
Abstract
Background
Type II collagenopathies are a spectrum of diseases and skeletal dysplasia is one of the prominent features of collagenopathies. Molecular defects of the COL2A1 gene cause type II collagenopathies that is mainly an autosomal dominant disease, whereas some rare cases with autosomal recessive inheritance of mode have also been identified.
Case presentation
The patient was a 5-year-old male with a short neck, flat face, epiphyseal dysplasia, irregular vertebral endplates, and osteochondritis. Sequencing result indicated NM_001844.4: c.3662C > T; p. (Ser1221Phe) a novel missense variant, leading to a serine-to-phenylalanine substitution. Sanger sequencing confirmed the variant compared to his parents and brother.
Conclusions
We identified a novel homozygous variant of the COL2A1 gene as the cause of type II collagenopathies in a Chinese male, enriching the spectrum of genotypes. This is the first case of type II collagenopathies inherited in an autosomal recessive manner in China and East Asia, and it is the first case that resulted from serine substitution in the world.
Similar content being viewed by others


Background
Type II collagenopathies are a series of diseases characterized by skeletal dysplasia [1]. The clinical symptoms are notably variable, including short stature, eyesight abnormality, hearing loss, kyphosis, and epiphyseal dysplasia. According to the variety and severity of their symptoms, patients are classified into 21 different phenotypes [2] such as Stickler syndrome type I (STL1, MIM#108300) and Spondyloepiphyseal dysplasia congenita (SEDC, MIM#183900), though clinical presentations of these phenotypes overlap considerably. Type II collagenopathies are resulted by mutation of COL2A1 (MIM #120140) and are inherited in an autosomal dominant manner. Recently, autosomal recessive inherence was identified in patients with type II collagenopathies: all of the patients were from West Asia [3,4,5,6], probably owing to a higher rate of consanguinity and extensive development of gene sequencing.
The COL2A1 gene, which is located in the 12q13.11 region, contains 54 exons and encodes the alpha-1 chain of procollagen type II [7]. It is specifically expressed in the vitreous, cartilage, inner ear, and intervertebral discs, which explains the typical clinical features of type II collagenopathies [8, 9]. The protein encoded by COL2A1 contains a typical triple-helical domain and a C-terminal region where variants have been found previously. The triple-helical domain comprises Gly-X-Y repeats, which is a typical feature of collagens. Most variants found in patients with type II collagenopathies are located in the triple-helical domain and Gly substitutions in this domain are usually related to severe phenotypes [2].
In the present study, whole-exome sequencing (WES) was performed on DNA sample obtained from one Chinese patient manifesting a short neck, flat face, epiphyseal dysplasia, irregular vertebral endplates, and osteochondritis. A novel homozygous variant of the COL2A1 gene was identified and the patient was diagnosed with type II collagenopathy. This is the first case of a patient with a COL2A1 homozygous variant in China and in East Asia. It is also the first case that resulted from a serine substitution worldwide.
Case presentation
The patient was a 5-year-old male with a birth length of 51 cm (+ 0.33SD) and a birth weight of 4000 g (+ 1.6SD). He is the second child of a non-consanguineous healthy couple and was delivered by caesarean section at 40 weeks. Development was normal before one and a half years of age; subsequently, lower-limb abnormality and short stature were noticed. Since three years of age, he has been suffered from consistent low bone mineral density (Z:1.6 ~ − 2.4, 1–5%) which could not be corrected by calcium and vitamin D supplementation. He occasionally complained of ankle ache after exercise. At 5 years of age, he was referred to our department due to the manifested bone abnormality.
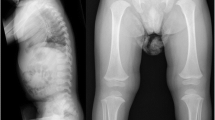
Physical examination showed a flat face, short neck, short lower limbs especially under the knees, brachydactyly, enlargement of the medium interphalangeal joints with normal height (105.8 cm, − 1.29SD) and weight (24 kg, + 1.87SD) (Fig. 1A–F). The patient had normal hearing, vision, cognitive development, and expression ability. Chest computed tomography and urinary system ultrasound were normal. X-ray of the spine showed that platyspondyly with irregular vertebral endplates of the lumbar vertebra and thoracic vertebra, and bony defects were observed on the top of T12 (Fig. 1G, H). X-ray of the knees and hand confirmed the results found on physical examination (Fig. 1I, J) and that of the pelvis revealed a heterogeneous bone structure of the proximal femur (Fig. 1K). Magnetic resonance imaging of the right ankle showed signs of osteochondritis (Fig. 1L, M).

Clinical photographs of the patient. A–C Facial characteristics including flat face and short neck. D, E Short lower limbs. F Brachydactyly and enlargement of medium interphalangeal joints. G, H Platyspondyly with irregular vertebral endplates. I, J Metaphyseal enlargement of medium interphalangeal and knee joints. L Heterogeneous bone structure of the proximal femur. K, M Osteochondritis of the right ankle
Laboratory investigations revealed normal levels of liver function, renal function, erythrocyte sedimentation rate, thyroid function, and levels of parathyroid hormone and serum calcium. Additionally, the patient showed elevated levels of alkaline phosphatase (184 IU/L, reference range: 45–129 IU/L) and serum inorganic phosphorus (1.8 mmol/L, reference range: 0.78–1.65 mmol/L). Further, myocardium zymogram examination revealed that creatine kinase isoenzyme-MB (4.8 μg/L, reference range: < 3.7 μg/L) and creatine kinase isoenzyme (193 U/L, reference range: 55–170 U/L) levels were above the normal.
The patient’s father was 173 cm, and his mother was 156 cm in height; both parents were healthy. He also had an 8-year-old healthy brother, with a height of 130.5 cm (Fig. 2A). The parents’ and the only brother's spines, pelvises, and knees were evaluated using X-ray and no abnormality was observed (Additional file 1: Figs. S1, S2, S3).

Verification and functional prediction of the c.3662C > T in the COL2A1 gene. A Pedigree of the family. B Sanger sequencing showed a homozygous missense variant in the patient, and the proband’s parents and brother were all heterozygous of the same locus. Black arrows, mutant base. C The position of the mutant residue, indicated in red, was highly conserved
WES was performed on the proband. Sequencing was performed and clusters were generated with an Illumina HiSeq 2000 system (Illumina, Inc.) and an Illumina cBot system (Illumina Inc., San Diego, CA, USA) respectively. The average read depth was 162.91X (Additional file 1: Table S1). All variants were annotated and filtered by Ingenuity Variant Analysis (Ingenuity Systems, Redwood City, CA, USA). Candidate variants were analyzed while skeletal dysplasia was selected as the main filtering symptom. Among all filtered variants, a homozygous missense variant in COL2A1 was confirmed and explained the patient’s condition (PM2 + PP3 + PP4), according to the guidelines recommended by the American College of Medical Genetics and Genomics (ACMG) (Additional file 1: Table S2). The novel variant NM_001844.4: c.3662C > T in COL2A1 led to a serine-to-phenylalanine substitution. Sanger sequencing indicated that the parents and brother were heterozygous for this variant (Fig. 2A, B).
Using in silico tools, we evaluated the pathogenicity of the variant of the COL2A1. The position of the variant is highly conserved in multiple species (Fig. 2C). Functional prediction indicated that the variant has a deleterious effect on the protein according to PolyPhen-2 (probably damaging, score = 0.99), SIFT (damaging, score = 0.003), and MutationTaster (disease causing, score = 1). To better assess the pathogenicity, a three-dimensional model was generated and examined using the I-TASSER server[10] (http://zhanglab.ccmb.med.umich.edu/I-TASSER) and Pymol v.1.8.4.0 software (https://www.pymol.org; Schrödinger, New York, NY, USA) respectively (Fig. 3). Normally, the amino acid residue serine is located in the C-terminal propeptide, which participates in the formation of an α-helix. The variant c.3662C > T is indicated to alter the hydrogen bond resulting in disruption of the normal protein structure. The patient was finally diagnosed with type II collagenopathy caused by a novel homozygous variant in COL2A1.

Three-dimensional structure model of the WT COL2A1 and p. (Ser1221Phe) mutant. The Triple helical domain and C-terminal propeptide are shown in yellow and white respectively. A Wild type protein: Serine at 1221 (red) interacts with Glycine at 1217 and Isoleucine at 1218. B Mutant protein: Serine at 1221 (red) interacts only with Proline at 1210
Discussion and conclusions
In the present study, since the observed phenotype was not completely typical for collagenopathy, WES was considered for diagnosis based on its wide coverage and not cost-incurring performance. We successfully identified a novel homozygous variant of the COL2A1 gene in a Chinese patient with type II collagenopathy. The variant c.3662C > T was located in exon 51 and could result in a serine-to-phenylalanine substitution in the C-terminal region. Both parents of the proband were heterozygous for this variant. The allele frequency of the variant was absent from the gnomAD database (http://gnomad.broadinstitute.org/) and the 1000 Genomes Project (http://www.1000genomes.org). To the best of our knowledge, this is the first report of a variant resulting in serine replacement in COL2A1. Additionally, pathogenicity confirmed by in silico studies and the highly conserved protein and nucleotide sequences implied that this variant was disease-causing. Though the variant is classified as a variant of uncertain significance according to ACMG guidelines, a high correlation between this variant and the mild phenotype of type II collagenopathy was observed.
To date, 514 variants of the COL2A1 gene have been reported in the human gene mutation database (http://www.hgmd.cf.ac.uk/ac/), including 241 missense variants, 95 splicing variants, 31 nonsense variants, 93 small deletions, 32 small insertions, and 22 other variants such as gross deletions/ insertions /duplications and complex rearrangements. As mentioned above, mutations in the COL2A1 gene are mainly autosomal dominant although an autosomal recessive inheritance manner was reported in recent years. All reported COL2A1 variants with autosomal recessive inheritance are shown in Fig. 4.

The schematic diagram of the distribution of 5 reported variants as well as c.3662C > T in the COL2A1 gene
To demonstrate a genotype–phenotype correlation, we collected the clinical information of all patients with homozygous missense variants (Table 1) and analyzed the data by domains (Additional file 1: Table S3). We also compared these data with that of heterozygous variants.
Vertebral abnormality, such as irregular vertebral endplates and platyspondyly could be observed in almost all patients with homozygous missense variants. Platyspondyly is also a common feature in different phenotypes of type II collagenopathy inherited in a heterozygous manner [2]. Eyesight abnormality, kyphosis, scoliosis, and waddling gait only occur in patients with mutations in the triple-helical domain. However, in cases with heterozygous mutations, these symptoms are not specifically associated with mutations in the triple-helical domain. Additionally, glycine substitution was highly relevant to SEDC [11]. Glycine substitution usually leads to an abnormal conformation or destabilization of the triple helix, thereby acting in a dominant‐negative way [2]. No glycine substitution has been reported in patients with homozygous variants to date.
Variants in the C-terminal region could also affect collagen formation. Patients with mutations in the C-terminal region present with milder phenotypes with some characteristic symptoms, probably because the variant has limited influence on the mature type II collagen [11]. Brachydactyly, especially of the middle and distal phalanges, seems to be the most common clinical feature of variants in the C‐terminal region [3, 12], which was also observed in the present case. Further, variants in the C-terminal propeptide are also associated with platyspondylic skeletal dysplasia Torrance type(MIM#151210) and with Spondyloperipheral dysplasia (SPPD, MIM#271700) [13]. Based on the present case, serine substitution in the C-terminal region seems to have no relation with severe phenotypes; however, there is not enough data reported on serine substitution at this time to make a definitive conclusion.
Our data collection and analysis may provide more insights into the phenotype of type II collagenopathy, especially for patients with homozygous missense variants. Tham et al. [5] reported the first patient with a homozygous variant and assumed the relationship between the bi-allelic variant of COL2A1 and SEDC. This was supported by Barat‐Houari et al. [4], who described a more severe patient with SEDC with homozygosity. However, Girisha et al. [3] then reported four patients with bi-allelic variants in COL2A1 which rarely caused SEDC. In type II collagenopathies dominantly inherited, more than 100 COL2A1 variants have been reported in patients diagnosed with SEDC, and most of the variants are located in the triple-helical domain (74% Gly replacements and 10% Arg-to-Cys substitutions) [14]. Our patient had mild abnormalities of the vertebrae with no hearing or ocular involvement, which shows that the case had a limited correlation with SEDC. Similar to autosomal dominant cases, different domains and amino acid substitutions should be considered. SEDC is not a homozygous variant-specific phenotype.
We also noticed that the patient’s height was within the normal range, whereas all of the previously reported cases had short stature at the last evaluation. Tham et al. [5] described detailed height information of a patient from birth to 11 years of age; height at birth was variable. Short stature in recessively inherited patients seems to become more severe as the patient gets older, indicating that height influence on the patient is probably cumulative. However, in patients with heterozygous mutations in the C-terminal propeptide, height clustered around the average level [13]. There is no specific relationship between height and domains in patients with homozygous variants. The proband’s height should be monitored closely.
It should be noted that all patients with homozygous variants were from consanguineous families in West Asia, except for our patient. Thus, the carrying rate of COL2A1 in Chinese may be underestimated.
In conclusion, this study reported the first Chinese patient with type II collagenopathy with autosomal recessive inheritance, thereby enriching the spectrum of genotypes. The patient presented with a mild phenotype. With the rapid development and application of sequencing technologies, we believe that more variants relating to milder phenotypes will be identified. However, direct functional evidence is lacking to prove the pathogenicity of all variants inherited in an autosomal recessive manner. The precise genotype–phenotype correlation and specific mechanisms remain unknown and require further study.
Availability of data and materials
The variant has been submitted to the NCBI ClinVar database whose accession number is SCV001755682. The raw sequence datasets generated during the current study are not publicly available because it is possible that individual privacy could be compromised but they are available from the corresponding author on reasonable request.
Abbreviations
- STL1:
-
Stickler syndrome type I
- SPPD:
-
Spondyloperipheral dysplasia
- SEDC:
-
Spondyloepiphyseal dysplasia congenital
- WES:
-
Whole-exome sequencing
- ACMG:
-
American College of Medical Genetics and Genomics
References
Kannu P, Bateman J, Savarirayan R. Clinical phenotypes associated with type II collagen mutations. J Paediatr Child Health. 2012;48(2):E38-43.
Barat-Houari M, et al. Mutation update for COL2A1 gene variants associated with type II collagenopathies. Hum Mutat. 2016;37(1):7–15.
Girisha KM, et al. Biallelic variants p.Arg1133Cys and p.Arg1379Cys in COL2A1: further delineation of phenotypic spectrum of recessive Type 2 collagenopathies. Am J Med Genet A. 2020;182(2):338–47.
Barat-Houari M, et al. Confirmation of autosomal recessive inheritance of COL2A1 mutations in spondyloepiphyseal dysplasia congenita: lessons for genetic counseling. Am J Med Genet A. 2016;170a(1):263–5.
Tham E, et al. Autosomal recessive mutations in the COL2A1 gene cause severe spondyloepiphyseal dysplasia. Clin Genet. 2015;87(5):496–8.
Al-Sannaa NA, et al. Spondylo-epiphyseal dysplasia in two sibs due to a homozygous splicing variant in COL2A1. Eur J Med Genet. 2020;63(12):104059.
Spranger J, Winterpacht A, Zabel B. The type II collagenopathies: a spectrum of chondrodysplasias. Eur J Pediatr. 1994;153(2):56–65.
Carter EM, Raggio CL. Genetic and orthopedic aspects of collagen disorders. Curr Opin Pediatr. 2009;21(1):46–54.
Barat-Houari M, et al. The expanding spectrum of COL2A1 gene variants IN 136 patients with a skeletal dysplasia phenotype. Eur J Hum Genet. 2016;24(7):992–1000.
Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5(4):725–38.
Zhang B, et al. Integrated analysis of COL2A1 variant data and classification of type II collagenopathies. Clin Genet. 2020;97(3):383–95.
Zankl A, et al. Dominant negative mutations in the C-propeptide of COL2A1 cause platyspondylic lethal skeletal dysplasia, torrance type, and define a novel subfamily within the type 2 collagenopathies. Am J Med Genet A. 2005;133a(1):61–7.
Terhal PA, et al. Mutation-based growth charts for SEDC and other COL2A1 related dysplasias. Am J Med Genet C Semin Med Genet. 2012;160c(3):205–16.
Nenna R, et al. COL2A1 gene mutations: mechanisms of spondyloepiphyseal dysplasia congenita. Appl Clin Genet. 2019;12:235–8.
Acknowledgements
The authors thank the family for participating and supporting this study.
Funding
This work was supported by the National Science Foundation for Young Scientists of China (Grant No.81900722), Pudong New Area Science and Technology Development Fund (Grant PKJ2018-Y46), and Shanghai Health and Family Planning Commission Fund (Grant 20204Y0346). The funders did not have any role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
XW designed the study; QZ drafted and revised the manuscript; RY revised the manuscript. QZ, QL, XL, BF, GC, and JW acquired, analyzed, and interpreted the data. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Ethics Committee of Shanghai Children's Medical Center (SCMCIRB-Y2019049), Shanghai Jiao Tong University School of Medicine. Written informed consent to participate was obtained from the patient’s parents.
Consent for publication
Written informed consent for publication of identifying images and other personal or clinical details was obtained from the patient’s parents. And the copy of the written consent is available for review by the editor of this journal.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Table S1. Variants filtering processes . Table S2. Significant variants found in the patient. Table S3. Clinical manifestations of patients with type II collagenopathies with homozygous mutations in different domains. Figure S1. Radiographs of the proband’s father. A, B. The cervical spine. C, D. The thoracic spine. F. The right pelvis. E, G. The lumbar spine. Figure S2. Radiographs of the proband’s mother. A, B. The Tervical spine. C, D. The thoracic spine. E, F. The right knee. G, H. The lumbar spine. Figure S3. Radiographs of the proband’s brother. A, B. The cervical spine. C, D. The thoracic and lumbar spine. E, F. The right knee.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, Q., Yao, R., Li, Q. et al. A novel homozygous variant of COL2A1 in a Chinese male with type II collagenopathy: a case report. BMC Med Genomics 14, 201 (2021). https://doi.org/10.1186/s12920-021-01048-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-021-01048-0