
Abstract
Background
SOX3 is essential for pituitary development normally at the earliest stages of development. In humans, variants of SOX3 can cause X-linked hypopituitarism with various clinical manifestations, with or without mental retardation.
Case presentation
We present an 8-year-old Chinese patient with congenital hypopituitarism who had a 6.180 Mb duplication on Xq26.3q27.1 including SOX3, F9, and eight other contiguous genes. The main complains of the boy was short stature. His height was 90.1 cm (− 5.87SDS), weight 11.5 kg (− 5.25SDS). He developed growth hormone (GH) deficiency, cryptorchidism and low thyroid function. Pituitary magnetic resonance imaging revealed the pituitary dysplasia. After diagnosis, levothyroxine was given for one month first, and the thyroid function basically returned to normal, but the growth situation did not improve at all. Then recombinant human GH was given, his height, growth rate and height SDS were improved significantly in the 2 years follow-up. The level of height SDS improved from − 5.87 SDS before treatment to − 3.27 SDS after the first year of treatment and − 1.78 SDS after the second years of treatment. Gonadal function and long-term prognosis of the patient still need further observation and follow-up.
Conclusions
This is the first case of Chinese male patient with multiple hypophysis dysfunction caused by SOX3 duplication, which will expand the range of phenotypes observed in patients with duplication of SOX3.
Similar content being viewed by others


Background
SOX (SRY related high mobility group box) genes encode many transcription factors with key regulatory roles in different developmental processes, such as embryogenesis and nervous system development [1]. SOX3 (OMIM#313430), a member of the SOX family, is essential for pituitary development and is expressed in stem and neuroepithelial progenitor cells at the earliest stages of development [2,3,4].
The variant of SOX3 often manifested as X-linked hypopituitarism with various clinical manifestations, including isolated GH deficiency (IGHD), congenital hypopituitarism (CH), etc., with or without mental retardation [5]. Studies have identified CH patients in families with GH deficiency and intellectual disability, which further confirms the important role of SOX3 in the development of the hypothalamic-pituitary axis [6].
Here, we report the first Chinese patient harboring duplication on Xq26.3q27.1 including SOX3 gene. He developed CH, but he did not present intellectual disability.
Case presentation
The Chinese boy was eight years old, borned by cesarean section. His birth weight was 2.5 kg, and the length was unknown. After birth, he was diagnosed with hypoxie-ischemic encephalopathy. Therefore, he was hospitalized locally for 40 days. During hospitalization, the fasting blood glucose was normal, the length of penis was not recorded, and no abnormalities were reported in the head magnetic resonance imaging (MRI). Milestones of motor development were normal (sit at 6–7 months, stand alone at 10–11 months and walk at 1.5 years). He had hernia surgery at the age of 4 years. His parents were non-consanguineous and phenotypically normal. Apart from a history of nephrolithiasis, his mother had no other special medical history records. His father is 172 cm tall (− 0.12 SDS), and his mother is 159 cm (− 0.30 SDS) tall. His target height was 171.5 ± 4 cm (− 1.2 SDS). His 1 year-old young sister was normal.
The main complains of the boy was short stature. His height was 90.1 cm (− 5.87SDS), weight 11.5 kg (− 5.25SDS). He had proportional short stature, slightly prominent forehead, wide eye distance, deep-set eyes, low bridge of nose, hypotrophy of the maxilla, and high and narrow arched palate (Fig. 1A1, A2). He has cryptorchidism on the right side. The left testis was 1 ml and the location was a little bit high. Using the WISC-IV to investigate the intelligence profile, and the results showed that his intelligence was normal (the total IQ was ≤ 92).
Hormone measurement revealed dysfunction of anterior pituitary. The results of GH response on insulin and arginine hydrochloride testing showed peak value of GH as 0.20 ng/mL. His serum concentrations of IGF1 was significantly lower than the normal range (15.51 ng/L, Ref. 119 ± 45 ng/L). LH and FSH was < 0.1 mU/mL and 0.31 mU/ml respectively. The level of TT3 is 1.14 nmol/L (Ref. 1.21–2.66 nmol/L), TT4 is 21.8 nmol/L (Ref. 67–163 nmol/L) and TSH is 1.66 mU/L (Ref. 0.6–4.5 mU/L). The abnormal low level of the above pituitary-related hormone was supportive of the diagnosis of central hypothyroidism and GH deficiency. His level of adrenocorticotropic hormone (ACTH) and cortisol were normal though.
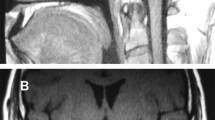
The pituitary MRI showed that he has emptysellasyndrome (2 mm) and low-density shadow in saddle area, indicated of pituitary dysplasia (Fig. 1B1, B2).

Physical examination results for the 8 -year- old patient. A1, A2 Facial appearance of the patient: slightly larger ears, low ear positions, external rotation, protrusion of ear lobes, thin lips, wide nose bridge, and low forehead hairline. B1, B2 Pituitary MRI of the patient revealed pituitary dysplasia
Whole‑exome sequencing (WES) and bioinformatics analysis
For this patient, the main clinical feature was short stature, his intelligence, speech and motor development were normal and his facial features were slightly abnormal. Hormone measurement revealed dysfunction of anterior pituitary. The results of GH response on insulin and arginine hydrochloride testing was supportive of the diagnosis of central hypothyroidism and growth hormone deficiency. So we identified the patient had multiple pituitary hormone deficiencies, therefore we selected WES for genetic detection firstly.
Genomic DNA from the proband and his parents was extracted from peripheral blood leucocytes by using the DNeasy kit (Qiagen). Whole exomes were captured (MyGenostics Inc., Beijing) and sequenced on Illumina NovaSeq 6000 series sequencer (PE150). We applied quality control filters to remove low-quality reads, and performed bioinformatics analysis using an in-house pipeline which included genome alignment with the Burrows-Wheeler Aligner. We annotated database-based MAF and 2015-ACMG Standards and Guidelines based pathogenicity for each generated genetic variant using an online system independently developed by Chigene. Variants with minor allele frequencies of < 0.05 in population databases (such as 1000 Genomes, dbSNP, ESP6500, in-house database (MyGenostics), and EXAC), expected to affect protein coding/splicing or present in the HGMD, would be included into the analysis. As for CNV calls, the total bases for each coding region were calculated using SAMtools, and the average mean depth of the CCDS regions were obtained by using the GATK 'DepofCoverage' command. Then use R to calculate the ratio of each sample compared to the average ratio of the other samples, and use ggplot to visualize the results. Ratios > 1.4 were designated as duplicates and < 0.6 were designated as deletions.
Genetic results
All the trios-based exomes by the high-throughput sequencing was analyzed (Fig. 2). For genetic screening of all exons, we screened for duplication of X-chromosomes in the patient and his mother. The duplicated segments is 6.180 Mb (Xq26.3 ~ q27.1, 133905314–140085817), involving ATP11C, CD40LG, F9, DHL1, GPR101, RBMX, SLC9A6, SOX3 and ZIC3.

Genetic Analysis diagram of the patient and his parents. The red circle shows 6.180 Mb duplication on Xq26.3q27.1 including SOX3, F9, and eight other contiguous genes. A represents the patient, B represents his father, and C represents his mother
Treatment
After diagnosis, the patient was first treated with levothyroxine. One month later, the thyroid function was restored to normal, but there was no improvement in height. After adjusting treatment to the recombinant human growth hormone (rhGH, 0.11U/kg/day), the height of the patient was significantly improved (Fig. 3). The growth rate was 2.8 cm/ year before treatment, 17.1 cm/year in the first year of treatment, and 11.9 cm/ year in the second year of treatment. The level of height SDS improved from − 5.87 SDS before treatment to − 3.27 SDS after the first year of treatment and − 1.78 SDS after the second years of treatment (Table 1). The child gained weight slowly during treatment, and 2 years later produced a 10th of the normal child's weight growth curve.

Growth situation of the patient during the 2-year treatment follow-up. The blue line presents a standard growth curve of Chinese boys (2–18 years old). Red and blue dots were the patient’s height and weight during the two-year treatment, respectively
During the 2-year treatment, the dose of rhGH was at 0.114–0.117U/kg/day. The IGF1 level was increased to 113–142 ng/ml. Blood glucose, insulin, ACTH and cortisol was monitored every 3 months, all within the normal range.
Discussion and conclusions
SOX3 gene, which belongs to SOX family Sry-related High Mobility Group Box, located at Xq27.1. It is a 1,341 nucleotides single exon gene. It is highly expressed throughout development in the pituitary and central nervous system (CNS), and is recognized to have a key role in regulating embryogenesis and CNS development [1]. It is also required for Rathke's capsule morphogenesis and hypothalamic-pituitary axis function [7].
SOX3 gene variants are associated with hypopituitarism in both animal and patients. Knockout mice experiments indicated that lack of SOX3 gene can lead to abnormal structure and dysfunction of hypothalamic pituitary axis and special abnormalities in the midline structure of the CNS [7, 8]. Human SOX3 variant can lead to congenital pituitary dysfunction (clinically manifested as one or several pituitary hormone deficiencies), neural tube defects and other symptoms.
Over-expression or under-expression of SOX3 can lead to similar clinical phenotypes. Clinical features varied from IGHD or combined pituitary hormone deficiency (CPHD) to panhypopituitarism, as well as anterior hypophysis dysplasia, posterior hypophysis ectopic, abnormal corpus callosum, with or without intellectual impairment [1, 9]. Recently, Arya et al. reported five male patients with Xq27.1 duplication, ranged from 323.8 KB to 11 Mb including SOX3 gene, diagnosed with congenital dysfunction of hypophysial. Three of them showed multiple pituitary hormone deficiency and two showed IGHD. All five patients presented with small penis or cryptorchidism, abnormalities of pituitary structure or other midline craniocerebral, such as corpus callosum dysplasia, hyaline septum deletion, etc. [10].
Another research reported an 8-year-old boy with 2.1 MB deletion on Xq27.1-q27.2, including the entire SOX3 gene, had mild mental retardation, language retardation, dysarticulation disorder, behavioral problems, mild facial abnormalities (slightly larger ears, low ear positions, external rotation, protrusion of ear lobes, thin lips, wide nose bridge, low forehead hairline, large hands), excess appetite and overeating. However, the level of GH, ACTH and TSH were at the normal range. Head MRI showed no obvious brain abnormalities at 6 years of age [11]. Dr. Stefano Stagi reported another case [1]. The boy was born as small for gestational age. He presented significantly short stature with lagging bone-age. He has complete growth hormone deficiency, central hypothyroidism, hypogonadism, and decreased ACTH level (but cortisol level was normal). It was different from other case reports that our patient do not have mental retardation or intellectual disability. In our case, the SOX3 gene duplication originated from his mother, but the mother had no symptoms of this disease, we speculate that it may be related to the random inactivation of the X chromosome. The mechanism of difference in clinical manifestation is unclear yet [9].
Additionally, the prolongation of polyalanine chain is one of the reasons for the decrease of SOX3 expression. Laumonnier et al. found that 11 alanine repetitions is associated with IGHD, mental retardation, and craniofacial deformity, and Woods et al. found that 7 alanine duplication is mainly manifested as panhypopituitarism and anterior pituitary hypoplasia [5, 6, 12]. Furthermore, incompleteness presence of SOX3 duplication was reported in literature. Lachlan et al. in 2004 reported a women with functional disomy at Xq27-qter, harboring SOX3, but she did not have any signs of GHD [13]. In another study, five women from one family with a 7.5 Mb duplication that including SOX3, all exhibited short stature. The author believed that the phenotypic variability may be caused by the altered levels of SOX3 due to incomplete inactivation of X chromosome variants [14].
Although the SOX3 gene is structurally similar to the SRY gene, it does not have a typical function in sex determination. The HMG DNA-binding domain of SOX3 is similar to that of the sex-determining gene SRY [15], however, the expression of the mouse homologous SOX3 gene is very low in developing mouse gonads, and SOX3 variants in mice or humans do not directly result in sexual determination abnormalities [16]. However, overexpression of SOX3 can up-regulate the expression of SOX9 through the synergistic effect with SF1 in the gonadal glands of developing XX mice, thus causing testicular development [17]. Grinspon et al. reported that gain-function variants of SOX3 lead to SRY negative 46, XX DSD [18]. There are two possible mechanisms about it: overexpression of anterior testis genes (e.g., SOX9) and underexpression of anterior ovarian/antitestis genes (such as WNT4 or RSPOI). In addition, XX male sex reversal has been reported to be associated with deletions and duplications in the upstream region of SOX3 [11, 17]. Although loss of function of SOX3 is not directly involved in sex determination, SOX3 is a necessary factor to spermatogenesis and gonadal function [19]. In our case, the loss function of SOX3 gene lead to the left cryptorchidism and right testicular dysplasia.
Over-expression of SOX3 was also a risk factor for neural tube defects (NTDs), one of the most common congenital defects caused by a combination of genetic and environmental factors. Over expression of SOX3 may prevent neural tube morphogenesis by undifferentiated neural cell precursors [9]. Duplication of SOX3 was found in patients with myelomeningococcal, hypophysis and male cognitive impairment [9, 20]. Uguen et al. reviewed 50 patients (44 patients, 6 fetuses) with Xq duplication ranged from 230 kb to 12.5 Mb, all included SOX3. Among them, 8 cases (6 fetuses, 2 patients) presented NTDs, indicated that the genetic character of NTDs was incomplete penetrance. In addition, SOX3 duplication can lead to other abnormalities. Stankiewicz et al. reported a 14-year-old girl with 7.5 Mb duplication on Xq26.2-q27.1 inherited from her mother, presented with dysmorphic features, scoliosis, hearing impairment and amenorrhea. Both the daughter and her mother have severe speech problems with stuttering and dyslalia [14].
So far, there have been no much reports on the treatment of patients with SOX3 duplication. Arya et al. summarize the general treatment of 4 patients with SOX3 duplication. However, the dose of the hormone and the change of their growth rate and height has not been reported [10].
In our case, after diagnosis, levothyroxine was given for one month first, and the thyroid function basically returned to normal, but the growth situation did not improve at all. Then rhGH was given, his height, growth rate and height SDS were improved significantly. Gonadal function and long-term prognosis of the patient still need further observation and follow-up. This is the first case of Chinese male patient with multiple hypophysis dysfunction caused by SOX3 duplication, which will expand the range of phenotypes observed in patients with duplication of SOX3.
Availability of data and materials
The SOX3 variant can be found in NCBI Nucleotide under the accession number NM_005634. The raw datasets generated and analysed during the current study are not publicly available in order to protect participant confidentiality. The datasets obtained during the current study are available from the corresponding author if the requirements are reasonable.
Abbreviations
- SOX :
-
SRY related high mobility group box
- IGHD:
-
Isolated GH deficiency
- CH:
-
Congenital hypopituitarism
- ACTH:
-
Adrenocorticotropic hormone
- MRI:
-
Magnetic resonance imaging;
- rhGH:
-
Recombinant human growth hormone
- CPHD:
-
Combined pituitary hormone deficiency
- NTDs:
-
Neural tube defects
References
Stagi S, Lapi E, Pantaleo M, Traficante G, Giglio S, Seminara S, et al. A SOX3 (Xq26.3–27.3) duplication in a boy with growth hormone deficiency, ocular dyspraxia, and intellectual disability: a long-term follow-up and literature review. Hormones (Athens). 2014;13(4):552–60.
Takagi M, Ishii T, Torii C, Kosaki K, Hasegawa T. A novel mutation in SOX3 polyalanine tract: a case of Kabuki syndrome with combined pituitary hormone deficiency harboring double mutations in MLL2 and SOX3. Pituitary. 2014;17(6):569–74.
Kelberman D, Dattani MT. Hypothalamic and pituitary development: novel insights into the aetiology. Eur J Endocrinol. 2007;157(Suppl 1):S3-14.
Dattani MT. Growth hormone deficiency and combined pituitary hormone deficiency: Does the genotype matter? Clin Endocrinol (Oxf). 2005;63(2):121–30.
Alatzoglou KS, Azriyanti A, Rogers N, Ryan F, Curry N, Noakes C, et al. SOX3 deletion in mouse and human is associated with persistence of the craniopharyngeal canal. J Clin Endocrinol Metab. 2014;99(12):E2702–8.
Laumonnier F, Ronce N, Hamel BCJ, Thomas P, Lespinasse J, Raynaud M, et al. Transcription factor SOX3 is involved in X-linked mental retardation with growth hormone deficiency. Am J Hum Genet. 2002;71(6):1450–5.
Bauters M, Frints SG, Van Esch H, Spruijt L, Baldewijns MM, de Die-Smulders CE, et al. Evidence for increased SOX3 dosage as a risk factor for X-linked hypopituitarism and neural tube defects. Am J Med Genet A. 2014;164A(8):1947–52.
Rizzoti K, Brunelli S, Carmignac D, Thomas PQ, Robinson IC, Lovell-Badge R. SOX3 is required during the formation of the hypothalamo-pituitary axis. Nat Genetics. 2004;36(3):247–55.
Hureaux M, Ben Miled S, Chatron N, Coussement A, Bessieres B, Egloff M, et al. SOX3 duplication: a genetic cause to investigate in fetuses with neural tube defects. Prenat Diagn. 2019;39(11):1026–34.
Arya VB, Chawla G, Nambisan AKR, Muhi-Iddin N, Vamvakiti E, Ajzensztejn M, et al. Xq27.1 duplication encompassing SOX3: variable phenotype and smallest duplication associated with hypopituitarism to date—a large case series of unrelated patients and a literature review. Horm Res Paediatr. 2019;92(6):382–9.
Helle JR, Baroy T, Misceo D, Braaten O, Fannemel M, Frengen E. Hyperphagia, mild developmental delay but apparently no structural brain anomalies in a boy without SOX3 expression. Am J Med Genet A. 2013;161A(5):1137–42.
Woods KS, Cundall M, Turton J, Rizotti K, Mehta A, Palmer R, et al. Over- and underdosage ofSOX3 is associated with infundibular hypoplasia and hypopituitar-ism. Am J Hum Genet. 2005;76(5):833–49.
Lachlan KL, Collinson MN, Sandford RO, van Zyl B, Jacobs PA, Thomas NS. Functional disomy resulting from duplications of distal Xq in four unrelated patients. Hum Genet. 2004;115(5):399–408.
Stankiewicz P, Thiele H, Schlicker M, Cseke-Friedrich A, Bartel-Friedrich S, Yatsenko SA, et al. Duplication of Xq262-q271, including SOX3, in a mother and daughter with short stature and dyslalia. Am J Med Genet A. 2005;138(1):11–7.
Alatzoglou KS, Kelberman D, Dattani MT. The role of SOX proteins in normal pituitary development. J Endocrinol. 2009;200(3):245–58.
Rosolowsky ET, Stein R, Marks SD, Leonard N. Marked phenotypic variable expression among brothers with duplication of Xq271 involving the SOX3 gene. J Pediatr Endocrinol Metab. 2020;33(3):443–7.
Sutton E, Hughes J, White S, Sekido R, Tan J, Arboleda V, et al. Identification of SOX3 as an XX male sex reversal gene in mice and humans. J Clin Invest. 2011;121(1):328–41.
Grinspon RP, Nevado J, Alvarez M, Rey GD, Castera R, Venara M, et al. 46XX ovotesticular DSD associated with a SOX3 gene duplication in a SRY-negative boy. Clin Endocrinol (Oxf). 2016;85(4):673–5.
Weiss J, Meeks JJ, Hurley L, Raverot G, Frassetto A, Jameson JL. Sox3 is required for gonadal function, but not sex determination, in males and females. Mol Cell Biol. 2003;23(22):8084–91.
Uguen A, Talagas M, Quemener-Redon S, Marcorelles P, De Braekeleer M. Duplication of SOX3 (Xq27) may be a risk factor for neural tube defects. Am J Med Genet A. 2015;167(7):1676–8.
Acknowledgements
We would like to thank the patient and his parents for participating in this work.
Funding
No funding was obtained for this study.
Author information
Authors and Affiliations
Contributions
XPL, YL, CQD, and FYW designed and organized the study. YL and XPL cared for the patient and supervised the study. ZGL and MNZ acquired the clinical data, prepared the samples from the family members and interpreted the genetic analyses. FYW and XY performed the genetic analyses. YL, CQD, and FYW wrote the manuscript that was edited by all other authors. XPL substantively revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethics Committee of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology (Approval Number: TJ-IRB20180703). All study procedures were conducted in accordance with the tenets of the Declaration of Helsinki. The patient and his parents provided written informed consent to participate in this study.
Consent for publication
Written informed consent to publish this case was obtained from the patient and his parents, including case description and medical data.
Competing interests
All authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Du, C., Wang, F., Li, Z. et al. Xq26.3-q27.1 duplication including SOX3 gene in a Chinese boy with hypopituitarism: case report and two years treatment follow up. BMC Med Genomics 15, 19 (2022). https://doi.org/10.1186/s12920-022-01167-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-022-01167-2