
Abstract
Background
Serum alpha-fetoprotein (AFP) is the approved serum marker for hepatocellular carcinoma (HCC) screening. However, not all HCC patients show high (≥ 20 ng/mL) serum AFP, and the molecular mechanisms of HCCs with normal (< 20 ng/mL) serum AFP remain to be elucidated. Therefore, we aimed to identify biological features of HCCs with normal serum AFP by investigating differential alternative splicing (AS) between HCCs with normal and high serum AFP.
Methods
We performed a genome-wide survey of AS events in 249 HCCs with normal (n = 131) and high (n = 118) serum AFP levels using RNA-sequencing data obtained from The Cancer Genome Atlas.
Results
In group comparisons of RNA-seq profiles from HCCs with normal and high serum AFP levels, 161 differential AS events (125 genes; ΔPSI > 0.05, FDR < 0.05) were identified to be alternatively spliced between the two groups. Those genes were enriched in cell migration or proliferation terms such as “the cell migration and growth-cone collapse” and “regulation of insulin-like growth factor (IGF) transport and uptake by IGF binding proteins”. Most of all, two AS genes (FN1 and FAM20A) directly interact with AFP; these relate to the regulation of IGF transport and post-translational protein phosphorylation. Interestingly, 42 genes and 27 genes were associated with gender and vascular invasion (VI), respectively, but only eighteen genes were significant in survival analysis. We especially highlight that FN1 exhibited increased differential expression of AS events (ΔPSI > 0.05), in which exons 25 and 33 were more frequently skipped in HCCs with normal (low) serum AFP compared to those with high serum AFP. Moreover, these events were gender and VI dependent.
Conclusion
We found that AS may influence the regulation of transcriptional differences inherent in the occurrence of HCC maintaining normal rather than elevated serum AFP levels.
Similar content being viewed by others


Background
Hepatocellular carcinoma (HCC) is the sixth most common cancer in the world and the fourth leading cause of cancer-related deaths [1]. The prognosis of HCC is mainly determined by the tumor stage at time of discovery. However, most patients do not complain of symptoms before the HCC has progressed considerably [2], and this makes early diagnosis of HCC difficult. Moreover, patients diagnosed at an advanced stage usually miss the optimal window for receiving curative treatment, and their prognosis is ultimately very poor [2, 3]. Thus, in order to improve the prognosis of HCC patients, it is important to detect HCC early and to apply curative treatment in the early stages.
It was reported that early diagnosis using liver ultrasonography (USG) and serum alpha-fetoprotein (AFP) reduced the mortality rate of HCC patients by 37% [4]. Based on this result, liver USG at 6-month intervals is generally recommended for surveillance in high-risk groups (liver cirrhosis or hepatitis B or C), either alone or in combination with serum AFP testing [5,6,7,8,9]. For serum AFP, the standard cut-off value for maintaining appropriate sensitivity and specificity in HCC surveillance was initially reported to be 20 ng/mL [10]. However, some HCC patients show serum AFP levels below 20 ng/mL [11] and thus can escape early diagnosis despite receiving HCC surveillance tests using USG and serum AFP. Recently, there has been doubt about the usefulness of AFP testing for early diagnosis of HCC [5,6,7] but its utility remains in areas where the prevalence of HCC is high, as reflected in some guidelines for HCC surveillance [8, 9]. Although several studies have been performed to evaluate the clinical features of patients or tumor characteristics in the cases of HCC with normal (< 20 ng/mL) serum AFP levels [12,13,14], the molecular mechanisms underlying this feature remain to be determined. Given that newer biomarkers have yet to be developed that are more satisfying in HCC surveillance, it is necessary to understand the molecular biology of these HCCs with normal serum AFP levels.
In this study, we performed a genome-wide analysis to identify differential alternative splicing (AS) events between HCCs with normal (< 20 ng/mL) and high (≥ 20 ng/mL) serum AFP levels using RNA-sequencing data for tumor tissues obtained from The Cancer Genome Atlas (TCGA) database. Furthermore, we investigated the potential functions of genes with identified AS events and their association with HCC development (Additional file 1: Figures S1−S5).
Methods
Study population
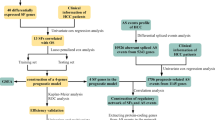
A total of 377 liver cancer patients were retrospectively analyzed, and their clinical data and tumor RNA sequencing data were obtained from TCGA (Fig. 1). The patients underwent surgical resection (n = 376) or liver transplantation (n = 1) for treatment of liver cancer between 1995 and 2013. Of these 377 patients, 15 were excluded due to having mixed types of hepatocholangiocarcinoma (n = 7), fibrolamellar carcinoma (n = 3), age of less than 18 years (n = 2), no available age data (n = 1), or recurred HCC (n = 2). Of the remaining 362 patients with primary HCC, 29 were also excluded due to the small number of the patients being Black or African American (n = 17), being American Indian or Alaska Indian (n = 2) or having no available data for race (n = 10). In addition, 84 patients were excluded due to having no available data for serum AFP (n = 78), RNA sequencing (n = 5), or duplication data entry (n = 1). Thus, 249 primary HCC patients with normal serum AFP (< 20 ng/mL, n = 131, normal/low AFP group) and elevated serum AFP (≥ 20 ng/mL, n = 118, high AFP group) were finally enrolled in this retrospective study (Additional file 2: Table S1). HCC was pathologically diagnosed in all patients.

Flowchart of patient enrollment and exclusions. Of the initial 377 patients, 249 were enrolled in this study
Clinical data
Clinical data for study subjects were obtained from TCGA; the collected data included age, gender, race, causes of HCC, presence of liver cirrhosis (LC), serum AFP, albumin, prothrombin time (PT), bilirubin, Child-Turcotte-Pugh (CTP) classification, and tumor stage (American Joint Committee on Cancer, AJCC). LC was pathologically diagnosed based on the Ishak fibrosis scoring system using surgically obtained peritumoral normal liver tissue [15, 16]. Because tumor tissues were recruited over a period of time, tumor stages as initially recorded were based on different versions of the AJCC staging system, ranging from the 4th to 7th; thus, in this study, all stages were adjusted to the AJCC 7th version.
Identification of alternative splicing events
RNA sequencing data, which was available for all enrolled patients (n = 249), was assessed to identify AS patterns in tumor tissues. We downloaded raw paired-end reads of RNA-seq Level 1 data (FASTQ files of study subjects) from the online Genomic Data Commons (GDC) data portal (http://gdac.broadinstitute.org/) in October 2018. We mapped the reads to the reference genome (i.e. release 75, GRCH 37.75 based on the hg19 reference sequence) using STAR v2.5 [17]. The proportions of uniquely mapped reads were greater than 80%. We detected AS events and quantified their expression as percent spliced in (PSI, ΔPSI or Δψ) using rMATs v3.2.5 [18]. PSI is a ratio of inclusion reads that indicates how efficiently sequences of interest are spliced into transcripts [19]. Analytically PSI can be represented as:
where IR is the number of inclusion reads and ER that of exclusion reads. ΔPSI is then the difference between the PSI values of the two groups under study. When identifying AS events differentially expressed between groups, ΔPSI > 5% with FDR < 0.05 was considered statistically significant [18]. All AS events were assessed: exon skipping (ES), intron retention (IR), alternative 3′ splicing site (A3SS), alternative 5′ splicing site (A5SS), and mutually exclusive exons (MXE). Gene-level expression in transcripts per million (TPM) was calculated by RSEM version 1.3.0 [20], and identification of differential gene expression was performed with the limma R package, version 3.38.3 [21], using as covariates AFP status, gender, and the presence or absence of vascular invasion.
Protein–protein interaction network analysis
Protein–protein interaction networks (PPINs) were created using genes from the STRING v.11.0 dataset (http://string-db.org/) [22]. In the absence of disconnected nodes, only the first gene interaction with a relaxed highest confidence > 0.7 was selected. PPINs were visualized using Cytoscape v3.7.2 [23].
Functional gene set enrichment analysis
Gene set enrichment analysis of the identified AS genes in terms of canonical pathways was conducted with the ConsensusPathDB-human database (CPDB) [24]. FDR < 0.05 was considered statistically significant.
Clinical association test
Within identified AS events, we investigated whether AFP-dependent splicing events had any relevant clinical features. First, we divided the 249 cases into two groups according to PSI values (i.e. low and high PSI) of AS exons through K-means clustering. With these two groups, we performed a Kaplan–Meier survival analysis to evaluate overall survival (OS) outcome according to PSI value. Other important clinical features of gender and vascular invasion were evaluated using Wilcoxon t-tests.
Statistical analysis
Patient baseline characteristics were described as mean ± standard deviation or as frequency. The significance of differences between categorical or continuous variables was determined by Chi-square test, Fisher’s exact test, or Student’s t test, as appropriate. Two-tailed p values of < 0.05 were considered statistically significant. In addition, to avoid false positives during multiple testing for DGE, we used a corrected q value obtained after FDR correction. A corrected FDR of < 0.05 was considered to be statistically significant. Statistical analyses of clinical data were performed using SPSS v19.0 (SPSS Inc, Chicago, IL, USA) [25]. All other analyses were conducted with R version 3.5.1 [26].
Results
Baseline characteristics of patients
The clinical characteristics of study subjects are shown in Table 1. The mean ages of normal and high AFP groups were 60 (standard deviation (SD) ± 13 years) and 59 years (SD ± 13 years), respectively (p = 0.317). The proportion of male patients was significantly higher in the normal AFP group (p = 0.005). Between the two AFP groups, there was no statistically significant difference in white/Asian race ratio, etiology, presence of LC, and AJCC 7th TNM stage (p values for all > 0.05). In the normal AFP group, vascular invasion was less likely than in the high AFP group (p = 0.008).
Comparability of AFP serum protein level and gene expression
In order to investigate whether AFP gene expression is comparable to serum levels of the corresponding protein product, AFP (ENSG00000081051) expression (TPM computed by RSEM) was compared to the serum AFP protein level (ng/mL, obtained from TCGA). To avoid bias due to high right-skew, the correlation coefficient was computed for log-transformed units. As shown in Fig. 2, as expected, AFP gene expression was positively correlated with AFP serum protein level (r = 0.73, p ≤ 2 × 10–16). Most of all, in high AFP patients, AFP is the most differentially expressed gene by absolute fold change and the six most significant (log value of fold change = 3.97, p = 1.72 × 10–9) when compared to low AFP patients (Fig. 3). Differential gene expression by AFP serum status is summarized in Additional file 3: Table S2.

AFP protein level is primarily explained by variation in AFP gene expression. Samples were grouped according to AFP status and fold changes calculated between “high AFP” and “normal AFP” samples. TPM, transcripts per million

Differential gene expression. Differential gene expression analysis was performed with the limma R package, version 3.38.3. Samples were grouped according to AFP status, and fold changes calculated between “high AFP” and “normal AFP” samples. Absolute fold changes of 2 with FDR = 0.05 are marked as blue lines. Significant genes are highlighted in red. The AFP gene is labeled. As expected, it shows the largest fold change of 3.97 (adjusted p = 1.72 × 10–9). Differential expression according to AFP status and additional covariates ‘vascular invasion’ and ‘gender’ are summarized in Additional file 3: Table S2
AS events differentially expressed between normal and high AFP groups
Having identified AFP-dependent gene expression (Fig. 2), we further investigated whether differences at the AS level were significant according to AFP level. We identified differential AS events between low and high AS groups using rMATs; these are summarized in Table 2, showing 161 differential AS events comprising 125 unique corresponding genes (ΔPSI > 0.05, FDR < 0.05). A more stringent cutoff of ΔPSI > 0.1 would have yielded only 16 AS events affecting 16 genes; we opted for the lower cutoff value in order to investigate more marginal differences between the two groups. This more detailed marginal signal comparison helps to investigate functional relationships between the groups. The distributions of each AS event type (ES, IR, A3SS, A5SS, and MXE) are summarized in Table 2 and detailed in Additional file 4: Table S3. As expected, ES and RI events were the most widely observed, followed by A5SS, A3SS, and MXE.
As stated above, we identified 125 genes with differential alternative splicing events in the context of AFP level. We then asked the question, “are these AFP level-dependent splicing events relevant to the mechanism underlying AFP levels in HCC patients?” We accordingly performed a multi-layered functional analysis at the network level combined with pathway analysis to interpret the relationship of molecular pathology to AFP level and performed a correlation test to evaluate the clinical implications of the identified AS events in HCC patients.
Functional analysis of genes with differentially expressed AS events
In an interaction network, proteins closer to each other are more likely to have related functions [27]. Proteins that are directly linked to a given protein (i.e. sharing a single network edge) are called "first interactors (first interacting groups)”. First interactors tend to be involved in the same disease or biological process [28]. We defined the first interactors of our 125 identified AS genes using the STRING database (relaxed highest confidence score ≥ 0.7) [29]. Of these, 31 genes were identified as having a known protein–protein interaction (PPI). The structure of this PPI network is illustrated in Fig. 4a. In order to investigate the functional association of the spliced genes with HCC molecular pathology, we further conducted functional enrichment analysis of the networked proteins over canonical pathways. Twenty genes from the PPI network (Additional file 1: Figure S1) were determined to belong to eight canonical pathways having significant enrichment, as illustrated in Fig. 4a. Most interestingly, protein products from two genes (FN1 and FAM20A) directly interact with AFP, with these genes belonging to the pathway “regulation of IGF transport and uptake by IGFBPs” and to “post-translational protein phosphorylation” or “extracellular matrix organization.”

Functional analysis of identified AS genes. a Canonical pathways enriched in the PPIN of AS genes. b Clinical implications of AS genes
Clinical association of differentially expressed AS events
The two AFP groups differed significantly in terms of gender and vascular invasion (VI) (Table 1), thus we tested the correlation of differentially expressed AS events with these variables. Among the 125 genes with differential AS events, 50 events (42 genes) and 34 events (27 genes) were gender and VI dependent, respectively as shown in Table 3 (more details in Additional file 5: Table S4). To further gain insight into the molecular connection between these factors and genetic pathways, we examined the gender- and VI-associated genes in the context of the entire 31genes PPIN, as shown in Fig. 4b. In the PPIN, six genes were associated with gender specifically; these consisted of three genes with ES events (FN1, GUSB, and CD44); two genes having RI events (RPP21 and SNRPA1); and one having both ES and MXE events (KHK). Moreover, seven genes were associated with VI specifically; these consisted of four genes with ES events (FN1, UGP2, DNAJC19, and RPS24); one gene having A5SS events (PQBP1); one gene with both ES and MXE events (KHK); and one gene having both ES and A5SS events (CHRD). All told, two genes (FN1 and KHK) were found to be associated with both gender and VI.
We additionally evaluated the presence in the PPIN of genes having differential AS that were associated with patient survival. Of 125 genes, 18 showed association with survival (Additional file 6: Table S5); of those, four genes (FN1, FAM20A, CHRD, and RPP21) were present in PPI networks. Of these 4 genes, we conducted survival analysis more in detail for FN1 and FAM20A, which were identified to directly interact with AFP in the present study (Fig. 4a). Notably, the estimated cumulative overall survival (OS) rate of patients with low levels of ES in FN1 and FAM20A were significantly higher than those of patients with high PSI for FN1 (p = 0.047) (Additional file 1: Figure S2A) and FAM20A (p = 0.024) (Additional file 1: Figure S2B), respectively.
For the two genes, we further analyzed the survival of patients according to higher PSI levels and gene-level expression. We grouped the 249 patients into high groups with PSI > 0.1 and 0.2 and low groups with PSI < 0.1 and 0.2 for each of two exons of FN1 and one exon of FAM20A, then tested for differences in survival. However, there were no survival differences in the context of PSI level (p values for all > 0.05). We also explored the survival of patients according to gene-level expression. The estimated cumulative overall survival (OS) rate of patients with low expression of FN1 was significantly higher than those of patients with high expression (p = 0.009) (Additional file 1: Figure S3A). However, there was no significant survival difference between patients with high and low expression of FAM20A (p = 0.489) (Additional file 1: Figure S3B).
Discussion
In this genome-wide analysis, we identified 125 genes with AS events that were differently expressed in the context of HCC with normal (low) serum AFP when compared to HCC with high serum AFP. Two of these AS genes, FN1 and FAM20A, had proteins that were identified to directly interact with AFP. Moreover, we found that in HCCs with normal serum AFP, FN1 exhibited more differential expression of AS events (ΔPSI > 0.05), with exons 25 and 33 of FN1 being more frequently skipped. Although the splicing level change is subtle, we believe that the observed splicing level change can be true as we found that gene-level expression did not differ based on AFP level. The present study is unique because we firstly imposed the possibility that AS may influence the regulation of transcriptional differences inherent in the occurrence of HCC with normal serum AFP level. These findings suggest that genome-wide analysis to identify differential AS events may be helpful for early diagnosis of HCCs showing normal (< 20 ng/mL) serum AFP. As FN1 is also known to be overexpressed in HCC [30], we further evaluated the differential expression of FNI transcript isoforms between low and high AFP groups. As shown in Additional file 1: Figure S4, there were nine transcript isoforms annotated as having a protein product (Ensembl version GRCh37 release 100) and expressed in our dataset, of which one transcript (ENST00000357867) showed slightly different fold change (1.48, p = 0.011). We also verified the reliability of the AS quantification using another popular tools, which is MISO [31]. We measured the PSI by MISO and the measured PSI value was compared to the PSI from rMAT. We found that the PSI values from these two different tools were highly correlated (median correlation r = 0.70), verifying the reliability of the AS quantification between different methods. Especially, PSI value of 33rd exon and 25th exon of FNI from two tools were also highly correlated (r = 0.90 and r = 0.93, respectively), Additional file 1: Figure S5.
Together with abdominal USG, AFP has been used as a screening method in populations at high risk for HCC development [5,6,7,8,9] as well as for non-biopsy HCC diagnosis [5, 32] and the assessment of treatment responses [33]. However, not all HCC tumors show high serum AFP levels [11, 34], and with a cut-off of 20 ng/mL, the sensitivity of this method for the diagnosis of HCC is about 60% [10]. Thus, only liver USG is recommended as a screening method for early diagnosis of HCC in the USA, Europe, and Asia–Pacific countries where the prevalence of HCC is relatively low [5,6,7]. On the other hand, in Korea and Japan, where the prevalence of HCC is high, it has been recommended to perform liver USG and serum AFP measurement together for HCC surveillance [8, 9]. In order to not miss HCC patients with normal serum AFP, it is necessary to identify the features of these HCCs. Previous studies have reported associations of tumor size or the presence of PVT [35], but even HCCs with low serum AFP can exhibit large size, suggesting other factors must be associated with serum AFP level in HCC. Therefore, it is necessary to understand the differential molecular pathology between cases with normal or higher serum AFP levels.
Until now, no data has been reported that explains the underlying pathophysiologic mechanisms of serum AFP levels in HCC at the transcriptional level. In the present study, in order to interpret the molecular pathophysiology of HCC in relation to serum AFP level, we evaluated the association of gene expression with serum AFP protein. We found that AFP is the most differentially expressed gene when considering AFP status, and furthermore identified 161 AS events (125 genes) whose occurrence differed significantly between normal and high AFP groups. Interestingly, in the present study, two genes (FN1 and FAM20A) were identified as encoding proteins that interact directly with AFP, and an ES event in FN1 was found to be closely associated with the development of HCCs having normal serum AFP.
Moreover, the present study identified an association of high PSI for an ES event in FN1 or FMA20A with poor prognosis of HCC patients, who underwent surgical resection. In light of these findings, FN1 and FAM20A should receive further experimental investigation to evaluate whether either can be a potential candidate for diagnostic biomarkers for early diagnosis of HCC in high-risk population for HCC, but with normal serum AFP, and be prognostic predictors of post-surgical outcomes for HCC in them. The present study did not directly analyze the functional association of FN1 and FAM20A with HCC prognosis, but our data may suggest AS to have an effect on the regulation of transcriptional differences with regard to the survival of HCC patients.
The gene FN1 encodes fibronectin (FN), a glycoprotein produced by hepatocytes and present in both plasma and cellular forms [36, 37]; its major functions are in cell adhesion, cell morphology, thrombosis, cell migration, and embryonic differentiation, as well as being part of the “extracellular matrix organization” pathway. FN is a modular protein composed of homologous repeats of three prototypical domains known as types I, II, and III [38]; of these, fibronectin type-III (FN3) is the largest and the most abundant in the protein [39,40,41,42].Previous studies have reported alternatively spliced mRNA variants of FN1, and these have been highly associated with HCC development [43, 44], but have not been investigated in the context of normal serum AFP level. In the present study, we found that two exons of FN1 were more frequently skipped in HCCs with normal serum AFP than in those with high serum AFP. Figure 5a depicts the skipping of exon 25 and exon 33 (ENST00000354785 transcript isoform), highlighted in yellow and cyan respectively, showing significant mean PSI difference associated with serum AFP (Fig. 5b, c, respectively). These exons encode part of the FN3 domain region (PF00041) of the protein product. Moreover, FN1 was also significantly associated with the clinical features of gender (25th exon only) and VI (33rd exon only) in HCC (Fig. 5d, e, respectively). While exon 25 was more skipped in male patients than female patients, exon 33 was more skipped in HCC without VI relative to cases with VI. These findings suggest that female patients with loss of FN3 domains due to skipping of exon 33 may be most likely to have HCCs with normal (low) serum AFP and without VI. Therefore, in these patients, HCC surveillance using liver USG with serum AFP testing may not be useful, and more careful inspection using liver CT or magnetic resonance imaging [45] may be needed for early detection of HCC.

Case study: FN1 gene. There are two exons more frequently skipped. a The gene overview, with skipped exons highlighted in yellow (25th exon, described in b, d and cyan (33rd, described in Panel C and E). The 25th (b) and 33rd exons (c) are more frequently skipped in low AFP HCC patients. Interestingly, the 25th exon tends to be more skipped in males compared to females (d), and the 33rd exon tends to be less skipped in VI (e)
In the present study, we investigated the relationship between AFP level and the functionality of the 125 genes having AS associated with HCC molecular pathology. Through functional enrichment analysis on the 31 genes having a known PPI with AFP, we identified eight enriched canonical pathways. Of particular note were FN1 and FAM20A, identified to directly interact with AFP and to be part of the pathways “regulation of IGF transport and uptake by IGFBPs” and “post-translational protein phosphorylation” or “extracellular matrix organization.” It has been reported that IGF expression is associated with HCC development [46,47,48]. IGFBPs bind IGF ligands with high affinity and modulate the half-life and bioavailability of IGF-I/III [49]. Moreover, IGFBP activities are closely regulated by post-translational modifications and IGFBP proteases [50,51,52,53,54]. These findings suggest that further understanding AS of FN1 may provide insight into whether these AFP level-dependent splicing events are relevant to the development of HCCs with normal serum AFP. However, no direct clinical data has been reported associating FAM20A with hepatocarcinogenesis, and thus, the association of this gene with HCC development needs to be validated in a further study.
There are some limitations in this study. First, this study was retrospectively analyzed due to the nature of the data, and thus selection bias was inevitable. Second, validation using blood samples of HCC patients was not performed. However, we applied well-defined bioinformatics analysis methods to identify differentially expressed AS events between HCCs with normal and high serum AFP levels. Nonetheless, the outcomes of this study need to be validated using a well-designed prospective study in the future.
Conclusion
In conclusion, in this genome-wide study, we found that AS of FN1 was differently expressed in HCC with normal serum AFP compared to those with high serum AFP. Moreover, we found that skipping of exon 33 of FN1 occurred more frequently in the normal AFP group than in the high AFP group, and less frequently in patients with VI. Interestingly, the PSI level of exon 33 is also associated with overall survival rate. Given that AS events associated with the development of HCC having normal AFP level has not been sufficiently investigated, we expect that our outcomes may provide helpful information for understanding the molecular characteristics of HCCs with normal serum AFP. In addition, if these results are validated in well-designed future studies, it may be possible to carry out individualized HCC surveillance in high-risk populations using a genome-wide study and consideration of serum AFP.
Availability of data and materials
All data generated or analyzed during this study are included in this published article (and its supplementary information files).
Abbreviations
- AFP:
-
Alpha-fetoprotein
- AJCC:
-
American Joint Committee on Cancer
- AS:
-
Alternative splicing
- CTP:
-
Child-Turcotte-Pugh
- DGE:
-
Differential gene expression
- GDC:
-
Genomic Data Commons
- HCC:
-
Hepatocellular carcinoma
- LC:
-
Liver cirrhosis
- OS:
-
Overall survival
- PT:
-
Prothrombin time
- PVT:
-
Portal Vein Thrombosis
- TCGA:
-
The Cancer Genome Atlas
- TNM:
-
Tumor node metastasis
- USG:
-
Ultrasonography
- VI:
-
Vascular invasion
References
Fitzmaurice C, Allen C, Barber RM, Barregard L, Bhutta ZA, Brenner H, Dicker DJ, Chimed-Orchir O, Dandona R, Global Burden of Disease Cancer Collaboration, et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: a systematic analysis for the global burden of disease study. JAMA Oncol. 2017;3(4):524–48.
Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362(9399):1907–17.
Llovet JM, Bru C, Bruix J. Prognosis of hepatocellular carcinoma: the BCLC staging classification. Semin Liver Dis. 1999;19(3):329–38.
Zhang BH, Yang BH, Tang ZY. Randomized controlled trial of screening for hepatocellular carcinoma. J Cancer Res Clin Oncol. 2004;130(7):417–22.
Heimbach JK, Kulik LM, Finn RS, Sirlin CB, Abecassis MM, Roberts LR, Zhu AX, Murad MH, Marrero JA. AASLD guidelines for the treatment of hepatocellular carcinoma. Hepatology. 2018;67(1):358–80.
European Association for the Study of the Liver. Electronic address EEE, European Association for the Study of the L: EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J Hepatol. 2018;69(1):182–236.
Omata M, Cheng AL, Kokudo N, Kudo M, Lee JM, Jia J, Tateishi R, Han KH, Chawla YK, Shiina S, et al. Asia-Pacific clinical practice guidelines on the management of hepatocellular carcinoma: a 2017 update. Hepatol Int. 2017;11(4):317–70.
Korean Liver Cancer Study Group (KLCSG), National Cancer Center, Korea (NCC). 2014 KLCSG-NCC Korea practice guideline for the management of hepatocellular carcinoma. Gut Liver. 2015;9(3):267–317.
Kudo M, Matsui O, Izumi N, Iijima H, Kadoya M, Imai Y, Okusaka T, Miyayama S, Tsuchiya K, Ueshima K, et al. JSH consensus-based clinical practice guidelines for the management of hepatocellular carcinoma: 2014 update by the Liver Cancer Study Group of Japan. Liver Cancer. 2014;3(3–4):458–68.
Trevisani F, D’Intino PE, Morselli-Labate AM, Mazzella G, Accogli E, Caraceni P, Domenicali M, De Notariis S, Roda E, Bernardi M. Serum alpha-fetoprotein for diagnosis of hepatocellular carcinoma in patients with chronic liver disease: influence of HBsAg and anti-HCV status. J Hepatol. 2001;34(4):570–5.
Colombo M. Screening for cancer in viral hepatitis. Clin Liver Dis. 2001;5(1):109–22.
Nomura F, Ohnishi K, Tanabe Y. Clinical features and prognosis of hepatocellular carcinoma with reference to serum alpha-fetoprotein levels. Analysis of 606 patients. Cancer. 1989;64(8):1700–7.
Carr BI, Guerra V. Low alpha-fetoprotein levels are associated with improved survival in hepatocellular carcinoma patients with portal vein thrombosis. Dig Dis Sci. 2016;61(3):937–47.
Hamamura K, Shiratori Y, Shiina S, Imamura M, Obi S, Sato S, Yoshida H, Omata M. Unique clinical characteristics of patients with hepatocellular carcinoma who present with high plasma des-gamma-carboxy prothrombin and low serum alpha-fetoprotein. Cancer. 2000;88(7):1557–64.
Everhart JE, Wright EC, Goodman ZD, Dienstag JL, Hoefs JC, Kleiner DE, Ghany MG, Mills AS, Nash SR, Govindarajan S, et al. Prognostic value of Ishak fibrosis stage: findings from the hepatitis C antiviral long-term treatment against cirrhosis trial. Hepatology. 2010;51(2):585–94.
Goodman ZD. Grading and staging systems for inflammation and fibrosis in chronic liver diseases. J Hepatol. 2007;47(4):598–607.
Dobin A, Gingeras TR. Mapping RNA-seq Reads with STAR. Curr Protoc Bioinform. 2015;51:11–9.
Shen S, Park JW, Lu ZX, Lin L, Henry MD, Wu YN, Zhou Q, Xing Y. rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc Natl Acad Sci U S A. 2014;111(51):E5593-5601.
Schafer S, Miao K, Benson CC, Heinig M, Cook SA, Hubner N. Alternative splicing signatures in RNA-seq data: percent spliced in (PSI). Curr Protoc Hum Genet. 2015;87(1):11.16.11-11.16.14.
Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011;12(1):323.
Smyth GK. Limma: linear models for microarray data. In: Bioinformatics and computational biology solutions using R and bioconductor. Berlin: Springer; 2005. p. 397–420.
Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607–13.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
Kamburov A, Stelzl U, Lehrach H, Herwig R. The ConsensusPathDB interaction database: 2013 update. Nucleic Acids Res. 2013;41(Database issue):D793–800.
SPSS I. SPSS Statistical Software: Release 19.0. Armonk: IBM Corporation; 2012.
Team RC. R: a language and environment for statistical computing. 2013.
Sharan R, Ulitsky I, Shamir R. Network-based prediction of protein function. Mol Syst Biol. 2007;3:88.
Hartwell LH, Hopfield JJ, Leibler S, Murray AW. From molecular to modular cell biology. Nature. 1999;402(6761 Suppl):C47-52.
Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45(D1):D362–8.
Kim H, Park J, Kim Y, Sohn A, Yeo I, Jong YuS, Yoon JH, Park T, Kim Y. Serum fibronectin distinguishes the early stages of hepatocellular carcinoma. Sci Rep. 2017;7(1):9449.
Katz Y, Wang ET, Airoldi EM, Burge CB. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat Methods. 2010;7(12):1009–15.
El-Serag HB, Kanwal F, Davila JA, Kramer J, Richardson P. A new laboratory-based algorithm to predict development of hepatocellular carcinoma in patients with hepatitis C and cirrhosis. Gastroenterology. 2014;146(5):1249–55.
Memon K, Kulik L, Lewandowski RJ, Wang E, Ryu RK, Riaz A, Nikolaidis P, Miller FH, Yaghmai V, Baker T, et al. Alpha-fetoprotein response correlates with EASL response and survival in solitary hepatocellular carcinoma treated with transarterial therapies: a subgroup analysis. J Hepatol. 2012;56(5):1112–20.
Giannini EG, Marenco S, Borgonovo G, Savarino V, Farinati F, Del Poggio P, Rapaccini GL, Anna Di Nolfo M, Benvegnu L, Zoli M, et al. Alpha-fetoprotein has no prognostic role in small hepatocellular carcinoma identified during surveillance in compensated cirrhosis. Hepatology. 2012;56(4):1371–9.
Carr BI, Akkiz H, Uskudar O, Yalcin K, Guerra V, Kuran S, Karaogullarindan U, Altintas E, Ozakyol A, Tokmak S, et al. HCC with low- and normal-serum alpha-fetoprotein levels. Clin Pract (Lond). 2018;15(1):453–64.
Tamkun JW, Hynes RO. Plasma fibronectin is synthesized and secreted by hepatocytes. J Biol Chem. 1983;258(7):4641–7.
Roy DC, Hocking DC. Recombinant fibronectin matrix mimetics specify integrin adhesion and extracellular matrix assembly. Tissue Eng A. 2013;19(3–4):558–70.
Petersen TE, Thogersen HC, Skorstengaard K, Vibe-Pedersen K, Sahl P, Sottrup-Jensen L, Magnusson S. Partial primary structure of bovine plasma fibronectin: three types of internal homology. Proc Natl Acad Sci U S A. 1983;80(1):137–41.
Aota S, Nomizu M, Yamada KM. The short amino acid sequence Pro-His-Ser-Arg-Asn in human fibronectin enhances cell-adhesive function. J Biol Chem. 1994;269(40):24756–61.
Aziz-Seible RS, Casey CA. Fibronectin: functional character and role in alcoholic liver disease. World J Gastroenterol. 2011;17(20):2482–99.
Singh P, Schwarzbauer JE. Fibronectin and stem cell differentiation-lessons from chondrogenesis. J Cell Sci. 2012;125(Pt 16):3703–12.
Veevers-Lowe J, Ball SG, Shuttleworth A, Kielty CM. Mesenchymal stem cell migration is regulated by fibronectin through alpha5beta1-integrin-mediated activation of PDGFR-beta and potentiation of growth factor signals. J Cell Sci. 2011;124(Pt 8):1288–300.
Jagirdar J, Ishak KG, Colombo M, Brambilla C, Paronetto F. Fibronectin patterns in hepatocellular carcinoma and its clinical significance. Cancer. 1985;56(7):1643–8.
Matsui S, Takahashi T, Oyanagi Y, Takahashi S, Boku S, Takahashi K, Furukawa K, Arai F, Asakura H. Expression, localization and alternative splicing pattern of fibronectin messenger RNA in fibrotic human liver and hepatocellular carcinoma. J Hepatol. 1997;27(5):843–53.
Kim HL, An J, Park JA, Park SH, Lim YS, Lee EK. Magnetic resonance imaging is cost-effective for hepatocellular carcinoma surveillance in high-risk patients with cirrhosis. Hepatology. 2019;69(4):1599–613.
Mazziotti G, Sorvillo F, Morisco F, Carbone A, Rotondi M, Stornaiuolo G, Precone DF, Cioffi M, Gaeta GB, Caporaso N, et al. Serum insulin-like growth factor I evaluation as a useful tool for predicting the risk of developing hepatocellular carcinoma in patients with hepatitis C virus-related cirrhosis: a prospective study. Cancer. 2002;95(12):2539–45.
Schirmacher P, Held WA, Yang D, Chisari FV, Rustum Y, Rogler CE. Reactivation of insulin-like growth factor II during hepatocarcinogenesis in transgenic mice suggests a role in malignant growth. Cancer Res. 1992;52(9):2549–56.
Breuhahn K, Vreden S, Haddad R, Beckebaum S, Stippel D, Flemming P, Nussbaum T, Caselmann WH, Haab BB, Schirmacher P. Molecular profiling of human hepatocellular carcinoma defines mutually exclusive interferon regulation and insulin-like growth factor II overexpression. Cancer Res. 2004;64(17):6058–64.
Baxter RC. Insulin-like growth factor (IGF)-binding proteins: interactions with IGFs and intrinsic bioactivities. Am J Physiol Endocrinol Metab. 2000;278(6):E967-976.
Mohan S, Baylink DJ. IGF-binding proteins are multifunctional and act via IGF-dependent and -independent mechanisms. J Endocrinol. 2002;175(1):19–31.
Lelbach A, Muzes G, Feher J. The insulin-like growth factor system: IGFs, IGF-binding proteins and IGFBP-proteases. Acta Physiol Hung. 2005;92(2):97–107.
Clemmons DR. Role of insulin-like growth factor binding proteins in controlling IGF actions. Mol Cell Endocrinol. 1998;140(1–2):19–24.
Arai T, Busby W Jr, Clemmons DR. Binding of insulin-like growth factor (IGF) I or II to IGF-binding protein-2 enables it to bind to heparin and extracellular matrix. Endocrinology. 1996;137(11):4571–5.
Jones JI, D’Ercole AJ, Camacho-Hubner C, Clemmons DR. Phosphorylation of insulin-like growth factor (IGF)-binding protein 1 in cell culture and in vivo: effects on affinity for IGF-I. Proc Natl Acad Sci U S A. 1991;88(17):7481–5.
Acknowledgements
The support and resources from the Center for High Performance Computing and Vice President’s Clinical and Translational Research Scholar Program at the University of Utah are gratefully acknowledged.
About this supplement
This article has been published as part of BMC Medical Genomics Volume 13 Supplement 11 2020: Data-driven analytics in biomedical genomics. The full contents of the supplement are available at https://bmcmedgenomics.biomedcentral.com/articles/supplements/volume-13-supplement-11.
Funding
The work is supported by development funding from Department of Biomedical Informatics, University of Utah School of Medicine, the Collaborative Genome Program for Fostering New Post-Genome Industry of the National Research Foundation (NRF) and Information Technology Research Center (ITRC) support program (IITP-2020-2018-0-01833) supervised by the Institute for Information & Communications Technology Planning & Evaluation (IITP) funded by the Ministry of Science and ICT (MSIT) (NRF-2017M3C9A6047623), and Inha University Research grant. Publication cost are funded by the development funding from Department of Biomedical Informatics, University of Utah School of Medicine. None of the funding body had a role in the study design, data collection, analysis, and interpretation of data, or in the writing of the manuscript.
Author information
Authors and Affiliations
Contributions
YJJ and YHL were responsible for the concept and design of the study, the acquisition, analysis and interpretation of the data, and the drafting of the manuscript. HMA, JC, SGH, JHS, and SYB helped with analysis and interpretation of the data, and the drafting of the manuscript. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
12920_2020_836_MOESM1_ESM.pdf
Additional file 1: Figure S1. Pathways significantly enriched in AS genes. Figure S2. Cumulative overall survival curve of patients according to PSI level in FN1 and FAM20A. Cumulative overall survival curve of patients according to PSI level of an exon skipping event in FN1 (A) and an exon skipping event in FAM20A (B). These events are marked in orange color in the Additional file 6: Table S5. Figure S3. Cumulative overall survival curve of patients according to FN1 and FAM20A gene expression. Cumulative overall survival curve of patients according to expression of an exon skipping event in FN1 (A) and an exon skipping event in FAM20A (B). K-means clustering was used to divide the cases into two groups according to the gene-level expression value calculated by RSEM. With these two groups, we performed a Kaplan–Meier survival analysis to evaluate overall survival (OS) outcome according to gene expression. Figure S4. Expression of FN1 mRNA transcripts between low and high AFP groups. We selected protein coding transcripts for which the median value of expression in total samples was more than zero, and compared expression between low (< 20 ng/mL) and high (> = 20 ng/mL) serum AFP groups with a t-test. Fold changes were calculated as the ratio of mean expression and tested by t-test. Figure S5. High correlation of PSI values between two different AS quantification tools. (A) A scatter plot of PSI level of 33rd exon in FN1 calculated from x-axis (rMATs) and y-axis (MISO). The correlation is 0.900. (B) A scatter plot of PSI level of 25th exon in FN1 calculated from x-axis (rMATs) and y-axis (MISO). The correlation is 0.928.
Additional file 2: Table S1.
249 TCGA patients enrolled in the study.
Additional file 3: Table S2.
Differential gene expression between the normal and high AFP groups.
Additional file 4: Table S3.
Significant AS events differentially expressed between the normal and high AFP groups.
Additional file 5: Table S4.
Correlation of differentially expressed AS events with gender and VI.
Additional file 6: Table S5.
Correlation of differentially expressed AS events with survival rate.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Jin, YJ., Aycheh, H.M., Han, S. et al. Differential alternative splicing between hepatocellular carcinoma with normal and elevated serum alpha-fetoprotein. BMC Med Genomics 13 (Suppl 11), 194 (2020). https://doi.org/10.1186/s12920-020-00836-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-020-00836-4