
Abstract
Background
Understanding gene regulation is important but difficult. Elucidating tissue-specific gene regulation mechanism is even more challenging and requires gene co-expression network assembled from protein–protein interaction, transcription factor and gene binding, and post-transcriptional regulation (e.g., miRNA targeting) information. The miRNA binding affinity could therefore be changed by SNP(s) located at the 3′ untranslated regions (3′UTR) of the target messenger RNA (mRNA) which miRNA(s) interacts with. Genome-wide association study (GWAS) has reported significant numbers of loci hosting SNPs associated with many traits. The goal of this study is to pinpoint GWAS functional variants located in 3′UTRs and elucidate if the genes harboring these variants along with their targeting miRNAs are associated with genetic traits relevant to certain tissues.
Methods
By applying MIGWAS, CoCoNet, ANNOVAR, and DAVID bioinformatics software and utilizing the gene expression database (e.g. GTEx data) to study GWAS summary statistics for 43 traits from 28 GWAS studies, we have identified a list of miRNAs and targeted genes harboring 3′UTR variants, which could contribute to trait-relevant tissue over miRNA-target gene network.
Results
Our result demonstrated that strong association between traits and tissues exists, and in particular, the Primary Biliary Cirrhosis (PBC) trait has the most significant p-value for all 180 tissues among all 43 traits used for this study. We reported SNPs located in 3′UTR regions of genes (SFMBT2, ZC3HAV1, and UGT3A1) targeted by miRNAs for PBC trait and its tissue association network. After employing Gene Ontology (GO) analysis for PBC trait, we have also identified a very important miRNA targeted gene over miRNA-target gene network, PFKL, which encodes the liver subunit of an enzyme.
Conclusions
The non-coding variants identified from GWAS studies are casually assumed to be not critical to translated protein product. However, 3′ untranslated regions (3′UTRs) of genes harbor variants can often change the binding affinity of targeting miRNAs playing important roles in protein translation degree. Our study has shown that GWAS variants could play important roles on miRNA-target gene networks by contributing the association between traits and tissues. Our analysis expands our knowledge on trait-relevant tissue network and paves way for future human disease studies.
Similar content being viewed by others
Background
A microRNA (miRNA), a noncoding RNA which contains about 22 nucleotides, plays a significant role on the regulation of gene expression. By binding to the 3′ untranslated regions (3′UTR) of the target messenger RNA (mRNA), which transfers the genetic information from DNA to the ribosome for protein synthesis in RNA polymerase, miRNA suppresses the translation of the targeting mRNA and/or can activate gene expression under certain conditions [1]. Due to those features and miRNA’s ability to control cell growth and differentiation, people believe that there is an association between miRNA’s deficiency or excess and human diseases [2].
Single nucleotide polymorphism or SNP is a change or mutation at a single position in a DNA sequence. The development of human diseases is also affected by SNP(s) on the basis of the observation that SNPs can cause synonymous and non-synonymous changes which affects amino acid sequence or protein function [3]. However, 3′ untranslated regions (3′UTR) of genes often contain variants which can change binding affinity of targeting miRNAs so that gene expression and protein translation will be affected. It’s important to pinpoint functional important variants in 3′UTRs and elucidate if the genes harboring these variants along with their targeting miRNAs are associated with genetic traits.
In many cancers, the gene PTEN is known to be regulated by miRNAs through competing endogenous RNA networks [4, 5]. Such miRNAs can also have other targeted genes containing SNPs that could contribute to cancers and/or human diseases by changing DNA sequences or influencing gene expression.
Genome-wide association studies (GWAS) has identified and analyzed millions of genetic risk variants which trigger the development of complex diseases [6], and it has reported significant numbers of loci hosting SNPs associated with many traits as well [7]. The disease causativeness annotation for non-coding variants (e.g. 3′UTR variants) identified from GWAS studies and clarification of their effects on miRNAs binding are challenging. MIGWAS (miRNA–target gene networks enrichment on GWAS) is an analytic pipeline to quantitatively evaluate tissue enrichment and elucidate complex biology of the genetic traits over miRNA-target gene networks which provide resources for the genetics of human complex traits, and thus contributes to a deeper understanding of miRNA’s influence on human diseases as well as drug discovery [7, 8].
Composite likelihood-based Covariance regression Network model (CoConet) is a network method for identification of trait-relevant tissues or cell types by incorporating tissue-specific gene co-expression networks [9]. CoCoNet further understands data from GWAS by demonstrating gene co-expression sub-networks which helps predict gene-level association effect sizes on and GWAS traits and diseases. Moreover, CoConet infers trait-relevant tissue based on tissue-specific gene co-expression patterns and proves that tissue-specific gene networks underlie disease etiology [9].
ANNOVAR [10] is an efficient software tool to utilize update-to-date information to annotate genetic variants based on their functional influence. SNPs reported from large scale association studies and sequencing projects can be annotated by ANNOVAR which can be utilized to report functional score and identify variants based on SNPs’ functional influence on genes [10].
The goal of our study is to identify causative SNPs which could change the binding affinity of miRNAs and genes, which could contribute to trait-tissue relevance over miRNA-target gene network.
Methods
We obtained summary statistics in the form of marginal z-scores for 43 traits from 28 GWAS studies [11]. These studies collect a wide range of complex traits and diseases that can be classified into six phenotype categories [12, 13]: anthropometric traits (e.g. height and BMI), hematological traits (e.g. MCHC and RBC), autoimmune diseases (e.g. CD and IBD), neurological diseases (e.g. Alzheimer's disease and Schizophrenia), metabolic traits (e.g. FG and HDL), and social traits (e.g. ever smoked and college completion). We removed SNPs within the major histocompatibility complex (MHC) region (Chr6: 25–34 Mb) following [14]. We then intersected the SNPs from all the studies and retained a common set of 622,026 SNPs for analysis. We paired the marginal z-scores from these studies with the SNP correlation matrix estimated using 503 individuals of European ancestry from the 1000 Genomes Project [15] for inference.
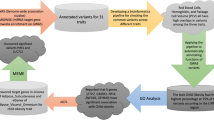
We employed MIGWAS [7], CoCoNet [9], and ANNOVAR [10] software packages to infer trait-relevant tissues based on next-generation sequencing omics data (e.g. GTEx data [16]) and annotated the variants obtained from GWAS and harbored by miRNAs targeted genes associated with traits. We also conducted functional analysis on miRNA targeted genes harboring 3′UTR variants with significant tissue-trait association using annotation tool DAVID [17, 18] for the Primary Biliary Cirrhosis (PBC) trait (Fig. 1).

Workflow of elucidating variants and miRNAs contributed to tissue-trait association
Results
Data acquisition
We obtained 43 traits from various published resources and classified them into six different categories with the total number of samples 3 × 106 (Table 1). Details of the information for the summary statistics of 43 traits from 29 GWAS studies are provided in Additional file 1: Table S1. The table lists the phenotype name, category, abbreviation, number of individuals, reference, and downloaded websites for each of the 43 traits.
MIGWAS Results
We applied MIGWAS to 43 traits (total samples = 3,035,223) and identified biologically relevant tissues. In particular, we identified that 94 out of 180 tissues have significant (p-value < 0.01) association with at least two traits and that 11 traits have significant association with more than 10 tissues (Fig. 2).

Association between tissues and traits identified by MIGWAS for 43 GWAS
We calculated p-value association between traits and all tissues using MIGWAS for 43 GWAS. Interestingly, it reported that PBC trait has the most significant p-value for all tissues (Fig. 3). A detailed association result from MIGWAS is shown in Additional file 2: Table S2.

Association between traits with all tissues identified by MIGWAS for 43 GWAS
We have identified total 332 miRNAs as candidate biomarkers of all traits. The miRNA distribution statistics for all reported traits by MIGWAS is shown in Fig. 4. We found that there are no miRNAs identified through MIGWAS overlapping across significant tissue-traits. We divided 31 traits into two groups that the significant trait group has 15 traits and the non-significant trait group has 16 traits. If the p-value of the trait is less than 0.01 in at least one tissue, it is considered as a significant trait. The non-significant trait Height has 60 miRNAs which is the most among all 31 traits. A list of detailed gene-miRNA pairs reported for all traits is shown in Additional file 3: Table S3.

The miRNA count distribution for all reported traits by MIGWAS (a includes significant traits whose p-value are less than 0.01 in at least one tissue, while b includes non-significant traits
CoCoNet output
We took candidate target genes identified by MIGWAS to retrieve Gene Level Effective Size of each gene (Ensemble gene ID) for the PBC trait. We used CoCoNet to calculate the association between traits and tissues in terms of log likelihood (Fig. 5).

Likelihood values between PBC trait and tissues (red ones have greater loglikelihood based on − 4536.7474 (the log(likelihood) for PBC))
ANNOVAR annotation
We examined the PBC trait showing the smallest association p-value reported by MIGWAS to see if there are any SNPs located in 3′UTR regions of genes targeted by miRNAs. To do so, we took the SNP list for the PBC trait and converted them to ANNOVAR variant annotated input format with ANNOVAR package. We obtained 9,890 annotated variants (5279 genes) exclusively located in 3′UTR region of these genes (Additional file 4: Table S4).
We crossed the 55 genes reported by MIGWAS with 5279 genes annotated by ANNOVAR and obtained 13 genes in common. The detailed information about these 13 genes is shown in Table 2.
PTEN associated variant analysis
Since PBC is a liver-related trait, we searched the TCGA LIHC dataset [19] and checked anti-correlated pairs for PTEN targeted miRNA: hsa-mir-590. We then used hsa-mir-590 to identify its targeted genes in the anti-correlated pair list and obtained 22 additional target genes. We then identified SNPs using UCSC Genome Browser for ten genes including PTEN with ClinVar variant information and reported the result in a new Additional file 5: Table S5.
Functional analysis
Gene Ontology (GO) analysis on 55 miRNA targeted genes harboring 3′UTR variants with significant tissue-trait association for PBC trait only showed that PFKL is located in a set of genes with term Acetylation (p-value < 0.05). This gene encodes the liver (L) subunit of an enzyme that catalyzes the conversion of d-fructose 6-phosphate to d -fructose 1,6-bisphosphate, which is a key step in glucose metabolism (glycolysis) [20].
The list of genes and their GO terms identified as the top cluster by DAVID is shown in Table 3.
We have also conducted a GO analysis for 22 genes targeted by hsa-mir-590 with target relationship of PTEN. Some enrichment analysis results are reported in Table 4. The detailed DAVID results are reported in Additional file 6: Table S6 and Additional file 7: Table S7.
Discussions
In our study, we used several cutting-edge bioinformatics tools (MIGWAS, CoCoNet and ANNOVAR) to dissecting SNPs reported for 43 traits from GWAS. The MIGWAS reported miRNA enrichment over target gene network for most (31) of traits. The CoCoNet was adopted to analyze the PBC trait for its significant tissue association over gene co-expression network. The ANNOVAR tool was employed to annotate 3′UTR variants class harbored by a list of miRNA target genes associated with Primary Biliary Cirrhosis (PBC) traits.
Although it may seem like the tools are interrelated and are three separate experiments, they are indeed connected. We first applied the tool MIGWAS to 43 traits and identified significant biologically relevant tissues. When the p-value association between traits and all tissues were calculated by using MIGWAS for 43 GWAS, we found that the PBC trait had the most significant p-value for all tissues. The CoCoNet experiment then utilized some of the results produced by MIGWAS to provide an output. Specifically, for the retrieved PBC trait result from the MIGWAS experiment, we used CoCoNet to analyze the tissues association over gene co-expression network. Again, we used the PBC trait result reported by MIGWAS in another tool: ANNOVAR. We analyzed the PBC trait for SNPs in 3′UTR regions by converting the SNP list into ANNOVAR variant annotated input, and we retrieved 5279 genes located in 3′UTR regions. We then crossed the 55 genes from MIGWAS with the 5279 genes from ANNOVAR and found 13 genes in common. The experiments might seem unrelated, but the results from MIGWAS analyzed by both CoCoNet and ANNOVAR shows that the 3 tools are indeed connected and are not interrelated.
In a previous study, the research found a strong association between PBC and rs231725 [21]. rs231725 is a SNP in the 3′ flanking region of CTLA4, a gene which has an impact on the risk of PBC [21]. Indeed, PTEN is associated with 9 traits (Crohn's disease [22], Inflammatory bowel disease [23], Rheumatoid arthritis [24], Systemic lupus erythematosus [25], Ulcerative colitis [23], Coronary artery disease [26], Type 2 diabetes [27], Alzheimer's disease [28], Autism [29]) out of our analyzed 43 traits.
Total 12 traits (Alzheimer’s disease, Coronary artery disease, Crohn’s disease, Ever Smoked, Fasting glucose, High density lipoproteins, Inflammatory bowel disease, Low density lipoproteins, Schizophrenia, Type 1 diabetes, Triglycerides, Ulcerative colitis) do not have MIGWAS association results reported in the analysis. It seems that liver tissue does not appear to have the smallest likelihood with PBC based on the total genes in human genome. We also ran CoCoNet with the targeted gene set of miRNAs enriched over the target network for PBC trait only to calculate loglikelihood, and the result stays the same.
The transcript expression of PFKL has the highest RPKM (Reads Per Kilobase of transcript, per Million mapped reads) in kidney samples in the RNA-seq which was performed from 4 human individuals in order to determine tissue-specificity of all protein-coding genes [30].
PFKL contains a transcript variant ATP-dependent 6-phosphofructokinase, liver type isoform a which represents the longer transcript and encodes the shorter isoform (a). The CD-Search shows the protein classification of this transcript variant, which is Eukaryotic_PFK domain-containing protein, and finds a specific hit, Eukaryotic_PFK, which shows a high confidence association between the query sequence and a domain model. Phosphofructokinase (PFK) is a key regulatory enzyme that that phosphorylates fructose 6-phosphate in glycolysis. It belongs to the PFK family, evolving from the bacterial PFKs by gene duplication and fusion events and then exhibiting complex behavior. PFK family also includes ATP-dependent phosphofructokinases (allosteric homotetramers) and pyrophosphate (PPi)-dependent phosphofructokinases (mostly dimeric and nonallosteric homotetramers). In addition, protein sequences of Opisthokonta are in the multiple sequence alignment in the Eukaryotic_PFK [30].
We found 13 genes, UGT3A, SYNGR, ASAP3, CD80, GXYLT1, IFNAR, KLHL22, POU2AF1, SFMBT2, SIRPB1, SIRPG, ZC3HAV1 and ZNF585B which are in common in the gene list applied from MIGWAS and ANNOVAR.
ASAP3 encodes a member of a subfamily of ADP-ribosylation factor(Arf) GTPase-activating proteins that its expression level affects cell proliferation and migration, and it called up-related in liver cancer 1 [31]. DDEFL1 (cloned ASAP3) is unregulated in hepatocellular carcinoma (HCC) through microarray analysis. The studies [32] showed that DDEFL1 had increased expression and had overexpression which increased colony formation in NIH3T3 cells and human hepatoma cell lines [32].
In the transcript expression of UGT3A1, liver samples have the second highest RPKM among 27 different tissues [33].
SFMBT2 are polycomb group proteins that bind to methylated lysins in histone tails. The formation of transcription-resistant higher-order chromatin structures at target genes are induced by it so that this gene can repress transcription [34]. SFMBT2 was cloned and designated as KIAA1617 by Nagase et al. [35], who obtained clones from fetal brain cDNA library and then sequenced them [35]. Kuzmin et al. [36] reported that SFMBT2 interacted with the YY1 (600013) transcription when SFMBT2 was expressed from the paternal allele in mouse blastocysts and in mouse embryonic tissues early in development, and later during mouse embryonic development [36].
ZC3HAV1 encodes a zinc finger protein that can prevent replication certain viruses and inhibit viral gene expression by targeting and eliminating viral mRNAs in the cytoplasm [37]. Yu et al. [38] identified ZC3HAV1 and called it ZAP in their analysis of gene expressed in human fetal liver [38]. Northern blot analysis of rat Zap that was cloned by Gao et al. [39] showed that mRNA was highly expressed in kidney and liver [39].
Conclusions
Our study tried to understand GWAS data through identifying candidate miRNA-gene pairs over miRNA-target gene network. Using several cutting-edge bioinformatical tools and databases and adopting visualization, we assessed trait-tissue relevance based on tissue-specific gene co-expression information including protein–protein interaction and transcription factor binding evidence. We annotated 3′UTR variants harbored by genes targeted by miRNAs expressed in tissues significantly associated with PBC trait. Our study provides evidence that the association between tissues and traits could be affected by the 3′UTR variants of genes which change binding affinity of targeting miRNAs. The analysis emphasizes the influence of variants on genetic traits and miRNA-targeting gene networks, and thus could contribute to additional studies and detections on human diseases. We think our study provided a valuable approach for elucidating 3′UTR variants which could contribute to genetic traits with tissue relevance in the context of miRNA influential human diseases.
Abbreviations
- miRNA:
-
Micro ribonucleic acid
- RNA:
-
Ribonucleic acid
- mRNA:
-
Messenger ribonucleic acid
- DNA:
-
Deoxyribonucleic acid
- SNP:
-
Single-nucleotide polymorphism
- 3′UTR:
-
Three prime untranslated region
- GWAS:
-
Genome-wide association study
- MIGWAS:
-
MiRNA–target gene networks enrichment on GWAS
- CoConet:
-
Composite likelihood-based Covariance regression Network model
- GO:
-
Gene Ontology
- PFK:
-
Phosphofructokinase
- TCGA:
-
The Cancer Genome Atlas
- LIHC:
-
Liver hepatocellular carcinoma
References
O’Brien J, Hayder H, Zayed Y, Peng C. Overview of MicroRNA biogenesis, mechanisms of actions, and circulation. Front Endocrinol (Lausanne). 2018;9:402.
Ardekani AM, Naeini MM. The role of microRNAs in human diseases. Avicenna J Med Biotechnol. 2010;2(4):161–79.
Moszynska A, Gebert M, Collawn JF, Bartoszewski R. SNPs in microRNA target sites and their potential role in human disease. Open Biol. 2017;7(4):170019.
de Giorgio A, Krell J, Harding V, Stebbing J, Castellano L. Emerging roles of competing endogenous RNAs in cancer: insights from the regulation of PTEN. Mol Cell Biol. 2013;33(20):3976–82.
Yang C, Wu D, Gao L, Liu X, Jin Y, Wang D, Wang T, Li X. Competing endogenous RNA networks in human cancer: hypothesis, validation, and perspectives. Oncotarget. 2016;7(12):13479–90.
Mills MC, Rahal C. A scientometric review of genome-wide association studies. Commun Biol. 2019;2:9.
Sakaue S, Hirata J, Maeda Y, Kawakami E, Nii T, Kishikawa T, Ishigaki K, Terao C, Suzuki K, Akiyama M, et al. Integration of genetics and miRNA-target gene network identified disease biology implicated in tissue specificity. Nucleic Acids Res. 2018;46(22):11898–909.
Okada Y, Muramatsu T, Suita N, Kanai M, Kawakami E, Iotchkova V, Soranzo N, Inazawa J, Tanaka T. Significant impact of miRNA-target gene networks on genetics of human complex traits. Sci Rep. 2016;6:22223.
Shang LS, Smith JA, Zhou X. Leveraging gene co-expression patterns to infer trait-relevant tissues in Genome-wide Association Studies. PLoS Genet. 2019;16(4):e1008734.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164.
Hao X, Zeng P, Zhang S, Zhou X. Identifying and exploiting trait-relevant tissues with multiple functional annotations in genome-wide association studies. PLoS Genet. 2018;14(1):e1007186.
Lu Q, Powles RL, Abdallah S, Ou D, Wang Q, Hu Y, Lu Y, Liu W, Li B, Mukherjee S, et al. Systematic tissue-specific functional annotation of the human genome highlights immune-related DNA elements for late-onset Alzheimer’s disease. PLoS Genet. 2017;13(7):e1006933.
Pickrell JK, Berisa T, Liu JZ, Segurel L, Tung JY, Hinds DA. Detection and interpretation of shared genetic influences on 42 human traits. Nat Genet. 2016;48(7):709–17.
Finucane HK, Bulik-Sullivan B, Gusev A, Trynka G, Reshef Y, Loh PR, Anttila V, Xu H, Zang C, Farh K, et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat Genet. 2015;47(11):1228–35.
Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65.
The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health abN, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript were obtained from UCSC genome browser (http://genome.ucsc.edu/cgi-bin/hgTables) on 10/16/2019.
da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009a;4(1):44–57.
da Huang W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009b;37(1):1–13.
Dai X, Ding L, Liu H, Xu Z, Jiang H, Handelman SK, Bai Y. Identifying interaction clusters for MiRNA and MRNA pairs in TCGA network. Genes (Basel). 2019;10(9):702.
Vora S, Seaman C, Durham S, Piomelli S. Isozymes of human phosphofructokinase: identification and subunit structural characterization of a new system. Proc Natl Acad Sci U S A. 1980;77(1):62–6.
Juran BD, Atkinson EJ, Schlicht EM, Fridley BL, Lazaridis KN. Primary biliary cirrhosis is associated with a genetic variant in the 3’ flanking region of the CTLA4 gene. Gastroenterology. 2008;135(4):1200–6.
Tokuhira N, Kitagishi Y, Suzuki M, Minami A, Nakanishi A, Ono Y, Kobayashi K, Matsuda S, Ogura Y. PI3K/AKT/PTEN pathway as a target for Crohn’s disease therapy (Review). Int J Mol Med. 2015;35(1):10–6.
Noren E, Almer S, Soderman J. Genetic variation and expression levels of tight junction genes identifies association between MAGI3 and inflammatory bowel disease. BMC Gastroenterol. 2017;17(1):68.
Li XF, Chen X, Bao J, Xu L, Zhang L, Huang C, Meng XM, Li J. PTEN negatively regulates the expression of pro-inflammatory cytokines and chemokines of fibroblast-like synoviocytes in adjuvant-induced arthritis. Artif Cells Nanomed Biotechnol. 2019;47(1):3687–96.
Al-Mayouf SM, AlTassan RS, AlOwain MA. Systemic lupus erythematosus in a girl with PTEN variant and transaldolase deficiency: a novel phenotype. Clin Rheumatol. 2020;39:3511–5.
Wang D, Wang Y, Ma J, Wang W, Sun B, Zheng T, Wei M, Sun Y. MicroRNA-20a participates in the aerobic exercise-based prevention of coronary artery disease by targeting PTEN. Biomed Pharmacother. 2017;95:756–63.
Yin L, Cai WJ, Chang XY, Li J, Zhu LY, Su XH, Yu XF, Sun K. Analysis of PTEN expression and promoter methylation in Uyghur patients with mild type 2 diabetes mellitus. Medicine (Baltimore). 2018;97(49):e13513.
Cui W, Wang S, Wang Z, Wang Z, Sun C, Zhang Y. Inhibition of PTEN attenuates endoplasmic reticulum stress and apoptosis via activation of pi3k/akt pathway in Alzheimer’s disease. Neurochem Res. 2017;42(11):3052–60.
Rademacher S, Eickholt BJ. PTEN in autism and neurodevelopmental disorders. Cold Spring Harb Perspect Med. 2019;9(11):a036780.
Information GIBMNLoMUNCfB: 2004—[cited 2020 04 08]. https://www.ncbi.nlm.nih.gov/gene/5211.
Ha VLBS, Inoue H, Vass WC, Campa F, Nie Z, de Gramont A, Ward Y, Randazzo P. ASAP3 is a focal adhesion-associated Arf GAP that functions in cell migration and invasion. J Biol Chem. 2008;283(22):14915–26.
Okabe H, Furukawa Y, Kato T, Hasegawa S, Yamaoka Y, Nakamura Y. Isolation of development and differentiation enhancing factor-like 1 (DDEFL1) as a drug target for hepatocellular carcinomas. Int J Oncol. 2004;24(1):43–8.
Gene [Internet]. Bethesda (MD): National Library of Medicine (US) NCfBI: 2004—[cited 2020 04 08]. https://www.ncbi.nlm.nih.gov/gene/133688.
Lee K, Na W, Maeng JH, Wu H, Ju BG. Regulation of DU145 prostate cancer cell growth by Scm-like with four mbt domains 2. J Biosci. 2013;38(1):105–12.
Nagase T, Kikuno R, Nakayama M, Hirosawa M, Ohara O. Prediction of the coding sequences of unidentified human genes. XVIII. The complete sequences of 100 new cDNA clones from brain which code for large proteins in vitro. DNA Res. 2000;7(4):273–81.
Kuzmin A, Han Z, Golding MC, Mann MR, Latham KE, Varmuza S. The PcG gene Sfmbt2 is paternally expressed in extraembryonic tissues. Gene Expr Patterns. 2008;8(2):107–16.
Ye P, Liu S, Zhu Y, Chen G, Gao G. DEXH-Box protein DHX30 is required for optimal function of the zinc-finger antiviral protein. Protein Cell. 2010;1(10):956–64.
Yu Y, Zhang C, Zhou G, Wu S, Qu X, Wei H, Xing G, Dong C, Zhai Y, Wan J, et al. Gene expression profiling in human fetal liver and identification of tissue- and developmental-stage-specific genes through compiled expression profiles and efficient cloning of full-length cDNAs. Genome Res. 2001;11(8):1392–403.
Gao G, Guo X, Goff SP. Inhibition of retroviral RNA production by ZAP, a CCCH-type zinc finger protein. Science. 2002;297(5587):1703–6.
Acknowledgements
The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The results published here are in whole or part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/. The data used for the analyses described in this manuscript were obtained from UCSC genome browser (http://genome.ucsc.edu/cgi-bin/hgTables) on 10/16/2019. We thank Anna Liu for providing file format conversion assistance, Chenjian Fu for assisting to debug Python scripts, and Cong Zeng for helping with paper review. We also thank Li Fang and Kai Wang for offering valuable ANNOVAR annotation advice.
About this supplement
This article has been published as part of BMC Medical Genomics Volume 13 Supplement 11 2020: Data-driven analytics in biomedical genomics. The full contents of the supplement are available at https://bmcmedgenomics.biomedcentral.com/articles/supplements/volume-13-supplement-11.
Funding
This research was partially supported from the University of Michigan to XZ. The funders had no role in the study design, data collection and analysis, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
BL performed the annotation analysis, produced figures, and participated to draft the manuscript. JD ran the variant pipeline analysis. JY generated PTEN ClinVar variant annotation result. YF conducted PTEN associated gene and function analysis. LS provided CoCoNet database files and offered the running advice. XZ provided the data and biological interpretation, and offered the study advice. YB designed and supervised the project, generated results and provided biological interpretation, and drafted the manuscript. All authors read, revise, and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Details of the summary statistics of 43 traits from 29 GWAS studies.
Additional file 2: Table S2.
Tissue association result from MIGWAS.
Additional file 3: Table S3.
Gene-miRNA target pairs identified by MIGWAS for each trait.
Additional file 4: Table S4.
Identified 9890 annotated variants exclusively located in 3′UTR region of 5279 genes.
Additional file 5: Table S5.
Identified SNPs for ten genes with ClinVar variant information.
Additional file 6: Table S6.
Genes and GO terms in the top cluster for 55 genes with significant tissue and PBC trait association identified by DAVID.
Additional file 7: Table S7.
Genes and GO terms in the top cluster for hsa-mir-590 with target relationship of PTEN.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, B., Dong, J., Yu, J. et al. Pinpointing miRNA and genes enrichment over trait-relevant tissue network in Genome-Wide Association Studies. BMC Med Genomics 13 (Suppl 11), 191 (2020). https://doi.org/10.1186/s12920-020-00830-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-020-00830-w