated in exons 18–22 (Fig. 4C), indicating that this extracellular C-terminal region is essential for PHEX protein function [36]. It has been reported that patients with variants at the C-terminal region or truncated proteins exhibited more severe HR phenotypes [35], and variants leading to truncated PHEX proteins generally corresponded to decreased tubular reabsorption of phosphate and 1,25(OH)2D [37]. Out of the four novel PHEX variants found in the present study, the first two (located in exon 9) resulted in a truncated PHEX product, the third (located in exon 9) resulted in amino acid alteration, and the fourth (located in exon 17) resulted in a truncated protein. Zhang et al. reported that the missense variants were clustered in exons 15, 17, 19, and 20 in PHEX [22], while the missense variant identified in this study was located in exon 9. This identifies exon 9 as an important novel player in the progression of HR.
The HR patients included in this study had a significantly shorter stature (ranging from − 4.26 to − 11.10 SD) than previously reported HR patients in Chinese cohort (− 2.70 ± 1.60 SD) [22]. Possible reasons for this are chronical nutritional deficiencies and extended periods of untreated disease progression due to patients’ living in remote rural areas with limited access to medical facilities. Noteworthy is that HR patients with a more severe phenotype often had a family history. As the majority of the HR patients recruited in this study also had suffered frequent fractures (Table 1), which can be misdiagnosed as osteogenesis imperfecta, clinical examinations combined with molecular testing are crucial for a precise diagnosis.
In this study, one compound heterozygous variant was found in SLC34A3 in family 9 (Fig. 2J, Table 1). SLC34A3 is expressed in tubule cells and is responsible for the maintenance of phosphate homeostasis [38]. In previous studies, phenotypic heterogeneity was found in patients with SLC34A3 variants: approximately 25% of HHRH patients did not exhibit rickets and half lacked renal calcification [39]. It was also shown that heterozygous carriers presented milder skeletal phenotypes than homozygous ones [40]. Typical HR symptoms were noted in the patient with SLC34A3 variants in this study, while the heterozygous carriers (I-1 and I-2) barely showed any noticeable phenotypes. A study with a large sample size is needed to obtain a comprehensive understanding of the genotype/phenotype correlation in HHRH.
Conclusion
In summary, we identified nine pathogenic variants in PHEX and one compound heterozygous variant in SLC34A3 in nine Chinese families with HR. These variants included four novel variants in PHEX and two novel variants in SLC34A3. The current study confirms the various variant types associated with HR and provides new insight that expands our understanding of the function of PHEX.
References
Amatschek S, Haller M, Oberbauer R. Renal phosphate handling in human—what can we learn from hereditary hypophosphataemias? Eur J Clin Investig. 2010;40(6):552–60.
Albright F, Butler AM, Bloomberg E. Rickets resistant to vitamin D therapy. Am J Dis Child. 1937;54(3):529–47.
Michałus I, Rusińska A. Rare, genetically conditioned forms of rickets: differential diagnosis and advances in diagnostics and treatment. Clin Genet. 2018;94(1):103–14.
Yuan L, Wu S, Xu H, Xiao J, Yang Z, Xia H, et al. Identification of a novel PHEX mutation in a Chinese family with X-linked hypophosphatemic rickets using exome sequencing. Biol Chem. 2015;396(1):27–33.
BinEssa HA, Zou M, Al-Enezi AF, Alomrani B, Al-Faham MS, Al-Rijjal RA, et al. Functional analysis of 22 splice-site mutations in the phex, the causative gene in x-linked dominant hypophosphatemic rickets. Bone. 2019;125:186–93.
Acar S, Demir K, Shi Y. Genetic causes of rickets. J Clin Res Pediatr Endocrinol. 2017;9(Suppl 2):88–105.
Ma SL, Vega-Warner V, Gillies C, Sampson MG, Kher V, Sethi SK, et al. Whole exome sequencing reveals novel PHEX splice site mutations in patients with hypophosphatemic rickets. PLoS ONE. 2015;10(6): e0130729.
Baroncelli GI, Toschi B, Bertelloni S. Hypophosphatemic rickets. Curr Opin Endocrinol Diabetes Obes. 2012;19(6):460–7.
Liu S, Guo R, Quarles LD. Cloning and characterization of the proximal murine Phex promoter. Endocrinology. 2001;142(9):3987–95.
Rowe PS. Regulation of bone-renal mineral and energy metabolism: the PHEX, FGF23, DMP1, MEPE ASARM Pathway. Crit Rev Eukaryot Gene Expr. 2012;22(1):61–86.
Liu S, Guo R, Simpson LG, Xiao Z-S, Burnham CE, Quarles LD. Regulation of fibroblastic growth factor 23 expression but not degradation by PHEX. J Biol Chem. 2003;278(39):37419–26.
Larsson T, Marsell R, Schipani E, Ohlsson C, Ljunggren O, Tenenhouse HS, et al. Transgenic mice expressing fibroblast growth factor 23 under the control of the α1 (I) collagen promoter exhibit growth retardation, osteomalacia, and disturbed phosphate homeostasis. Endocrinology. 2004;145(7):3087–94.
Nesbitt T, Fujiwara I, Thomas R, Xiao ZS, Quarles LD, Drezner MK. Coordinated maturational regulation of PHEX and renal phosphate transport inhibitory activity: evidence for the pathophysiological role of PHEX in X-linked hypophosphatemia. J Bone Miner Res. 1999;14(12):2027–35.
Liu S, Zhou J, Tang W, Jiang X, Rowe DW, Quarles LD. Pathogenic role of Fgf23 in Hyp mice. Am J Physiol Endocrinol Metab. 2006;291(1):E38-49.
Tenenhouse HS, Beck L. Renal Na(+)-phosphate cotransporter gene expression in X-linked Hyp and Gy mice. Kidney Int. 1996;49(4):1027–32.
Carpenter TO, Imel EA, Holm IA, Jan de Beur SM, Insogna KL. A clinician’s guide to X-linked hypophosphatemia. J Bone Miner Res. 2011;26(7):1381–8.
Gaucher C, Walrant-Debray O, Nguyen T-M, Esterle L, Garabédian M, Jehan F. PHEX analysis in 118 pedigrees reveals new genetic clues in hypophosphatemic rickets. Hum Genet. 2009;125(4):401–11.
Guven A, Al-Rijjal RA, BinEssa HA, Dogan D, Kor Y, Zou M, et al. Mutational analysis of PHEX, FGF23 and CLCN5 in patients with hypophosphataemic rickets. Clin Endocrinol. 2017;87(1):103–12.
Mumm S, Huskey M, Cajic A, Wollberg V, Zhang F, Madson KL, et al. PHEX 3′-UTR c.* 231A> G Near The Polyadenylation Signal Is A Relatively Common, Mild, American Mutation That Masquerades As Sporadic or X-linked recessive hypophosphatemic rickets. J Bone Miner Res. 2015;30(1):137–43.
Kang Q, Xu J, Zhang Z, He J, Lu L, Fu W, et al. Three novel PHEX gene mutations in four Chinese families with X-linked dominant hypophosphatemic rickets. Biochem Biophys Res Commun. 2012;423(4):793–8.
Gu J, Wang C, Zhang H, Yue H, Hu W, He J, et al. Targeted resequencing of phosphorus metabolism-related genes in 86 patients with hypophosphatemic rickets/osteomalacia. Int J Mol Med. 2018;42(3):1603–14.
Zhang C, Zhao Z, Sun Y, Xu L, JiaJue R, Cui L, et al. Clinical and genetic analysis in a large Chinese cohort of patients with X-linked hypophosphatemia. Bone. 2019;121:212–20.
Kawahara T, Watanabe H, Omae R, Yamamoto T, Inazu T. A novel PHEX mutation in Japanese patients with X-linked hypophosphatemic rickets. Case Rep Genet. 2015;2015(2015): 301264.
Li H, Ji C, Zong X, Zhang Y. Height and weight standardized growth charts for Chinese children and adolescents aged 0 to 18 years. Zhonghua Er Ke Za Zhi. 2009;47(07):487–92.
Li L, Mao B, Li S, Xiao J, Wang H, Zhang J, et al. Genotypic and phenotypic characterization of Chinese patients with osteogenesis imperfecta. Hum Mutat. 2019;40(5):588–600.
Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10(6):845–58.
Sabbagh Y, Boileau G, Campos M, Carmona AK, Tenenhouse HS. Structure and function of disease-causing missense mutations in the PHEX gene. J Clin Endocrinol Metab. 2003;88(5):2213–22.
Li S-S, Gu J-M, Yu W-J, He J-W, Fu W-Z, Zhang Z-L. Seven novel and six de novo PHEX gene mutations in patients with hypophosphatemic rickets. Int J Mol Med. 2016;38(6):1703–14.
Dixon PH, Christie PT, Wooding C, Trump D, Grieff M, Holm I, et al. Mutational analysis of PHEX gene in X-linked hypophosphatemia. J Clin Endocrinol Metab. 1998;83(10):3615–23.
Liao H, Zhu HM, Liu HQ, Li LP, Liu SL, Wang H. Two novel variants of the PHEX gene in patients with X-linked dominant hypophosphatemic rickets and prenatal diagnosis for fetuses in these families. Int J Mol Med. 2018;41(4):2012–20.
Durmaz E, Zou M, Al-Rijjal RA, Baitei EY, Hammami S, Bircan İ, et al. Novel and de novo PHEX mutations in patients with hypophosphatemic rickets. Bone. 2013;52(1):286–91.
Lin X, Zhu Y, Luo J, Huang J. Genetic analysis of three families with X-linked dominant hypophosphatemic rickets. J Pediatr Endocrinol Metab. 2018;31(7):789–97.
Huang Y, Mei L, Pan Q, Tan H, Quan Y, Gui B, et al. Novel de novo nonsense mutation of the PHEX gene (p. Lys50Ter) in a Chinese patient with hypophosphatemic rickets. Gene. 2015;565(1):150–4.
Tyynismaa H, Kaitila I, Näntö-Salonen K, Ala-Houhala M, Alitalo T. Identification of fifteen novel PHEX gene mutations in Finnish patients with hypophosphatemic rickets. Hum Mutat. 2000;15(4):383–4.
Holm IA, Nelson AE, Robinson BG, Mason RS, Marsh DJ, Cowell CT, et al. Mutational analysis and genotype-phenotype correlation of the PHEX gene in X-linked hypophosphatemic rickets. J Clin Endocrinol Metab. 2001;86(8):3889–99.
Filisetti D, Ostermann G, von Bredow M, Strom T, Filler G, Ehrich J, et al. Non-random distribution of mutations in the PHEX gene, and under-detected missense mutations at non-conserved residues. Eur J Hum Genet. 1999;7(5):615–9.
Morey M, Castro-Feijóo L, Barreiro J, Cabanas P, Pombo M, Gil M, et al. Genetic diagnosis of X-linked dominant hypophosphatemic rickets in a cohort study: tubular reabsorption of phosphate and 1, 25 (OH) 2 D serum levels are associated with PHEX mutation type. BMC Med Genet. 2011;12(1):116.
Lorenz-Depiereux B, Benet-Pages A, Eckstein G, Tenenbaum-Rakover Y, Wagenstaller J, Tiosano D, et al. Hereditary hypophosphatemic rickets with hypercalciuria is caused by mutations in the sodium-phosphate cotransporter gene SLC34A3. Am J Hum Genet. 2006;78(2):193–201.
Dasgupta D, Wee MJ, Reyes M, Li Y, Simm PJ, Sharma A, et al. Mutations in SLC34A3/NPT2c are associated with kidney stones and nephrocalcinosis. J Am Soc Nephrol. 2014;25(10):2366–75.
Hasani-Ranjbar S, Ejtahed H-S, Amoli MM, Bitarafan F, Qorbani M, Soltani A, et al. SLC34A3 intronic deletion in an Iranian kindred with hereditary hypophosphatemic rickets with hypercalciuria. J Clin Res Pediatr Endocrinol. 2018;10(4):343–9.
Guo X, Chen F, Gao F, Li L, Liu K, You L, et al. CNSA: a data repository for archiving omics data. Database (Oxford). 2020;2020:baaa055.
Chen FZ, You LJ, Yang F, Wang LN, Guo XQ, Gao F, et al. CNGBdb: China National GeneBank DataBase. Yi Chuan. 2020;42(8):799–809.
Acknowledgements
The authors are grateful to Dr. Sara Sprangers for her critical reading on the manuscript. We also thank Dr. Pengqi Xu for his help on protein analysis. We thank all of the HR patients and their families for their participation in this study.
Funding
This study was funded by grants from the National Key Research and Development Program of China (2016YFE0128400) and CAMS Innovation Fund for Medical Sciences (CIFMS, 2021‐I2M‐1‐051, 2016-I2M-3-003).
Author information
Authors and Affiliations
Contributions
YC, YY, and QW performed the experiment and prepared the manuscript. XR provided the samples from Family 3. SL performed 3D structure analysis of the proteins. LL and WX collected samples from the patients. XG contributed to the experimental work. TY, SI, and ZW were responsible for data analysis. XZ designed and supervised this research. All authors performed critical reading of the manuscript and approved its final version. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Institutional Review Board (IRB) of the Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences, Beijing, China (015–2015). Written informed consent to participate was obtained from the participants themselves or their guardians. All methods were performed in accordance with the Declaration of Helsinki.
Consent for publication
Written informed consent for the publication of this manuscript was obtained from all participants and the legal guardians of children aged under 18.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Supplementary materials.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Cao, Y., You, Y., Wang, Q. et al. Identification of six novel variants from nine Chinese families with hypophosphatemic rickets. BMC Med Genomics 15, 161 (2022). https://doi.org/10.1186/s12920-022-01305-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-022-01305-w