
Abstract
Background
Mutations in the telomerase reverse transcriptase (TERT) promoter region have been proposed as novel mechanisms for the transcriptional activation of telomerase. Two recurrent mutations in the TERT promoter, C228T and C250T, are prognostic biomarkers. Herein, we directly compared the commercially available iTERT PCR kit with NGS-based deep sequencing to validate the NGS results and determine the analytical sensitivity of the PCR kit.
Methods
Of the 2032 advanced solid tumors diagnosed using the TruSight Oncology 500 NGS test, mutations in the TERT promoter region were detected in 103 cases, with 79 cases of C228T, 22 cases of C250T, and 2 cases of C228A hotspot mutations. TERT promoter mutations were detected from 31 urinary bladder, 19 pancreato-biliary, 22 hepatic, 12 malignant melanoma, and 12 other tumor samples.
Results
In all 103 TERT-mutated cases detected using NGS, the same DNA samples were also tested with the iTERT PCR/Sanger sequencing. PCR successfully verified the presence of the same mutations in all cases with 100% agreement. The average read depth of the TERT promoter region was 320.4, which was significantly lower than that of the other genes (mean, 743.5). Interestingly, NGS read depth was significantly higher at C250 compared to C228 (p < 0.001).
Conclusions
The NGS test results were validated by a PCR test and iTERT PCR/Sanger sequencing is sensitive for the identification of the TERT promoter mutations.
Similar content being viewed by others


Background
Mutations in the telomerase reverse transcriptase (TERT) promoter region are frequently observed in specific types of human cancers, leading to enhanced expression of telomerase. Genome-wide association studies have identified multiple variants at the TERT locus, which are associated with the lengths of telomeres and risk of several cancers [1, 2] strongly suggesting that this locus is a common susceptibility locus for many human cancers. The most remarkable advancement in improving our understanding of the genetic role of TERT in human cancer was the landmark finding of mutations in the promoter region of the TERT gene in melanoma using whole-genome sequencing [3, 4]. These mutations have also been reported in other human cancers, such as bladder cancer and glioblastoma [5, 6]. In human cancers, there are two common recurrent mutations in the TERT promoter region, which are located at two hotspots: chr5, 1,295,228 (GRCh37/hg19 by Entrez Gene) C>T (C228T) and 1,295,250 (GRCh37/hg19 by Entrez Gene) C>T (C250T), corresponding to the positions 124 and 146 bp upstream of the TERT translation start site, respectively [7]. Transcriptional activation of TERT via mutation in the promoter region or other mechanisms limits the production of active telomerase in many human cancers [8]. The prognostic power of the TERT promoter mutation highlights its potential use as an important biomarker to predict the aggressive clinical behavior in melanoma, glioma, medulloblastoma, bladder cancer, thyroid cancer, urogenital cancer, and laryngeal cancer [9,10,11]. TERT promoter mutation is associated with worse prognosis in melanoma, glioma, meningioma, thyroid cancer, and bladder cancer [12,13,14,15,16,17,18] and is also associated with a high risk of malignant transformation and progression to advanced stages in hepatocellular carcinoma [19, 20].
TERT promoter mutations in clinical samples are diagnosed using Sanger sequencing and next-generation sequencing (NGS) [21,22,23]. Recent advancements in DNA isolation and NGS methods have facilitated the sensitive detection of TERT mutations in the formalin-fixed, paraffin-embedded (FFPE) tumor tissues. Although only a small percentage (~ 3%) of human DNA is GC rich, the promoter region consists of GC-rich cis-elements [24]. Similarly, the TERT promoter region is rich in GC (> 80%), making the DNA of the affected patients less amenable to amplification. Given that the amplification of templates with GC-rich regions is more difficult than those with non-GC-rich regions using the polymerase chain reaction (PCR) [25, 26] and NGS also shows a very low read depth in this region compared to others [27], we attempted to validate the TERT promoter mutations detected by NGS with a combination of conventional PCR and Sanger sequencing methods. For this purpose, we used a commercially available iTERT PCR kit to detect the mutations at the two hotspots in the TERT promoter region using 103 NGS-verified cases.
Methods
Patients samples
In this study, we used a total of 103 cases diagnosed with TERT promoter mutations at the C228T and C250T hotspots using the TruSight Oncology (TSO) 500 NGS test in the Department of Pathology and Translation Genomics of Samsung Medical Center between November 2019 and March 2021. To obtain the negative predictive value (NPV), we added 100 TERT wild type cancers from colon (n = 34), urinary tract (n = 1), melanoma (n = 4), liver (n = 2), pancreatobiliary tract (n = 17), soft tissue (n = 14), and stomach (n = 28). This study was performed in accordance with the Institutional Review Board guidelines of Samsung Medical Center (IRB 2020-06-045-001) for data analysis and investigational treatments. All patients provided informed consent to participate in this study.
DNA extraction
Tumors were micro-dissected from most of the samples, except for small samples that were used for the extraction of genomic DNA. Genomic DNA was isolated from the FFPE tissue sections (generally measuring 6–10 mm) and purified using the AllPrep DNA/RNA FFPE Kit (Qiagen, Venlo, Netherlands) [28]. The Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA) was used for DNA concentration determination and 120 ng of input DNA was used for library preparation following modification of the manufacturer’s instructions [29]. The DNA integrity number, which is a measure of the size of the DNA fragments and consequently the quality of the DNA, was determined using the Genomic DNA ScreenTape (Agilent Technologies, Santa Clara, CA) on an Agilent 2200 TapeStation system (Agilent Technologies).
Library preparation, sequencing, and data analysis
A library was prepared using a hybrid capture-based TSO 500 gene library preparation kit (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions. Briefly, the DNA was fragmented using Covaris S2 (Covaris, Woburn, MA, USA) to generate DNA fragments of 90–250 bp, with a target peak of approximately 180 bp. Next, the samples underwent end repair and A-tailing before unique molecular identifier ligation. Then, amplification was performed to add the index sequences for sample multiplexing. Two hybridization/capture steps were performed. Finally, the libraries were pooled, denatured, and diluted to the appropriate loading concentrations. The sequenced data were then analyzed to identify the clinically relevant classes of genomic alterations, including the single nucleotide variants (SNVs), copy number variants, small insertions and deletions (indels), and rearrangements/fusions. In the TSO 500 analysis, unique molecular identifiers determined the unique coverage at each position and reduced the background noise caused by sequencing and deamination artifacts in the FFPE samples. Results of SNVs and small indels with a variant allele frequency (VAF) of less than 2% were eliminated. Data outputs exported from the TSO 500 pipeline (Illumina) [30] were annotated using the Ensembl Variant Effect Predictor (VEP) annotation engine [30], with information from several databases, such as the Single Nucleotide Polymorphism Database (dbSNP), Genome Aggregation Database (gnomAD; genome and exome sequencing), 1000 genomes project database, ClinVar database, Catalogue Of Somatic Mutations In Cancer (COSMIC) database, Reference Sequence (RefSeq) database, and Ensembl and alignment to the hg19 human reference genome GRCh37 version (http://genome.ucsc.edu/). Mutation allele frequencies below predefined thresholds were considered to be wild-type.
iTERT PCR and Sanger sequencing
PCR was performed using an iTERT Mutation Detection Kit (GENINUS Inc., Seoul, Korea), according to the manufacturer's instructions. The PCR reactions were assembled on ice and preincubated at 94 °C for 15 min, followed by 40 cycles at 94 °C for 20 s, 58 °C for 40 s, 72 °C for 1 min, and a final extension at 72 °C for 5 min using a C1000 Touch Thermal Cycler Kit (Bio-Rad, Hercules, CA). Bidirectional sequencing was performed using the BigDye Terminator v.3.1 Kit (Applied Biosystems, Foster City, CA, USA) on an ABI 3130xL Genetic Analyzer. The results were marked as mutation-positive if a mutation was detected in both the forward and reverse DNA strands [31]. Positive controls were included in each sequencing run: normal human guide DNA (gDNA) (wild-type) and cancer cell (e.g., the C228T‐positive MDA-MG-231 cell line)-derived genomic DNA that yielded the expected TERT promoter sequences in each case.
Statistical analysis
Statistical analyses were performed using GraphPad Prism v.8.0 (GraphPad Software, CA, USA). Visualization of the genetic alterations was conducted using the R-package. All statistical analyses were performed using the SPSS software v.24.0 (IBM Corp., Armonk, NY). The general characteristics and demographic parameters were compared using Fisher's exact test and other quantitative data were analyzed using paired t-tests.
Results
NGS with TSO 500
TERT promoter mutations were detected in 103 (5.1%) out of 2032 cases and consisted of 79 (77%) C228T, 22 (21%) C250T, and 2 (2%) C228A mutations. Of these 103 cases, the TERT promoter mutations were detected in urinary bladder tumor (31/47, 66%), pancreato-biliary (19/127, 15%), hepatocellular carcinoma (22/41, 54%), and malignant melanoma (12/90, 13%). The tumor mutation burden was found to be high in 25 cases with the TERT promoter mutations. The precise characteristics of the tumors with TERT promoter mutations are shown in Table 1.
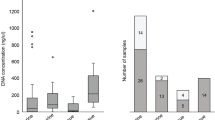
With NGS tests, the average sequencing read depth was 300, which was higher than the depth requirements (≥ 150). The average read depth of the TERT promoter region was 320.4 (range, 31–1223; median, 254), which was significantly lower than that of the other genes (mean, 743.5; range, 238–1416; median, 757) (p < 0.001) (Fig. 1a–c). The average sequencing read depth of the TERT promoter regions at C228 and C250 were 261.73 ± 19.13 (range, 31–779; median, 236.0) and 536.41 ± 66.73 (range, 69–1223; median, 468.50), respectively. Interestingly, NGS read depth was higher at C250 compared to C228 with statistical significance (p < 0.001) (Fig. 1d). The average VAFs in the C228T and C250T mutations of the TERT promoter region were 31.8% (range, 7.7–70.9%) and 32.0% (range, 8.7–85.1%), respectively.

The average depth of sequencing coverage in the telomerase reverse transcriptase (TERT) promoter region a and other genes b. There was a statistically significant decrease in the sequencing read depth in the TERT promoter region than the other genes c. Sequencing read depth was significantly higher at C250 compared to C228 d
iTERT PCR and Sanger sequencing
In 103 cases harboring the TERT promoter mutations, iTERT PCR and Sanger sequencing were performed using the same DNA left over after the NGS test. The iTERT PCR test showed 100% sensitivity and specificity for the detection of TERT promoter mutations and achieved 100% positive predictive value (PPV) and NPV. The peak heights of the wild-type and mutant alleles detected by Sanger sequencing varied and correlated very well with the VAFs detected using NGS (Fig. 2). Although the mean read depths were relatively smaller in the TERT promoter region than in the other regions, we found that the peak heights of mutant alleles in Sanger sequencing correlated well with the VAFs, suggesting that read depths have very little effects on the detection of TERT promoter mutations. In addition to the validation of NGS results with Sanger sequencing in the TERT promoter region, we also established the efficacy of the iTERT PCR kit.

Results of iTERT polymerase chain reaction and Sanger sequencing in the representative cases. According to the variant allele frequencies of the TERT promoter mutation, there was good correlation among the heights of the mutant peaks
Discussion
Two hotspot mutations, C228T and C250T, in the TERT promoter region have been proposed as novel mechanisms for the activation of telomerase in malignant cells, and act as important biomarkers for predicting aggressive clinical behavior in various types of cancer [9]. However, the GC-rich sequences within the TERT promoter region make their DNA less amenable to PCR amplification. In the present study, we used the commercially available iTERT PCR kit to simultaneously validate the NGS results and explore the analytical sensitivity of the PCR kit. In 103 samples diagnosed with hotspot mutations in the TERT promoter region using NGS tests, the same DNA was also tested with the iTERT PCR kit, which verified the presence of the same mutations with 100% agreement. Although the read depth of the TERT promoter region was smaller than that of other genes, the peak heights of mutant alleles in Sanger sequencing correlated with the VAFs of the NGS test, suggesting that the read depth has little impact on the detection of TERT promoter mutations.
Telomeres are composed of "TTAGGG" repeats at the end of chromosomes and the telomere length plays a critical role in multiple human diseases, including cancer [9]. The TERT promoter mutations were found to be the most common point mutations in several types of cancer, including 60–100% of glioblastoma [5, 10, 32, 33], 22–71% of melanoma [4, 15, 34], 29–100% of bladder cancer [3, 35,36,37,38], and 29–65% of hepatocellular carcinoma [39,40,41] cases (Table 2). To date, the C228T and C250T hotspot mutations have been identified in over 50 distinct types of cancer, and they are responsible for the activation of the TERT promoter region and TERT gene transcription [3, 4].
Interestingly, we found that NGS read depth was higher at C250 compared to C228 with statistical significance although GC contents around C228 and C250 were similar (76.9% and 78.3%) and the exact molecular mechanism underlying our results are unknown. TERT promoter mutations, C228T and C250T, were heterozygous and mutually exclusive, but both mutations result in the generation of an 11-bp identical sequence, 5′-CCCCTTCCGGG-3′. Although low read depth of C228T TERT promoter mutation, we confirmed same Sanger sequencing results.
In the present study, we detected the TERT promoter mutations in 5.1% of all tested cases by NGS and the majority of these mutations were C228T and C250T. We also identified two C228A mutations from colon cancer samples. The TERT promoter mutations were mainly detected in urinary bladder cancer (66%), hepatocellular carcinoma (54%), pancreato-biliary cancer (15%), and malignant melanoma (13%), and the overall incidence was similar to that reported previously [3, 6,7,8, 35, 42]. As most of the patients whose samples were used for NGS exhibited advanced stages of the disease with aggressive tumor behavior [7], we did not compare the prognostic differences between the patients with and without TERT promoter mutations in the present study. The clinicopathological characteristics of the TERT promoter mutations in brain [27] and thyroid tumors [43] have been previously reported by researchers at our institute.
To identify any problems associated with the amplification of GC-rich genes (and/or using GC-rich primers) [26, 44, 45], we focused on the read depth of the NGS test as well as the performance of the commercially available PCR kit in the present study. We found that although the read depth was small in the GC-rich TERT promoter region, mutations were detected in the samples by NGS and these results were further validated by Sanger sequencing. It is well known that the sensitivity of different NGS workflows can vary between clinical laboratories, particularly based on the bioinformatic pipeline used and the types of variants that the pipeline is designed and validated to detect. Therefore, carefully evaluating the coverage of NGS remains vital [46]. For many clinical laboratories adopting NGS as a diagnostic platform, detection of low-VAF somatic mutations is a challenge [47]. Even at a high read depth, NGS shows a rapid drop in detection accuracy of low-VAF somatic mutations [48,49,50].
In the present study, although the average read depth of the TERT promoter region was significantly lower than that of the other genes, we observed that the average VAFs in the C228T and C250T mutations of the TERT promoter region were more than 30% and the lowest VAF was 7.7%. These results suggest that mutations in the TERT promoter region are shared by many tumor cells and make the TERT promoter mutation accurate with relatively low read depth in the GC-rich TERT promoter region in NGS. Moreover, high VAFs in the TERT promoter mutation enabled high PPV and NPV using the iTERT PCR kit.
Several cancers are reported to harbor frequent mutations in the TERT promoter region [7]. Moreover, the simple and inexpensive iTERT PCR kit successfully demonstrated the TERT promoter mutations detected by NGS in all tested cases, even with miniscule amounts (~ 10 ng/μl) of DNA (Table 3). Therefore, we validated the NGS results with the gold standard PCR test and found that the iTERT PCR test is sensitive for the identification of the TERT promoter mutations in solid cancers. Based on these observations, we can suggest the iTERT PCR test as a simple, cheap, easily accessible, and effective alternative to NGS that can be widely used for the detection of TERT promoter mutations in diagnostic laboratories.
Availability of data and materials
Reference genome (hg19) used in this study can be obtained from the UCSC databases (https://hgdownload.soe.ucsc.edu/). The datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Abbreviations
- NGS:
-
Next-generation sequencing
- TERT:
-
Telomerase reverse transcriptase
- PCR:
-
Polymerase chain reaction
- TSO:
-
TruSight Oncology
- VAF:
-
Variant allele frequency
- TD:
-
Total read depth
- TV:
-
Tumor volume
References
Bojesen SE, Pooley KA, Johnatty SE, Beesley J, Michailidou K, Tyrer JP, et al. Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nat Genet. 2013;45:371–84, 84e1–2.
Codd V, Nelson CP, Albrecht E, Mangino M, Deelen J, Buxton JL, et al. Identification of seven loci affecting mean telomere length and their association with disease. Nat Genet. 2013;45:422–7, 7e1–2.
Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, et al. TERT promoter mutations in familial and sporadic melanoma. Science. 2013;339:959–61.
Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013;339:957–9.
Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA Jr, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci USA. 2013;110:6021–6.
Liu X, Wu G, Shan Y, Hartmann C, von Deimling A, Xing M. Highly prevalent TERT promoter mutations in bladder cancer and glioblastoma. Cell Cycle. 2013;12:1637–8.
Hafezi F, Perez Bercoff D. The solo play of TERT promoter mutations. Cells. 2020;9:66.
Yuan X, Larsson C, Xu D. Mechanisms underlying the activation of TERT transcription and telomerase activity in human cancer: old actors and new players. Oncogene. 2019;38:6172–83.
Bell RJ, Rube HT, Xavier-Magalhaes A, Costa BM, Mancini A, Song JS, et al. Understanding TERT promoter mutations: a common path to immortality. Mol Cancer Res. 2016;14:315–23.
Nonoguchi N, Ohta T, Oh JE, Kim YH, Kleihues P, Ohgaki H. TERT promoter mutations in primary and secondary glioblastomas. Acta Neuropathol. 2013;126:931–7.
Macerola E, Loggini B, Giannini R, Garavello G, Giordano M, Proietti A, et al. Coexistence of TERT promoter and BRAF mutations in cutaneous melanoma is associated with more clinicopathological features of aggressiveness. Virchows Arch. 2015;467:177–84.
Biczok A, Kraus T, Suchorska B, Terpolilli NA, Thorsteinsdottir J, Giese A, et al. TERT promoter mutation is associated with worse prognosis in WHO grade II and III meningiomas. J Neurooncol. 2018;139:671–8.
Sahm F, Schrimpf D, Olar A, Koelsche C, Reuss D, Bissel J, et al. TERT promoter mutations and risk of recurrence in meningioma. J Natl Cancer Inst. 2016;66:108.
Descotes F, Kara N, Decaussin-Petrucci M, Piaton E, Geiguer F, Rodriguez-Lafrasse C, et al. Non-invasive prediction of recurrence in bladder cancer by detecting somatic TERT promoter mutations in urine. Br J Cancer. 2017;117:583–7.
Griewank KG, Murali R, Puig-Butille JA, Schilling B, Livingstone E, Potrony M, et al. TERT promoter mutation status as an independent prognostic factor in cutaneous melanoma. J Natl Cancer Inst. 2014;106:66.
Nagore E, Heidenreich B, Rachakonda S, Garcia-Casado Z, Requena C, Soriano V, et al. TERT promoter mutations in melanoma survival. Int J Cancer. 2016;139:75–84.
Populo H, Boaventura P, Vinagre J, Batista R, Mendes A, Caldas R, et al. TERT promoter mutations in skin cancer: the effects of sun exposure and X-irradiation. J Invest Dermatol. 2014;134:2251–7.
Rachakonda PS, Hosen I, De Verdier PJ, Fallah M, Heidenreich B, Ryk C, et al. TERT promoter mutations in bladder cancer affect patient survival and disease recurrence through modification by a common polymorphism. Proc Natl Acad Sci USA. 2013;110:17426–31.
Kolquist KA, Ellisen LW, Counter CM, Meyerson MM, Tan LK, Weinberg RA, et al. Expression of TERT in early premalignant lesions and a subset of cells in normal tissues. Nat Genet. 1998;19:182–6.
Lee HW, Park TI, Jang SY, Park SY, Park W-J, Jung S-J, et al. Clinicopathological characteristics of TERT promoter mutation and telomere length in hepatocellular carcinoma. Medicine. 2017;96:66.
Salgado C, Roelse C, Nell R, Gruis N, van Doorn R, van der Velden P. Interplay between TERT promoter mutations and methylation culminates in chromatin accessibility and TERT expression. PLoS ONE. 2020;15:e0231418.
Stasik S, Salomo K, Heberling U, Froehner M, Sommer U, Baretton GB, et al. Evaluation of TERT promoter mutations in urinary cell-free DNA and sediment DNA for detection of bladder cancer. Clin Biochem. 2019;64:60–3.
Chang KP, Wang CI, Pickering CR, Huang Y, Tsai CN, Tsang NM, et al. Prevalence of promoter mutations in the TERT gene in oral cavity squamous cell carcinoma. Head Neck. 2017;39:1131–7.
Wilson AG, Symons JA, McDowell TL, McDevitt HO, Duff GW. Effects of a polymorphism in the human tumor necrosis factor alpha promoter on transcriptional activation. Proc Natl Acad Sci USA. 1997;94:3195–9.
McDowell DG, Burns NA, Parkes HC. Localised sequence regions possessing high melting temperatures prevent the amplification of a DNA mimic in competitive PCR. Nucleic Acids Res. 1998;26:3340–7.
Mamedov TG, Pienaar E, Whitney SE, TerMaat JR, Carvill G, Goliath R, et al. A fundamental study of the PCR amplification of GC-rich DNA templates. Comput Biol Chem. 2008;32:452–7.
Lee H, Lee B, Kim DG, Cho YA, Kim JS, Suh YL. Detection of TERT promoter mutations using targeted next-generation sequencing: overcoming GC bias through trial and error. Cancer Res Treat. 2021;6:66.
Kang SY, Kim KM. Highly sensitive duplex MSI test and BAT40 germline polymorphism. APMIS. 2021;129:607–15.
Cho YA, Lee H, Kim DG, Kim H, Ha SY, Choi YL, et al. PD-L1 expression is significantly associated with tumor mutation burden and microsatellite instability score. Cancers (Basel). 2021;13:66.
Pestinger V, Smith M, Sillo T, Findlay JM, Laes JF, Martin G, et al. Use of an integrated pan-cancer oncology enrichment next-generation sequencing assay to measure tumour mutational burden and detect clinically actionable variants. Mol Diagn Ther. 2020;24:339–49.
Lee D, Cho YH, Kang SY, Yoon N, Sung CO, Suh YL. BRAF V600E mutations are frequent in dysembryoplastic neuroepithelial tumors and subependymal giant cell astrocytomas. J Surg Oncol. 2015;111:359–64.
Mosrati MA, Malmstrom A, Lysiak M, Krysztofiak A, Hallbeck M, Milos P, et al. TERT promoter mutations and polymorphisms as prognostic factors in primary glioblastoma. Oncotarget. 2015;6:16663–73.
Johanns TM, Fu Y, Kobayashi DK, Mei Y, Dunn IF, Mao DD, et al. High incidence of TERT mutation in brain tumor cell lines. Brain Tumor Pathol. 2016;33:222–7.
Vinagre J, Almeida A, Populo H, Batista R, Lyra J, Pinto V, et al. Frequency of TERT promoter mutations in human cancers. Nat Commun. 2013;4:2185.
Hurst CD, Platt FM, Knowles MA. Comprehensive mutation analysis of the TERT promoter in bladder cancer and detection of mutations in voided urine. Eur Urol. 2014;65:367–9.
Wang K, Liu T, Ge N, Liu L, Yuan X, Liu J, et al. TERT promoter mutations are associated with distant metastases in upper tract urothelial carcinomas and serve as urinary biomarkers detected by a sensitive castPCR. Oncotarget. 2014;5:12428–39.
Allory Y, Beukers W, Sagrera A, Flandez M, Marques M, Marquez M, et al. Telomerase reverse transcriptase promoter mutations in bladder cancer: high frequency across stages, detection in urine, and lack of association with outcome. Eur Urol. 2014;65:360–6.
Nguyen D, Taheri D, Springer S, Cowan M, Guner G, Mendoza Rodriguez MA, et al. High prevalence of TERT promoter mutations in micropapillary urothelial carcinoma. Virchows Arch. 2016;469:427–34.
Nault JC, Calderaro J, Di Tommaso L, Balabaud C, Zafrani ES, Bioulac-Sage P, et al. Telomerase reverse transcriptase promoter mutation is an early somatic genetic alteration in the transformation of premalignant nodules in hepatocellular carcinoma on cirrhosis. Hepatology. 2014;60:1983–92.
Cevik D, Yildiz G, Ozturk M. Common telomerase reverse transcriptase promoter mutations in hepatocellular carcinomas from different geographical locations. World J Gastroenterol. 2015;21:311–7.
Kawai-Kitahata F, Asahina Y, Tanaka S, Kakinuma S, Murakawa M, Nitta S, et al. Comprehensive analyses of mutations and hepatitis B virus integration in hepatocellular carcinoma with clinicopathological features. J Gastroenterol. 2016;51:473–86.
Vinagre J, Nabais J, Pinheiro J, Batista R, Oliveira RC, Goncalves AP, et al. TERT promoter mutations in pancreatic endocrine tumours are rare and mainly found in tumours from patients with hereditary syndromes. Sci Rep. 2016;6:29714.
Kim TH, Kim YE, Ahn S, Kim JY, Ki CS, Oh YL, et al. TERT promoter mutations and long-term survival in patients with thyroid cancer. Endocr Relat Cancer. 2016;23:813–23.
Green MR, Sambrook J. Polymerase chain reaction (PCR) amplification of GC-rich templates. Cold Spring Harb Protoc. 2019;6:66.
Muralidharan K, Yekula A, Small JL, Rosh ZS, Kang KM, Wang L, et al. TERT promoter mutation analysis for blood-based diagnosis and monitoring of gliomas. Clin Cancer Res. 2021;27:169–78.
Lincoln SE, Hambuch T, Zook JM, Bristow SL, Hatchell K, Truty R, et al. One in seven pathogenic variants can be challenging to detect by NGS: an analysis of 450,000 patients with implications for clinical sensitivity and genetic test implementation. Genet Med. 2021;23:1673–80.
Kim J, Kim D, Lim JS, Maeng JH, Son H, Kang HC, et al. The use of technical replication for detection of low-level somatic mutations in next-generation sequencing. Nat Commun. 2019;10:1047.
Xu H, DiCarlo J, Satya RV, Peng Q, Wang Y. Comparison of somatic mutation calling methods in amplicon and whole exome sequence data. BMC Genomics. 2014;15:244.
Stead LF, Sutton KM, Taylor GR, Quirke P, Rabbitts P. Accurately identifying low-allelic fraction variants in single samples with next-generation sequencing: applications in tumor subclone resolution. Hum Mutat. 2013;34:1432–8.
Roberts ND, Kortschak RD, Parker WT, Schreiber AW, Branford S, Scott HS, et al. A comparative analysis of algorithms for somatic SNV detection in cancer. Bioinformatics. 2013;29:2223–30.
Borah S, Xi L, Zaug AJ, Powell NM, Dancik GM, Cohen SB, et al. Cancer. TERT promoter mutations and telomerase reactivation in urothelial cancer. Science. 2015;347:1006–10.
Huang DS, Wang Z, He XJ, Diplas BH, Yang R, Killela PJ, et al. Recurrent TERT promoter mutations identified in a large-scale study of multiple tumour types are associated with increased TERT expression and telomerase activation. Eur J Cancer. 2015;51:969–76.
Acknowledgements
Not applicable.
Funding
This work was supported by the Basic Science Research Program via the National Research Foundation of Korea (NRF), funded by the Ministry of Science and Information and Communication Technology (ICT) (NRF-2017R1A2B4012436), and by a grant from the Korea Health Technology R&D Project via the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (Grant numbers: HR20C0025 and HI21C1137).
Author information
Authors and Affiliations
Contributions
SYK, K-TJ and K-MK designed and supervised the study. SYK, DGK, HK, YAC, SYH, GYK, K-TJ and K-MK collected tissue samples and clinical data and performed histopathological examination. SYK and K-MK analyzed the data. SYK, DGK, HK, YAC, SYH, GYK, K-TJ and K-MK conducted the experiments. SYK and K-MK wrote the draft. SYK, GYK, K-TJ and K-MK revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethical approval and consent to participate
The study was approved by the ethics committee of the Samsung Medical Center Institutional Review Board (IRB 2020-06-045-001). This study was performed in accordance with the Institutional Review Board guidelines of Samsung Medical Center (IRB 2020-06-045-001) for data analysis and investigational treatments. All patients provided informed consent to participate in this study.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kang, S.Y., Kim, D.G., Kim, H. et al. Direct comparison of the next-generation sequencing and iTERT PCR methods for the diagnosis of TERT hotspot mutations in advanced solid cancers. BMC Med Genomics 15, 25 (2022). https://doi.org/10.1186/s12920-022-01175-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-022-01175-2