
Abstract
Background
Graves’ disease(GD) has a tendency for familial aggregation, but it is uncommon to occur in more than two generations. However, little is known about susceptibility genes for GD in the three-generation family.
Methods
DNA were extracted from three-generation familial GD patient with a strong genetic background in a Chinese Han population. The Whole Exome Sequencing (WES) was utilized to screen the genome for SNVs associated with GD and the Sanger Sequencing was used to confirm the potential disease-causing genes.
Results
In the case study, there were five patients with Graves’ disease(GD) from a three-generation family. The SNVs of MAP7D2(c. 452C > T: p. A151V), SLC1A7(c. 1204C > T: p. R402C), TRAF3IP3(c. 209A > T: p. N70I), PTPRB(c. 3472A > G: p. S1158G), PIK3R3(c. 121C > T: p. P41S), DISC1(c. 1591G > C: p. G531R) were found to be associated with the familial GD and the Sanger sequencing had confirmed these variations. Furthermore, PolyPhen-2 score showed that the variants in TRAF3IP3, PTPRB, PIK3R3 are more likely to change protein functions.
Conclusion
The MAP7D2, SLC1A7, TRAF3IP3, PTPRB, PIK3R3, DISC1 may be the candidate susceptibility genes for familial GD from a three generations family.
Similar content being viewed by others


Background
Graves’ disease is a thyroid-related autoimmune disorder which is caused by a complex interaction between susceptibility genes and multiple environmental factors. Previous familial and twin studies had shown that there was an association of genetic factors with Graves’ disease in 79% cases of Graves’ Disease which influenced the familial clustering in GD [1]. People with other autoimmune diseases such as type 1 diabetes or rheumatoid arthritis are also more likely to suffer from this disease. In addition, past reports reported that smoking increases the chance of Graves’ Disease. Other causes of Graves’ Disease may also include stress, infection, or childbirth. The prevalence of overt hyperthyroidism ranges from 0.2% to 1.3% in in the general population [2]. In China, the prevalence of hyperthyroidism is about 1.3% [3]. Graves’ disease is a multisystem syndrome including hypermetabolic syndrome, diffuse goiter, eye signs, skin lesions, and thyroid acropathy. The basic treatments of Graves’ disease are antithyroid drug treatment, radionuclide iodine treatment, surgical treatment and interventional embolization treatment. Family linkage analysis, candidate gene method and genome-wide association analysis (GWAS) have identified a greater number of Graves’ disease susceptibility loci as well. In GWAS, the existing sequence variations are identified from the whole human genome and the variations that related to the disease are screen out. GWAS method has allowed many previously undiscovered genes and chromosomal regions to be detected which help provide many clues to the pathogenesis of complex diseases. However, all the variants that have been discovered have little to the heritability of GD. Therefore, different approaches were applied in this case study to identify the more susceptibility loci.
In addition to twins study, the familial GD is the ideal object of study on contribution of genetic heritability to complex disease. Findings from family linkage analysis indicated that the 5q31-q33, 6p, 7q, 8q, 10q, 12q, 14q and 20q regions were related to GD susceptibility [4,5,6]. Subsequent research had verified that there was a linkage of familial GD to HLA gene and CTLA-4 gene. However, linkage analysis has not addressed the need for fine gene mapping in complex diseases and linkage analysis also requires a large familial sample size to localize the pathogenic genes by observing whether the genetic markers are co-segregated with the disease. In contrast, Whole Exome Sequencing can explore the susceptibility genes with a small sample size. Whole Exome Sequencing is an efficient strategy for fine mapping to determine the exact location of variant [7, 8]. In the case study, WES was utilized to screen disease-causing genes from a three-generation familial GD patient with a strong genetic background of Chinese Han.
Methods
Study participants
A three-generation family from Zhanjiang, Guangdong Province had been targeted for the case study. There were 25 people in the family tree including 13 males and 12 females (Fig. 1.). Five members were diagnosed with GD (Case 2 had type 2 diabetes simultaneously). Two healthy people in the first-degree relatives of the family were set as control group at the same time. Both the cases and control group were confirmed based upon medical history, physical examination, and results of thyroid function examination. All the five GD patients had typical clinical manifestation of hyperthyroidism such as heart and hands trembling. Four of the patients resorted to drug (Methimazole) for anti-thyroid treatment and one other patient resorted to I131 for anti-thyroid treatment. As of the month before the article was submitted, it was confirmed that the control group was still free of Graves' disease. Table 1 has the details of the participants.

Pedgree of study family
Thyroid function examination and susceptibility gene screening
Detection of thyroid hormones and TRAb by chemiluminescence method(Cobase411) was used and the reference range of indicators are: FT3: 2.3–6.8 pmol/L; FT4: 10–23.5 pmol/L; TSH: 0.34 ~ 4.0mIU/L; TRAb: 0 ~ 1.75 IU/L. For preparation of DNA, genomic DNA from EDTA-treated peripheral blood was extracted according to DNA extraction kit manual (Tiangen Biochemical Technology Co., Ltd.)
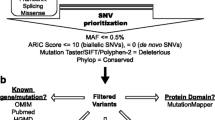
The Agilent SureSelect Human All Exome Kit(Agilent) was used for exon capture. The sequencing processes were exon capture, hybrid library cleaning and purification, PCR amplification of exon DNA library, library quality detection, sequencing, and data analysis. The PCR reaction conditions of PCR Amplification was initially 30 s at 98 °C, 10 cycles of 98 °C for 10 s, 10 cycles of 60 °C for 30 s, 10 cycles of 72 °C for 30 s, 10 cycles of 72 °C for 5 min at 72 °C, followed by a final extension of 4 °C. The exome region was sequenced by illumine hiseq2500, and GATK standard procedure was adopted to calibrate the initial data. Quality control of raw reads was performed with fastqc. Transition and Transversion (SNV) and Insertion and Deletion (InDel) of each sample were detected though VarScan and GATK HaplotypeCaller. In addition, SNV stands for single nucleotide variants and SNP stands for single nucleotide polymorphism. Both concepts refer to single nucleotide changes, but SNPs are generally two-state and SNV has no such restriction. In addition, if the frequency of the single-base variation in a species reaches a certain level, it is called SNP, and if the frequency is unknown (for example, only found in an individual), it is called SNV. Sanger sequencing was used to confirm the genotype variant of 6 genes (MAP7D2, SLC1A7, TRAF3IP3, PTPRB, PIK3R3, DISC1) that were in all the participants.
Results
The quality of raw data
The sequencing quality Q value was used to evaluate the sequencing error rate of the base. The base quality value Q20 indicated that the error rate was 1%. Similarly, the base quality value Q30 indicated that the error rate was 0.1%. In the case study, the data revealed that the sequencing quality of the seven samples were high (Table 2).
The base average coverage depth of all samples was larger than 100 × which meant that the detected SNV was reliable.
SNV/InDel detection and annotation of 7 samples
The SNV/InDel locus that was discovered with both VarScan and GATK methods was to be of high quality. If they were not discovered by both VarScan and GATK methods, the locus was medium quality. There were 144,169 high quality SNV/InDel locus involved in this study (Table 3).
The variations carried by the patient were identified as susceptibility genes of GD in the three-generation family, including MAP7D2(c. 452C > T: p. A151V), SLC1A7(c. 1204C > T: p. R402C), TRAF3IP3(c. 209A > T: p. N70I), PTPRB(c. 3472A > G: p. S1158G), PIK3R3(c. 121C > T: p. P41S), DISC1(c. 1591G > C: p. G531R). All SNV/InDel sites in the case study were rare variants according to several known population databases (Freq_Alt1000, Kaviar_20150923, ESP6500, gnomAD). In addition, Chinese Millionome Database (CMDB, https://db.cngb.org/cmdb/) were applied for the comparative analysis. CMDB contain considerable variation and their allele frequency information came from 141,431 unrelated healthy Chinese individuals (Phase I results). SNV function analysis indicated that all the SNV of MAP7D2, SLC1A7, TRAF3IP3, PTPRB, PIK3R3, DISC1 were of nonsynonymous variations. The genotype of the 6 genetic variations from the 7 samples by Sanger sequencing was the same with the results from the whole exome sequencing (Fig. 2). The PCR premiers of six genetic variants were shown in Table 4. Based on PolyPhen-2 prediction and the amino acids conservation analysis in orthologous species, the rs555004337 in TRAF3IP3, rs186466118 in PTPRB, and rs115181807 in PIK3R3 were likely to affect the protein function (Table 5 and Fig. 3). These genes involved in the Biological Process, Molecular Function, Cellular Component, and KEGG pathway were showed in an Additional file 1.

The Sanger sequencing of variations for the 7 participants

Amino acids conservation in orthologous species for the variants
Discussion
GD is an autoimmune disease with complex etiology. With the extensive development of GWAS research, many GD susceptibility genes have been identified such as HLA, CTLA4, PTPN22, and TSHR [9]. Gene mutations may affect the antigen presentation, T cell signal transduction, B cell antibody production, thyroid hormone, and thyroid-related apoptosis which may lead to the occurrence of GD. The gene mutation effects provide a theoretical basis for GD's precise diagnosis and treatment. However, the current impact of GD susceptibility gene polymorphism on the expression of corresponding proteins is still unknown and research on the interaction between genes is limited in elucidating the role of gene polymorphism in disease pathogenesis.
In the case study, it presents a rare familial GD case in 5 patients in a three-generation family. The five patients are consistent with the general characteristic of GD patients which is that the GD is prone to attack women at the age of 30–60 [10]. All the members of the three-generation family came from the same district of Zhanjiang city and they have been living in similar environment which guarantees the consistency of environmental factors in this case study. Although the etiology of GD is complex and clear identification of potential factors for GD has not been completed, it is widely recognized that the genetic determinants such as HLA, CTLA-4, PTPN22, and CD40 have contributed to the risk of GD [11]. However, no variation of these former identified genes was found, but the following variations of MAP7D2, SLC1A7, TRAF3IP3, PTPRB, PIK3R3, DISC1, and SUPT20HL were found in the familial GD. Furthermore, the variations in PTPRB, PIK3R3, and TRAF3IP3 were predicted to have alter the functions of the encoded protein. Familial GD of multigeneration is important for heritable studies because it avoids the genetic heterogeneity factor, so this case study may explain the genetic cause of the familial clustering of GD.
MAP7D2 (MAP7 domain containing 2) is located on X chromosome. MAP7D2 is specifically expressed in human brain tissue which has impact on the behavioral traits and cognition in human. MAP7D2 is also associated with sex-biased mental illnesses [12]. Previous studies has shown that gender predisposition to GD is associated with X chromosome inactivation (XCI) migration [13]. Thus, the MAP7D2 study is likely to provide important general information about the reason why women are more vulnerable to GD. SLC1A7 and DISC1 are also susceptibility genes for mental illnesses. In one research, Keith A. Young et al. discovered that DISC1 gene played a vital role in post-traumatic stress disorder (PTSD) severity of US military veterans [14]. In another research, Fujita K, et al. revealed that SLC1A7 gene expression in peripheral blood leukocytes was responsible for the association between socioeconomic status and depressive mood in healthy adults [15]. We speculated that SLC1A7 and DISC1 are involved in regulating the symptoms of GD such as nervousness and irritability
The protein encoded by PTPRB belongs to the family of protein tyrosine phosphatases (PTP). The activation of PTK (protein tyrosine kinase) was regulated through the binding of SH2 domains from PI3K(PIK3R3 gene encode). The balance of tyrosine protein phosphorylation was regulated by PTP/PTK which participated in cell signal transduction, cell growth regulation, differentiation, metabolism, transcription, immune responses, etc. Researches have demonstrated that the significant role of PTKs and PTPs were to modulate the tyrosine phosphorylation-dependent signaling pathways which were critical for the effector of NK cell and Neutrophil cell [16, 17]. TRAF3IP3 is also highly expressed in CD34 + CD38 + CD7 + common lymphoid progenitors (CLPs) Furthermore, CD34 + CD38 + CD7 + cells have the capacity to differentiate into B/NK/T cell which implies that TRAF3IP3 possibly may play a role in lymphoid development [18]. The overactivation of the T/B cell was regulated by CTLA-4 and CD40 gene variants which has been confirmed in the pathogenesis of autoimmune diseases including GD. Therefore, further studies are necessary to figure out whether the variations of PTPRB, PIK3R3 and TRAF3IP3 are involved in the dysfunction of thyroid autoimmune.
The case study is about the susceptibility genes of a single three-generation family of Graves disease, but there are insufficient samples of similar families to verify the results of this study. Also, the frequency of susceptibility genes screened in this study has not been further verified in the population of patients with sporadic Graves disease and the relationship between these gene mutations and sporadic Graves disease is uncertain. Finally, the susceptibility genes screened this time need to be further studied at the protein molecular level to further determine the biological significance of these mutations.
Conclusion
To summarize, the WES was applied to establish an association between MAP7D2, SLC1A7, TRAF3IP3, PTPRB, PIK3R3, DISC1 genes and the familial GD of a three-generation family. The findings in this cast studies are clues for further study and more verification and function researches are needed to explore these genes related to GD susceptibility.
Availability of data and materials
The datasets analysed during the current study are available in the Genome Sequence Archive for human repository (https://bigd.big.ac.cn/gsa-human/), under the accession code: HRA000505.
Abbreviations
- GD:
-
Graves’ disease
- WE:
-
Whole exome sequencing
References
Brix TH, Kyvik KO, Christensen K, Hegedüs L. Evidence for a major role of heredity in Graves’ disease: a population-based study of two Danish twin cohorts. J Clin Endocrinol Metab. 2001;86:930–4.
Taylor PN, Albrecht D, ScholzGlobal A, Gutierrez-Buey G, Lazarus JH, Dayan CM, Okosieme OE. epidemiology of hyperthyroidism and hypothyroidism. Nat Rev Endocrinol. 2018;14:301–16.
Lu, Z.; Zhong, N. Internal Medicine(M). Edition 7. Beijing; People's Publishing House. 2008; 712–721.
Sakai K, Shirasawa S, Ishikawa N, Ito K, Tamai H, Kuma K, Akamizu T, Tanimura M, Furugaki K, Yamamoto K, Sasazuki T. Identification of susceptibility loci for autoimmunethyroid disease to 5q31-q33 and Hashimoto’s thyroiditis to 8q23- q24 by multipoint affected sib-pair linkage analysis in Japanese. Hum Mol Genet. 2001;10:1379–86.
Jin Y, Teng W, Ben S, Xiong X, Zhang J, Xu S, Shugart YY, Jin L, Chen J, Huang W. Genome-wide scan of Graves’disease: evidence for linkage on chromosome 5q31 in Chinese Han pedigrees. J Clin Endocrinol Metab. 2003;88:1798–803.
Tomer Y, Ban Y, Concepcion E, Barbesino G, Villanueva R, Greenberg DA, Davies TF. Common and unique susceptibility loci in Graves and Hashimoto diseases: results of whole-genome screening in a data set of 102 multiplex families. Am J Hum Genet. 2003;73:736–47.
Glessner JT, Bick AG, Ito K, Homsy J, Rodriguez-Murillo L, Fromer M, Mazaika E, Vardarajan B, Italia M, Leipzig J, DePalma SR, Golhar R, Sanders SJ, Yamrom B, Ronemus M, Iossifov I, Willsey AJ, State MW, Kaltman JR, White PS, Shen Y, Warburton D, Brueckner M, Seidman C, Goldmuntz E, Gelb BD, Lifton R, Seidman J, Hakonarson H, Chung WK. Increased frequency of de novo copy number variants in congenital heart disease by integrative analysis of nucleotide variation array and exome sequence data. Circ Res. 2014;115:884–96.
Brastians PK, Amaro TW, Manley PE, Jones RT, Dias-Santagata D, Thorner AR, Lawrence MS, Rodriguez FJ, Bernardo LA, Schubert L, Sunkavalli A, Shillingford N, Calicchio ML, Lidov HG, Taha H, Martinez-Lage M, Santi M, Storm PB, Lee JY, Palmer JN, Adappa ND, Scott RM, Dunn IF, Laws ER Jr, Stewart C, Ligon KL, Hoang MP, Van HP, Hahn WC, Louis DN, Resnick AC, Kieran MW, Getz G, Santagata S. Exome sequencing identifies BRAF variations in papillary craniopharyngiomas. Nat Genet. 2014;46:161–5.
Simmonds MJ. GWAS in autoimmune thyroid disease: redefining our understanding of pathogenesis. Nat Rev Endocrinol. 2013;9:277–87.
Hemminki K, Li X, Sundquist J, undquist, K. . The epidemiology of Graves’ disease: evidence of a genetic and an environmental contribution. J Autoimmun. 2010;34:307–13.
Tomer Y. Mechanisms of autoimmune thyroid diseases: from genetics to epigenetics. Annu Rev Pathol. 2014;9:147–56.
Davies W. Genomic imprinting on the X chromosome: implications for brain and behavioral phenotypes. Ann N Y Acad Sci. 2010;1204(Suppl):E14–9.
Simmonds MJ, Kavvoura FK, Brand OJ, Newby PR, Jackson LE, Hargreaves CE, Franklyn JA, Gough SC. Skewed X chromosome inactivation and female preponderance in autoimmune thyroid disease: an association study and meta-analysis. J Clin Endocrinol Metab. 2014;99:E127-131.
Young KA, Morissette SB, Jamroz R, Meyer EC, Stanford MS, Wan L, Kimbrel NA. 5-HTTLPR and DISC1 risk genotypes for elevated PTSD symptoms in US military veterans. World Psychiatry. 2017;16:109–10.
Fujita K, Kuwano Y, Saijo S. Negative perception of socioeconomic status with depressive mood down-regulates expression of PPBP and SLC1A7 genes in peripheral blood leukocytes. Cogent Psychol. 2017;2017:4.
Vivier E, Nunès JA, Vély F. Natural killer cell signaling pathways. Science. 2004;306:1517–9.
Kruger J, Butler JR, Cherapanov V, Dong Q, Ginzberg H, Govindarajan A, Grinstein S, Siminovitch KA, Downey GP. Deficiency of Src homology 2-containing phosphatase 1 results in abnormalities in murine neutrophil function: studies in motheaten mice. J Immunol. 2000;165(10):5847–59.
Hoebeke I, De Smedt M, Stolz F, Pike-Overzet K, Staal FJ, Plum J, Leclercq G. T-, B- and NK-lymphoid, but not myeloid cells arise from human CD34(+)CD38(-)CD7(+) common lymphoid progenitors expressing lymphoid-specific genes. Leukemia. 2007;21:311–9.
Acknowledgements
We thanks to Shanghai Genesky Biotechnologies Inc. for providing WES technical support.
Funding
Our research was financially supported by grants from China Postdoctoral Science Foundation Grant(Grant NO. 2019 M652859). The funding bodies only provided the financial means to buy reagents for experiments, and the funding bodies played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
All authors contributed significantly to this study. ZQH and WGL were involved in designed the study, wrote and revised the paper. MSL and HW performed the experiment. YTC and ZHH retrieved the literature, analysed data. WL and AL collected patients blood sample and and clinical data. WGL supervised the research. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Informed written consent was obtained from all participants. In this study, no cases was under the age of 16. Ethical approval was provided by the the Ethics Committee of the Affiliated Hospital of Guangdong Medical University.
Consent for publication
Written informed consent for publication of the case was obtained for each participant. In our study, one case was under 18 and the written informed consent for publication was obtained from his parents.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1
. Gene function analysis.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hu, Z., Li, W., Li, M. et al. Screening of Graves' disease susceptibility genes by whole exome sequencing in a three-generation family. BMC Med Genomics 14, 46 (2021). https://doi.org/10.1186/s12920-020-00865-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-020-00865-z