
Abstract
Background
A variety of DNA-based methods have been applied to identify genetic markers of attention deficit hyperactivity disorder (ADHD), but the connection to RNA-based gene expression has not been fully exploited.
Methods
Using well defined cohorts of discordant, monozygotic twins from the Michigan State University Twin Registry, and case-controlled ADHD cases in adolescents, the present studies utilized advanced single molecule RNA sequencing to identify expressed changes in whole blood RNA in ADHD. Multiple analytical strategies were employed to narrow differentially expressed RNA targets to a small set of potential biomarkers of ADHD.
Results
RNA markers common to both the discordant twin study and case-controlled subjects further narrowed the putative targets, some of which had been previously associated with ADHD at the DNA level. The potential role of several differentially expressed genes, including ABCB5, RGS2, GAK, GIT1 and 3 members of the galactose metabolism pathway (GALE, GALT, GALK1) are substantiated by prior associations to ADHD and by established mechanistic connections to molecular pathways relevant to ADHD and behavioral control.
Conclusions
The convergence of DNA, RNA, and metabolic data suggests these may be promising targets for diagnostics and therapeutics in ADHD.
Similar content being viewed by others

Background
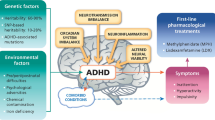
Attention deficit hyperactivity disorder (ADHD) is the most common psychiatric disorder in childhood and adolescence, affecting roughly 5% of youth worldwide [1]. Although heritability is substantial [2], environmental modulation is well known and environmental influences are of keen interest. Further, ADHD is a complex disease, with no single gene showing an overwhelming effect, except in very rare cases [2]. Further, ADHD is likely to be etiologically complex and to reflect multiple underlying etiology types [3]. Thus, ADHD is potentially a collection of disorders, which could involve inherited DNA variations, somatic epigenetic changes during neural differentiation, and environmental modifiers. A variety of genetic technologies have been applied to help identify either biomarkers or clues to causative mechanism, including GWAS, exome sequencing, and others [4].
Physiologically, numerous avenues have been examined for a potential role in ADHD. One theory holds that ADHD involves changes in the glutaminergic neurotransmitter systems, a major excitatory pathway in the brain, however genome-wide SNP analysis has so far failed to identify glutamate receptor DNA variants as a significant association with ADHD [5]. Several lines of evidence, including the utility of drugs such as methyphenidate, amphetamines, and atomoxetine has suggested that deficits in the dopaminergic and adrenergic systems could underlie the neurochemical basis of ADHD [6].
While a major focus has been on neurodevelopmental pathways, there are reasons to be cognizant of potentially important changes in non-neural endocrine systems. The strong linkage of ADHD with male gender has raised potential connections to steroid hormones, but it is difficult to exclude sex-linked genetic elements, and/or cultural perceptions related to the diagnosis of ADHD [7].
It has recently been suggested that RNA sequencing may be informative for identifying ADHD-related biomarkers, based on positive findings in a small paired analysis in 3 multiplex cases [8]. Additionally, targeted and candidate gene studies have stimulated interest in miRNAs in ADHD [9]. The present studies employed a broad and relatively unbiased strategy to obtain exploratory data for hypothesis generation using two types of ADHD cohorts analyzed by high-precision, genome-wide single molecule RNA sequencing.
The present studies employed cutting-edge RNA sequencing technology to very accurately quantify transcript levels across the entire transcriptome, for both coding, and non-coding transcripts. The RNA profiling approach was applied to stabilized whole blood samples drawn from two well-characterized cohorts: (a) children matched on ADHD and non-ADHD status and, (b) identical twins that were discordant for ADHD traits. The results provide a deep and broad map of RNA changes potentially related to ADHD, and can be combined with other ‘omic data, such as genome-wide association studies (GWAS), DNA methylation, and proteomic analysis to help identify new biomarkers, and secondarily, new clues to biological processes related to ADHD.
Methods
Participants
Non-twin case-controls
Case-controls were selected from the Oregon ADHD Longitudinal Study Cohort (for illustrative prior papers see [10, 11]. For that cohort, families were recruited for a case–control study of ADHD and non-ADHD, by soliciting community volunteers with public advertisements and mass mailings using commercial mailing lists. The local Institutional Review Board approved the studies. Parents provided written informed consent for their minor children under the age of 16; minor children provided written informed assent. All families completed a full multi-informant, multi-method screening process to establish eligibility and diagnostic group assignment for ADHD, non-ADHD, as well as comorbid disorders.
After screening, the research team conducted a diagnostic evaluation using standardized, well-normed rating scales from parents and teachers, parent semi-structured clinical interview, child intellectual testing, and clinical observation. Best-estimate research diagnoses and final eligibility were established by a team of two experienced clinicians (a child psychiatrist and a child psychologist), who independently assigned final diagnoses and comorbid disorders including ADHD, oppositional defiant disorder (ODD), and any lifetime mood disorder (major depression, dysthymia, or other), as reported herein. History of seizures or head injury, parent–teacher rating discrepancy making diagnosis uncertain, psychosis, mania, current major depressive episode, Tourette’s syndrome, autism, and estimated IQ < 80 were a basis for exclusion in the present study.
From 2144 volunteers, 850 eligible children were identified. The group used in the current study were a subset selected because they (a) clearly met Diagnostic and Statistical Manual of Mental Disorders-5 (DSM-5) criteria for ADHD or non-ADHD comparison group (rather than subthreshold), (b) had no prior history of psychiatric medication, (c) were Tanner stage 1 or 2 by parent report on the Pubertal Development Questionnaire, (d) were not a sibling of another child in the cohort; and, (e) were willing to give a blood sample. This process provided a set of non-twin case-controls (n = 48: 24 ADHD, 24 controls) for whom Paxgene-stabilized, frozen blood samples were submitted for RNA sequencing. After RNA sequencing, 23 ADHD and 21 case controls were available, as described in Table 1.
Twins
Twins were recruited from the Michigan State University Twin Registry (MSUTR; [12, 13]. The registry has over 30,000 twin pairs between the ages of 3 and 55. For the current study, recruitment was carried out via anonymous mailings to all MSUTR, identical (monozygotic, MZ) twin families with twins age 7–17 years old. Volunteering families were then screened for eligibility. They were restricted for the current study to (a) never-medicated youth, (b) age 7–17 (it proved impractical to restrict to pre-pubertal due to the relative rarity of discordant pairs), (c) no major medical illness, autism, or neurological condition in the screen record (later confirmed by clinical interview), (d) believed to be MZ (later confirmed by genotyping), (e) eligible for outreach (limited to accommodate multiple studies accessing the registry).
Those who responded then completed a phone screen for eligibility and, if eligible, were scheduled for a home visit. At the home visit, a trained staff member completed a clinical interview, drew blood into an RNA Paxgene stabilizer tube, and collected saliva into an Oragene tube for DNA isolation. Self- and parent-report rating scales were also collected, as described below.
The number of discordant pairs was limited by two major factors (a) about 1/3 of twin births are monozygotic, (b) ADHD has high heritability, so discordant pairs are rare. The final samples available for statistical analysis after clinical screening, RNA sequencing, and data quality checks is shown in Table 1, with additional recruitment details provided in Additional file 1: Data S1. After RNA sequencing, there were 16 discordant pairs meeting quality control metrics for analysis. In both cohorts, the size of the final groups was determined by the number of available samples meeting inclusion/exclusion criteria, and then by successful RNAseq analysis.
Measures
ADHD evaluation
ADHD was evaluated by the following measures: parent completed the Conners' Rating Scales-3rd Edition short form [14]; Strengths and Difficulties Questionnaire long form including the impairment module (SDQ) [15]; the ADHD Rating Scale (ADHD-RS) [16], and a semi-structured clinical interview (Kiddie Schedule for Affective Disorders and Schizophrenia, KSADS) administered by a Master’s-degree level clinician trained to reliability with an outside trainer (EB) [17]. For the twin data, these data were used to evaluate consensus ADHD status by two of the authors (JN,EB) blind to the RNA data. For the case–control data, teacher ratings were also available and consensus diagnosis was arrived at by an independent clinical team as described in the online appendix.
Definition of discordance
Twins were considered discordant if they met the following criteria:
-
(a)
At least a 3-symptom separation on the ADHD-RS or KSADS, with one twin below diagnostic threshold and never identified with ADHD; or
-
(b)
At least a 10 point T score difference on the ADHD-RS, or
-
(c)
One was previously diagnosed and treated and the other was not (and the untreated twin did not meet ADHD criteria by our measures), or
-
(d)
Any combination of these criteria.
ADHD discrepancy score
To create a quantitative estimate of degree of discordance, the following four variables were created:
-
(a)
Absolute difference between higher and lower scoring twin on an ADHD–RS (by parent) raw score (range 0–27) obtained at the same time as the RNA blood tube was collected, and rated over the past 6 months for (i) inattention, (ii) hyperactivity-impulsivity, and (iii) total. The total is reported here.
-
(b)
For the fourth variable, each twin pair was ranked on “degree of discordance” based on considering all available data including multiple time points rating scales (Conners, ADHD-RS, SDQ), the KSADs, and comorbid and substance use conditions. This was achieved by consensus rankings by JN and EB blind to the RNA data.
Whole blood RNA isolation
For complete methods, see Additional file 1: Data S1. Whole blood RNA was isolated from the Paxgene blood collection tube using the Paxgene Blood Isolation kit and the Qiagen QIACube Automation System. The RNA was quantified by optical absorbance at 260 and 280 nm using the NanoDrop 1000. The resulting nucleic acids were treated with TurboDNAse to remove residual DNA and then depleted of ribosomal RNA (rRNA) using the Illumina Ribo-Zero rRNA Removal Kit (H/M/R). RNA concentration was measured using the absorbance at 280 and 260 nm (Nanodrop).
Single molecule sequencing
First strand cDNA synthesis was carried out with 50–100 ng RNA at 95 °C for 5 min using 50 ng/µL random hexamers (Invitrogen #51,709) and 1 µL of 10 mM dNTP mix (Invitrogen #Y02256). 3′ Poly A tailing was achieved with terminal transferase (NEB #M0315). Cleaned samples were denatured and hybridized to the poly dT surface of SeqLL flow cells. Samples were sequenced using SeqLL’s True Single Molecule Sequencing (tSMS) technology that allows for RNA sequencing without requiring PCR amplification or library preparation ligation steps.
Read alignment and quantification
Data output from the sequencer is in raw short read format (SRF) files. SRF files were processed using the HeliSphere Bioinformatics package, first converting to SMS format for alignment. SMS reads were trimmed for leading T homopolymers and were filtered for reads with a minimal length of 25 bases after trimming. Trimmed reads were aligned to the HG38 human genome (GRCh38) supplemented with the complete ribosomal repeat unit (GenBank Accession U13369.1) using the HeliSphere BASIC analysis pipeline.
Statistical analysis of differentially expressed genes
From the aligned reads, a variety of analytical approaches were employed in order to identify differentially expressed genes (DEGs) which passed one or more filtering strategies. To minimize the impact of any one statistical method, the sample set was analyzed by 9 methods, deriving from 5 types of analysis: using the EdgeR Bioconductor package [18, 19] using either dispersion analysis (comEdgeR), or generalized regression analysis (glmEdgeR); with TCC package [20] using either edgeR (tccEdgeR), DESeq [21] (tccDESeq) or DESeq2 [22] (tccDESeq2) methods for differential expression analysis; with voom [23] from limma [24]; and also with baySeq [25] and ALDEx2 [26] packages. To find the most commonly identified DEGs, the results of each analysis were ranked by the resulting p value likelihood of a difference between groups adjusted for multiple testing using the Benjamini–Hochberg method. To constrain the size of the DEG list, it was predetermined to select the top 100 from each method and then combine them to achieve a single ranked list across methods. These were then ranked by the number of times a given DEG appeared in each of the 9 lists.
A second general strategy that has proven useful, due its simplicity and absence of assumptions about the distribution of RNAseq data, is to adjust the transcript counts only by correction for the transcript size and total informative reads. The RPKM calculation compensates for the size of the transcript, and for the total number of reads acquired. The number of reads aligned to each transcript are divided by the size of transcript in thousands of base pairs (per K), and then divided by the total number of informative reads obtained for that subject (in Millions), yielding RPKM. To further constrain the DEG list to transcripts with detectable expression, the total aligned read counts for the HG38 genome (GRCh38, n = 195,187) were filtered to include transcripts present at > 0.01 RPKM in 70% of the samples of at least one diagnostic group, leaving ~ 95 K working transcripts for analysis (see Fig. 1 for scatter plot of RPKM expression per transcript).

RNA sequencing of whole blood from ADHD Cases versus Matched Controls. Stabilized whole blood was sequenced, aligned, and then counted into UCSC-defined transcripts, expressed as reads per K of transcript per M of total assigned reads per subject (RPKM). Transcripts were filtered for > 0.01 RPKM in 70% of samples in at least 1 of the 2 groups, resulting in 95,511 transcripts remaining. The RPKM level of each transcript (colored squares) is expressed as the mean in Controls (X axis) versus the mean in ADHD cases (Y axis) for each of the transcripts, and plotted on log2 scale. The central green line is a slope of 1, bounded on each side by twofold change lines. Black line is the actual fit of the data distribution, defined by the equation in the upper left
Results
Clinical parameters of the study subjects
Case controlled cohort
A total of 100 never-medicated children on whom Paxgene stabilized blood was available were used to compose 24 pairs of ADHD affected or unaffected case controls. They were selected so that each ADHD case was matched by age and gender to an unaffected control. From these 24 pairs, 23 ADHD and 21 controls were successfully RNA sequenced to pre-specified criteria of read depth. The characteristics of the groups are shown in Table 1.
Discordant twins cohort
An initial cohort of 50 never-medicated, potentially discordant identical twin pairs was identified and then narrowed for clinical criteria focusing on a high degree of discordance in the severity of ADHD symptoms. A set of 24 pairs of twins with strong to moderate discordance were identified and 16 pairs had sufficient RNA quality and yield, and successful RNAseq data for further analysis (Table 1).
Analysis of differentially expressed genes
Case controlled cohort
The average yield of nucleic acids was 5.4 ug/tube with an average 260/280 ratio of 2.3 and a BioAnalyzer RIN RNA quality index of 8.97. The tSMS method produced a very broad profiling of ribosome-depleted RNA transcripts in stabilized whole blood. By averaging all subjects in each group, filtering out low expressing transcripts of < 0.01 RPKM, and comparing average RPKM for each transcript between groups, it is observed that tSMS yielded linear quantification (slope = 0.98) of gene expression over ~ 22 log2 orders of magnitude for more than 95,511 transcripts (Fig. 1).
Case Controlled Differentially expressed genes (DEGs). Employing the 9 filters approach, 391 transcripts were ranked in the top 100 by at least one method. Of these, two transcripts were identified by all 9 methods, and 5 transcripts were identified by 8 of the 9 methods. Note that although this reduces the likelihood that their differential expression is dependent on only one analytical approach, it does not fully eliminate potential type I error because these are not completely independent methods (Fig. 2). The full 391 gene list by p value in the 9 methods can be found in Additional file 2: Table S1. Jaccard similarity analysis reveals the best concordance between methods edgeR, DESeq and DESeq2. The most discordant results were obtained with BaySeq (Additional file 3: Data S2). The 2 transcripts passing all 9 methods with a corrected p < 0.05 are difficult to interpret: IGLV2-8 (↑2.0X in ADHD) is an immunoglobin lambda chain V-II region, and its expression appeared related to at least 10 other DEGs that were either HLA or IgG-related. More difficult to interpret is RNU1-94P (↑2.0X), which is a small nuclear protein pseudogene. The five transcripts passing 8 filters included 4 with better annotation: ABCB5, CWC27, IFI35, and AHNAK:

Schematic representation of analytical strategy and top hits. Two separate cohorts: ADHD Cases vs Control and ADHD Discordant Monozygotic Twins were analyzed for whole blood RNA levels by RNAseq. The data was analyzed by 9 distinct methods (i.e. EdgeR/com/glm/tcc, DESeq, etc.) and then differentially expressed genes (DEG) identified by multiple methods resulted in 391 transcripts for Case/Control of which 7 passed 8 or more filters. For discordant twins, 385 transcripts were identified of which 10 passed 8 or more filters. Comparing the 393 and 385 lists identified 5 transcripts identical in both cohorts
ABCB5 (↑1.9X in ADHD) is a member of the multi-drug resistance (MDR) family of transporters. ABCB5 has been mechanistically linked to glucose, phospholipid, and amino acid transport [27], and copy number variants have been linked to childhood obesity [28]. ADHD is also associated with increased risk of obesity, with mechanisms unknown [29].
CWC27 (↓1.4X) is a spliceosomal trans-peptidyl-isomerase that has been associated with retinal abnormalities and developmental disorders, including neurological defects, in children with mutations [30].
IFI35 (↑1.4X) is an interferon-induced protein that acts as a ‘damage-associated molecular pattern (DAMP)’ [31], and thus could indicate some type of inflammatory source in ADHD. Substantial circumstantial data suggests inflammation may play a role in ADHD [32].
AHNAK (↑1.3X) is a particularly interesting target because it is a giant 680 kD neuroblast differentiation-associated protein, that has been associated with a range of relevant neurological disorders including bipolar disorder [33], depressive-like behaviors in knockout mice [34], ß-adrenergic regulation of the cardiac CaV1.2 calcium channel [35], and a variety of immune functions. The AHNAK family member, AHNAK2 (↑1.9X), is also identified on this list, with DEG identification by 2 of the analytic methods.
The fifth transcript, RP11-35015.2 (↓1.6X) is a poorly annotated transcript that lies within intron 1 of the IGF1 receptor (IGF1R), and thus, difficult to more clearly understand.
The complete list of 391 selected (Additional file 2: Table S1) includes other interesting transcripts, such as BACE2 (↓1.6X) and MED6 (↑1.5X). However, we proceeded to further narrow this case–control list by virtue of analyzing the cohort of discordant twins, and then determining whether any systematic patterns of similarity emerged.
Discordant twins cohort
tSMS of 16 discordant twin pairs produced transcript profiling of similar breadth and linearity as observed in the case–control study (Additional file 4: Fig. S1). Using an essentially identical analytical approach to the case controls (Fig. 2), the results of RNAseq from monozygotic ADHD-discordant twins were subjected to 9 analytical approaches and then the top 100 transcripts from each were ranked by their presence on the 9 lists. The resulting list of 385 transcripts can be found in Additional file 5: Table S2.
Discordant twins DEGs: A total of 10 transcripts passed 8 of 9 filters and contains transcripts with close similarity to several of the case–control DEGs (Fig. 2). These high-ranking transcripts present potential hypotheses for further study with regard to ADHD, as follows:
ARL6IP5 (↓1.4X) is an ADP ribosylation factor-like GTPase 6-interacting protein, but is also known as JWA, a homologous gene of the glutamate-transporter-associated protein 3–18 (GTRAP3-18), and addicsin. ARL6IP5/JWA is expressed at high levels in the hippocampus and ARL6IP5/JWA knockout mice showed spatial cognitive potentiation and enhanced neurite growth in newborns [36]. Conditional astrocytic ARL6IP5/JWA null mice demonstrates a role as a neuroprotective factor against dopaminergic neuronal degeneration [37]. ARL6IP5/JWA has been associated with increased expression in the amygdala after chronic morphine treatment [38], and with morphine dependence via the delta opioid receptor [39].
CCDC107 (↓1.9X) is closely related to CCDC132 and CCDC84 found in the case control study. While relatively little is known about these coiled coil family members, coiled coil helix proteins (e.g. Chchd2) have been implicated in ADHD-like mouse models [40].
CCND1, cyclin D1 (↓2.9X), is related to CCNC and CCNL2 from the case control studies. While the cyclins are largely studied in relationship to cell cycle control, they can serve a variety of regulatory functions in cells.
DBF4B, DBF4 Zinc Finger B (↑2.5X), has been extensively studied as an activator of the Mcm2-7 helicase, a partner to Cdc7 kinase, and thus important for the initiation of DNA replication. Potentially of interest, it has also been associated with autism spectrum disorders via a semaphorin 5A (SEMA5A) eQTL network [41].
Dual specificity phosphatase 4 (DUSP4, ↓3.2X) is a family member to DUSP6 from the case–control study, and has recently been described as a control element in the suprachiasmatic clock network via modulation of vasoactive intestinal peptide signaling to ERK1/2 [42].
FAM159A (↑1.7X) has counterparts FAM104A, FAM134B, FAM157C, FAM162A, and FAM213B as differentially expressed in the case–control study. Little is known about FAM159A, but FAM134B, aka RETREG1 reticulophagy regulator 1, has a substantial literature connecting it with various functions including autophagy, and sensory neuropathy in humans [43] and Border Collies [44]. Inhibition of FAM134A causes impaired proteostasis in the endoplasmic reticulum due to the accumulation of misfolded proteins, which has been implicated in vascular dementia [45]. FAM162A is associated by GWAS to a gene-by-alcohol dependence interaction study of risky sexual behaviors and so it could be related to behavioral control [46]. Coincidentally, ADHD is associated with increased sexual risk taking [47].
RARS2 (↓1.6X) is the arginyl-tRNA synthetase gene that has been associated with a spectrum of neurological disorders including myoclonic epilepsy, mental retardation, spasticity, and extrapyramidal features [48]. Patients with RARS2 mutations exhibit early onset hypotonia, epileptic seizures, encephalopathy, and feeding difficulties in a syndrome termed pontocerebellar hypoplasia type 6 (PCH6) [49].
RN7SL454P (↓1.95X), has counterparts RN7SL423P and RN7SL687P as DEG in the case control cohort. It appears to be a small non-coding transcript, intronic to the dynein axonemal heavy chain 17 gene on chromosome 17 (DNAH17), but with no known function.
SIK3.IT1 (↑2.1X) is salt-inducible kinase 3, with known relations to sleep and circadian rhythm, and to glucose and lipid homeostasis, steroidogenesis, and adipogenesis [50].
SPICE1(↑1.4X) is a spindle and centriole-associated protein, which might relate to DBF4B and CCND1 in regards to cell cycle control. Computational screening identifies it as an aurora kinase substrate and it is known to cooperate with CEP120 in centriole elongation. Interestingly, SIK3 also interacts with aurora A, aurora B, and polo-like kinases, and SIK3 repression enhances the antimitotic effect of aurora inhibition [50]. Likewise, CCND1 has known interactions with aurora kinases [51]. The coiled coil proteins, potentially including CCDC107, are commonly associated with the centrosome maturation and aurora kinases [42], suggesting several possible coregulatory scenarios for SPICE1, CCND1, DBF4B, and SIK3 in ADHD, potentially in a non-mitotic, but centriolar/aurora kinase-mediated control of gene expression.
DEGs common to both cohorts
From the 385 DEG list compiled from 9 analyses of the discordant twins (Additional file 5: Table S2), 6 transcripts are identical to the 391 DEG case–control results obtained by similar methods (Additional file 2: Table S1). While 6 identical matches between 2 different cohorts of ADHD subjects could occur by random chance (Fisher’s exact text p = 0.318), it does suggest that these transcripts may merit further analysis as hypotheses in future studies.
HLA.DQB1.AS1, as the name implies, is an antisense transcript to the HLA-DQB1 locus on chromosome 6, which is elevated about twofold in ADHD cases. As noted previously, particularly in the case–control study, a substantial group of transcripts were HLA or IgG related, implying that some type of immune defect is at work. Because whole blood is being profiled, one must be cautious about an over-representation of immune-related transcripts (which are very plentiful in whole blood), but conversely, one cannot dismiss immune involvement as noted earlier. The potential role of inflammatory factors in ADHD has been raised over the years and is supported by various circumstantial data as recently reviewed [32], and recent analysis suggests the potential role of HLA loci in neurodevelopmental disorders such as ASD, and to a lesser degree ADHD [52].
In the same vein, IGHV3-74 is the variable region heavy chain transcript involved in antigen recognition by encoding IgM antibodies. While speculative, increased levels in the 2 cohorts could suggest some type of immune or autoimmune activity in ADHD.
The regulator of G protein signaling RGS2 is increased in both cohorts. RGS2 has diverse actions including promoting the translation of stress-associated proteins ATF4 and CHOP via an eIF-2B inhibitory domain [53]. Of potential importance, RGS2 variants have been associated with childhood adversities as predictors of anxious and depressive responses [54], as well as the regulation of nicotine-induced anxiolytic activity in mice, and cocaine-induced rewarding effects [55, 56]. Likewise, RGS2 is thought to mediate the anxiolytic effects of oxytocin [57], and affects T cell activation, anxiety, and male aggressive behavior [58]. RGS2 knockout mice exhibit increased fear learning, spatial learning, and neophobia [59]. Further, RGS2 modulates the activity and internalization of the dopamine D2 receptor in neuroblastoma cells [60], and has been implicated in dopamine receptor signaling during amphetamine self-administration [61].
Of potential interest is Park7 RNA (DJ-1), which is extensively investigated as related to early onset Parkinson’s Disease [62]. Based on some symptomatic similarities between Parkinson’s and ADHD, especially impulsivity [63], it was suggested there may be shared underlying causative factors. However, the circulating plasma protein levels of Park7 were not associated with ADHD in 125 ADHD patients versus 66 healthy controls [64], although whole blood RNA levels were not assessed.
Changes in RNU1-14P, which is a small nuclear pseudogene, is quite difficult to interpret, as is the RP11-661A12 transcript, though the latter is potentially an upstream ORF or alternate 5′ exon for the zinc finger CCCH-type containing 3 (ZC3H3) transcript, which is involved in nuclear adenylation and export of mRNAs [65].
Pathway analysis
An additional set of transcripts had very similar isoforms reported in the case control results, for example, MED7 vs MED6, CLIC2 vs CLIC1, JRK vs JRKL. While there is no assurance that these close family members perform similar functions, it is worth considering whether a similar pattern is reflected. A list of 66 of these overlapping transcripts was submitted for an unbiased analysis using pre-curated relationships between the gene products (Ingenuity Pathway Analysis). Several plausible relationships are identified in a manner that could identify latent variables that might account for a substantial subset of the transcript variations. Statistically, the top pathway identified centered around the well-characterized Akt/Insulin/PI3kinase/NfKB axis, as shown in Fig. 3. Underlying changes in glucose to insulin signaling could drive broader changes into the MED6/MED7/CCNC pathway as well as VAMP8/VAMP3/MMP/NDUF pathway. A second, and related, high scoring pathway is the Erk pathway, which would explain the CCND1/CDKN2B/CDKN2C/CDKN1C/RGS2 changes, and also the S100A12/S100A4/S100A8/CAPN1/DUSP4/DUSP6 transcript alterations (Fig. 4).

Pathway analysis of differentially expressed genes common to twins and case–control studies: AKT Pathway. Differentially expressed transcripts identified by both the case–control and twin studies were compared to a pre-curated database of biological pathways to determine whether any pathways were disproportionately affected. The top ranking hit, the AKT pathway, is shown schematically with gene products as polygons connected by lines indicating their known relationships. Transcripts identified in the present analysis are shown in color, with other transcripts in the pathway shown in gray

Pathway analysis of differentially expressed genes common to twins and case-controls: Erk Pathway. Differentially expressed transcripts identified by both the case–control and twin studies were compared to a pre-curated database of biological pathways to determine whether any pathways were disproportionately affected. The top ranking hit, the AKT pathway is shown schematically with gene products as polygons connected by lines indicating their known relationships. Transcripts identified in the present analysis are shown in color, with other transcripts in the pathway are shown in gray
RPKM analysis of the case–control and discordant twin datasets
In the context of a hypothesis-generating, exploratory study such as this, the prior analysis using 9 DEG methods may risk missing biologically important pathways in favor of statistical rigor. The datasets were re-analyzed using an RPKM threshold of 0.01, and combined fold-change (> 1.5) and p value (< 0.01, uncorrected) filtering approach that has proven useful in prior biomarker studies [66, 67]. This triple filter identified 524 transcripts in the case–control study (Additional file 6: Table S3), and 505 transcripts in the twin cohort (Additional file 7: Table S4). By filtering for transcripts that were common to both datasets at the gene symbol level, 14 transcripts were identified, but 3 could be excluded because the direction of the changes were in the opposite direction. The remaining 11 transcripts, common by both their presence and direction in both twins and case controls are potentially interesting.
ACP2 (↑2.0X twins) is a lysosomal acid phosphatase that is known to play a vital role in the removal of mannose-6-phosphate residues. ACP2 has known or suspected roles in several neurodevelopmental disorders, as emphasized by mutations in Acp2 causing severe cerebellar and neurodegenerative diseases [68]. Integrated analysis of GWAS and expression data identified ACP2 as a loci related to prepulse inhibition, a measure of sensorimotor gating that is known to be affected in several psychiatric disorders [69].
ALKBH6 (↑2.8X twins) is potentially important because, while relatively little is known about it, by analogy to its homolog ALKBH5, it is likely to function as a methyl-N6-adenosine (m6A) demethylase [70]. While there are extensive investigations into DNA modifications, such as CpG methylation, as a mode of genetic regulation, a quickly escalating literature suggests that defects in RNA modifications are a contributing factor in neurodevelopmental [71], and other disorders [72].
ASPSCR1 (↑2.6X twins), is a UBX domain containing tether for SLC2A4, which has a known fusion protein to TFE3 that is involved in certain cancers. However, better known as TUG, it has important roles as an interactor with the glucose transporter GLUT4, with regulatory activity over insulin-regulated aminopeptidase (IRAP) and vasopressin secretion [73]. While complex, vasopressin has been associated with ADHD by virtue of its known relation to social behaviors, and has been investigated as a potential therapy [74].
CLYBL (↑2.6X twins) is citrate lyase beta-like transcript, which encodes a malate/ß-methylmalate synthase with known effects on Vitamin B12 levels [75]. Vitamin B12 was thought to have a role in ADHD, but supplementation studies have not reported consistent beneficial effects [74]. The role of malate/ß-methylmalate in human physiology is incompletely studied, but methyphenidate treatment in rats causes significant changes in the citrate, malate, and isocitrate synthetic enzyme levels in the brain [76].
GAK (↑2.1X twins), cyclin G associated kinase, is potentially interesting in relation to ADHD. GAK (auxilin-2) has known involvement in synaptic function and neurological diseases [77], and is associated by GWAS with overlapping properties of Parkinson’s Disease and autoimmune diseases [78]. GAK was elevated in both cohorts (Fig. 5a) and in 14/16 of the discordant twin pairs, often in fairly striking fashion (Fig. 5b). GAK mRNA expression across a range of human tissues shows relatively high expression in the cerebellum, about twice the level observed in whole blood (Additional file 4: Fig. S2, GTEX).

GAK mRNA expression in ADHD case-controls and discordant twins. a Average transcript levels between groups in the case–control (n = 22 per group) and the discordant twins (n = 16 per group). mRNA levels expressed on log2 scale. b Detailed levels of GAK mRNA expression in the ADHD-discordant, monozygotic twin pairs (RPKM)
GALE (↑2.9X twins), UDP-galactose-4-epimerase, is one of 3–4 key enzymes in the synthesis and utilization of galactose, and changes in the other members of this family, especially GALT and GALK, were noticeably affected in the ADHD cases, with all 3 of these enzymes in the galactose processing pathway being elevated in the ADHD-affected twins (Fig. 6).

Differentially Expressed Genes in the Galactose Metabolism Pathway. Mean expression level of GALT (uc011lop.2, EC 2.7.7.12), GALE (uc057dhf.1, EC 5.1.3.2), and GALK1 (uc002jpk.4, EC 2.7.1.6) expressed in RPKM are shown for the control, unaffected twin, and the ADHD-affected twin. Bars are mean ± S.E.M
GIT1 (↑2.6X twins) was elevated > twofold in the ADHD subjects in both the discordant twin and case–control cohorts (Fig. 7, upper panel). Several striking coincidences draw attention to GIT1 as potential target. First, of the 15 known GIT1 isoforms, the changes in both cohorts seems largely restricted to a single isoform (uc060djr.1), which was elevated in 12 of 16 discordant twin pairs (Fig. 7, lower panel). GIT1 SNPs were previously associated with ADHD by genome-wide association studies (GWAS) studies that employ a relatively unbiased view of known genetic variation [79], although other cohorts did not support this association [80]. Fine mapping identifies an intronic SNP in GIT1 which causes reduced expression of GIT1 RNA and protein [81]. Strikingly, GIT1 is extensively spliced (Fig. 8), and the intronic SNP localizes to within 20 bp of 3′ terminus of the uc060djr.1 isoform identified in the present RNAseq analysis (Fig. 7). GIT1 knockout mice have ADHD-like traits including a shift in the neuronal excitation/inhibition balance associated with a decreased glial GABA intensity [4], and behavioral correction with methyphenidate and amphetamine [79]. Mechanistically, GIT1 is thought to play an important role in neurite outgrowth [82], synapse formation [83], and the turnover of ß2-adrenergic and other G-protein coupled receptors [84]. GIT1 is expressed at relatively high levels (tenfold above blood) in most brain regions, tibial nerve, and the testes (Additional file 4: Fig. S3, GTEX). While we cannot rule out a type I error, the present data suggests GIT1 merits further consideration as a factor in ADHD.

GIT1 in case controls and discordant twins. Upper panel: Average GIT1 mRNA levels by RNAseq in case–control study and the discordant twins study (Uc060djr.1 variant, mean RPKM as % control + S.E.M.). Lower Panel: GIT1 mRNA levels (Uc060djr.1 variant, mean RPKM) in 16 pairs of monozygotic twins discordant for ADHD severity

GIT1 Locus. Upper panel: Broad view of the GIT1 locus on 17q11.2 (chr17:29,569,429-29,593,667) showing several of the common splice variants, including the uc060djr.1 variant (red arrow). The region highlighted by red bar is magnified in the lower panel to show the position of the ADHD-linked SNP near the 3′ end of the uc060djr.1 transcript variant
STAM2 (↑1.5X twins), signal transducing adaptor molecule 2, is a member of the endosome-associated ESCRT-0 complex that is highly expressed in neurons, especially in the cerebral and cerebellar cortex, hippocampus, and medial habenula [85]. STAM2 regulates signaling via Jak2 and Jak3, which are directly involved in c-myc induction of IL-2 [86].
The remaining targets identified in both cohorts are more difficult to interpret. ERCC6L2 (↑2.9X twins) is excision-repair like 2, which has known relevance in cancer, but is difficult to connect with ADHD. HDLBP (↑1.3X twins) is highly relevant to high density lipoprotein metabolism, but has only a tenuous connection to ASD by virtue of a 2q27 deletion that causes reduced expression of HDLBP and 2 other genes [87]. IDS (↑1.4X twins), iduronate 2-sulfatase, is highly studied in Hunter syndrome mucopolysaccharidosis [88], but has no known relation to ADHD. UBE2J2 (↑2.5X twins) directs the ubiquitination of hydroxylated amino acids in the ER, but has no reported connection to ADHD or other developmental disorders [89].
Correlation between ADHD discordance and gene expression discordance
To further narrow candidate gene expression to potentially important correlates of ADHD, we moved from a categorical to a dimensional analysis of ADHD severity, building on evidence that ADHD functions like a trait in the population [90,91,92]. As explained earlier, ADHD severity scores (based on parent ADHD-RS raw scores) between the identical twins were compared to create a ‘discrepancy score’ for the twins. These scores were then ranked, with highest discrepancy (most different) being ranked 1, and then correlated to the difference in gene expression (fold change) between the paired twins, for the 505 RPKM list of transcripts (Additional file 7: Table S4). In an ideal scenario, the fold change would inversely correlate to the rank discrepancy (high fold change in gene expression, i.e. 10, associates with lowest numerical rank, ie 1, most discrepant). Negative correlations of r > -0.4 were observed for several transcripts of interest (boxed yellow, Additional file 8: Table S5), and closer inspection suggests they might have potential relevance to ADHD.
Among the highly correlated DEGs, RN7SKP194 (r = -0.60) bears some general similarity to the RN7SL454P target identified by the 9 filter approach, and discussed above. Both of these small nuclear pseudogenes are likely to have as yet unknown regulatory functions [93]. SRP14, signal recognition particle 14, is potentially interesting because it is 5–sevenfold lower in the ADHD twins, and it is part of a larger riboprotein complex thought to regulate translational arrest during protein synthesis in dendrites [94]. GMFG, glia maturation factor gamma, is almost 16-fold lower in the ADHD twins and affects a diverse range of cell types. MICU1, elevated threefold in ADHD twins, encodes a Ca+2-sensing, regulatory subunit of the mitochondrial uniporter, and mutations in MICU1 cause a range of symptoms that include progressive extrapyramidal signs, learning disabilities, and fatigue [95]. Among positively correlated transcripts, whereby the most discordant pairs showed the least fold-change in expression was GIT1 with r = 0.585 and an average increase of 2.4 fold in the ADHD twins. Unfortunately, the changes in RNA levels are generally not perfectly correlated with changes in protein expression (r = ~ 0.6) [96], and so this unexpected relationship may not be a significant impediment to GIT1′s relevance to ADHD.
Comparison to prior genetic studies
Prior exome sequencing of sporadic ADHD cases compared to sibling/parent triads identified ~ 8 interesting targets [4]. Of those, exome mutations in TBC1D9 are a relatively close match to RNAseq expression changes in TBC1D17 observed in the present discordant twin pairs. This suggest a closer look at this family of proteins, likely to be important in vesicle transport, may be warranted. A second possible match is between exome mutations in WDR83, and expression changes in WDR45B in discordant twins, and WDR74 in case control subjects. Also, we observed some similarity to transcripts identified in ADHD by Liao et al. [97], whereby transcripts MED8 and ARTN had suggestive p values in our analysis.
Discussion
Among the strengths of the present approach are the unique and well-characterized ADHD cohorts. In particular, monozygotic, but discordant twins present a powerful genetic model for comparison, and here demonstrated some intriguing similarity in expression pattern with a case–control cohort. They open the possibility of understanding environmental influences while largely controlling for genotype. An important methodological detail is that the RNAseq analysis examines the expression pattern on a relatively high resolution scale to the level of transcript isoforms. Other common RNAseq analytic platforms tend to aggregate expression to ‘gene level’ expression as a single transcript, which has the effect of masking changes in alternatively spliced transcripts. The case of GIT1 is an excellent example of where very specific changes in one splice variant might have high relevance to the disorder in question.
Limitations to the present study principally derive from the observational nature of the studies, a necessarily small samples for MZ discordant twins, and the necessity to use peripheral blood RNA, as opposed to a tissue more proximal to the presumed neural influences on ADHD. Further, the mRNA profiling gives us a very comprehensive view of the transcriptome, albeit at a specific point in time, and without strict control of the mental or physical state of the participants. Of course, causality is indeterminate: we cannot evaluate, for instance, whether changes in the galactose pathway ‘cause’ ADHD, or somehow result from the increased activity or altered diet or other behaviors of the children. A third option, which must be considered, is that both ADHD and galactose changes could result from changes in a different pathway, or from coincidental differences in ADHD teens, such as diet, drugs, or activity. While we can exclude ADHD medications as a source of variations herein, it is difficult to exclude other type of nutritional or nutraceutical differences.
Technically speaking, the RNAseq approach is intrinsically limited by the known genome and transcriptomes that are used to align and interpret the reads. Every RNA profiling method has unique ‘gaps’ and biases that can influence the outcomes, and thus it should not be surprising if a different RNAseq method identified other different differentially expressed transcripts. Further, the RNAseq of whole blood allows for the possibility that the types of cells present in blood at the time of sampling differ from patient to patient, or group to group. Because blood cell counts were not available on the subjects at the time of the blood draw, we were unable to identify such differences or adjust for them. Additionally, a valuable approach to understanding this large data set would be to conduct co-expression analysis, which is a logical next step that might reveal systematic changes not apparent from the current analysis [98,99,100].
While the present results therefore should be seen as preliminary, the nature of this work is largely unprecedented and therefore it is valuable to note that several patterns were identified that are suggestive as hypotheses for further investigation. Collectively, the results affirm some prior targets, such as GIT1, that were identified by DNA-based technologies, as also relevant to ADHD at the RNA level. Several new pathways are brought to light as potentially productive ground for further exploration. Based on a variety of lines of evidence, it would be quite unlikely if there is a single etiologic cause for ADHD, and the present results demonstrate that none of the RNA transcript changes were observed to occur in all of the youth. It is interesting to speculate that changes in GAK or GIT1, which showed quite strong changes in some, but not all subjects, could indicate particular genetic/epigenetic subtypes of ADHD. The cohort sizes obviously were not powered for a detailed subtype analysis.
An intriguing future direction would explore the possible role of the galactose pathway in ADHD, either as a modulator of core energy sensing via the insulin/AKT/NFkB/mediator pathway (Fig. 3), or as a regulator of galactosylation of key factors in the neurotransmitter pathway, as highlighted by consistent changes in GALE/GALK/GALT. Galactose metabolism could be related to energy sensing and inflammation via the well-established glucose/lactose/galactose connection to the immune/inflammatory pathways that is key to the obesity/insulin resistance/inflammation connection (reviewed in [101]). Additionally, galactose modification to proteins alters their inflammatory potential and is thought to be a key component of ‘inflammaging’ [102]. Potentially the most obvious effect of altered galactose metabolism would be the direct effect on the transport of dopamine to the brain, via galactose modification of dopamine [103]. Dopamine itself is poorly absorbed in the brain, but galactosylated dopamine has increased transit across the blood–brain barrier, and in mouse models increases attention without reducing activity [104]. Given a well-documented relationship between dopaminergic dysfunction and ADHD [105], it is quite plausible that perturbation of the galactose pathway in humans could produce an ADHD-like syndrome.
Conclusions
These results are the most extensive discordant MZ study of RNAseq expression in ADHD. The results, while preliminary, suggest several interesting hypotheses for further study.
Availability of data and materials
Expression-level de-identified data is available at Gene Expression Omnibus (GEO Accession GSE159104) for any investigators seeking to reproduce the results, or conduct new analysis within the bounds of the IRB-approved work. For the purpose of respecting the privacy of the participants, and their permission to use their samples only for specific research projects, sequence-level data is available through dbGAP (linked in GEO) upon appropriate permissions from Dr. Joel Nigg (niggj@ohsu.edu). The HG38 human genome used for alignment is available via UCSC (https://hgdownload.soe.ucsc.edu/goldenPath/hg38/bigZips/), and the ribosomal sequences are available at Genbank (https://www.ncbi.nlm.nih.gov/nuccore/U13369.1).
Abbreviations
- ADHD:
-
Attention-deficit hyperactivity disorder
- DSM-5:
-
Diagnostic and Statistical Manual of Mental Disorders-5
- DEG:
-
Differentially expressed genes
- GWAS:
-
Genome-wide association studies
- KSADS:
-
Kiddie Schedule for Affective Disorders and Schizophrenia
- MSUTR:
-
Michigan State University Twin Registry
- MZ:
-
Monozygotic
- ODD:
-
Oppositional defiant disorder
- RNAseq:
-
RNA sequencing
- RPKM:
-
Reads per thousand bases of transcript per million transcripts sequenced
- SDQ:
-
Strengths and Difficulties Questionnaire
- SRF:
-
Short read format
- tSMS:
-
True Single Molecule Sequencing
References:
Polanczyk G, de Lima MS, Horta BL, Biederman J, Rohde LA. The worldwide prevalence of ADHD: a systematic review and metaregression analysis. Am J Psychiatry. 2007;164(6):942–8.
Faraone SV, Larsson H. Genetics of attention deficit hyperactivity disorder. Mol Psychiatry. 2019;24(4):562–75.
Karalunas SL, Fair D, Musser ED, Aykes K, Iyer SP, Nigg JT. Subtyping attention-deficit/hyperactivity disorder using temperament dimensions: toward biologically based nosologic criteria. JAMA Psychiatry. 2014;71(9):1015–24.
Kim YS, Woo J, Lee CJ, Yoon BE. Decreased glial GABA and tonic inhibition in cerebellum of mouse model for attention-deficit/hyperactivity disorder (ADHD). Exp Neurobiol. 2017;26(4):206–12.
Huang X, Wang M, Zhang Q, Chen X, Wu J. The role of glutamate receptors in attention-deficit/hyperactivity disorder: from physiology to disease. Am J Med Genet B Neuropsychiatr Genet. 2019;180(4):272–86.
Sharma A, Couture J. A review of the pathophysiology, etiology, and treatment of attention-deficit hyperactivity disorder (ADHD). Ann Pharmacother. 2014;48(2):209–25.
Pinares-Garcia P, Stratikopoulos M, Zagato A, Loke H, Lee J. Sex: a significant risk factor for neurodevelopmental and neurodegenerative disorders. Brain Sci. 2018;8(8):154.
Lorenzo G, Braun J, Munoz G, Casarejos MJ, Bazan E, Jimenez-Escrig A. RNA-Seq blood transcriptome profiling in familial attention deficit and hyperactivity disorder (ADHD). Psychiatry Res. 2018;270:544–6.
Srivastav S, Walitza S, Grunblatt E. Emerging role of miRNA in attention deficit hyperactivity disorder: a systematic review. Atten Defic Hyperact Disord. 2018;10(1):49–63.
Miller LL, Gustafsson HC, Tipsord J, Song M, Nousen E, Dieckmann N, Nigg JT. Is the Association of ADHD with socio-economic disadvantage explained by child comorbid externalizing problems or parent ADHD? J Abnorm Child Psychol. 2018;46(5):951–63.
Nigg JT, Gustafsson HC, Karalunas SL, Ryabinin P, McWeeney SK, Faraone SV, Mooney MA, Fair DA, Wilmot B. Working memory and vigilance as multivariate endophenotypes related to common genetic risk for attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2018;57(3):175–82.
Burt SA, Klump KL. The Michigan State University Twin Registry (MSUTR): an update. Twin Res Hum Genet. 2013;16(1):344–50.
Klump KL, Burt SA. The Michigan State University Twin Registry (MSUTR): genetic, environmental and neurobiological influences on behavior across development. Twin Res Hum Genet. 2006;9(6):971–7.
Conners CK. Conners’ rating scales: revised technical manual. New York: Multi-Health Systems; 2003.
Goodman R. Psychometric properties of the strengths and difficulties questionnaire. J Am Acad Child Adolesc Psychiatry. 2001;40(11):1337–45.
DuPaul G, Power T, Anastopoulos A, Reid R. ADHD Rating Scale—IV: checklists, norms, and clinical interpretation. New York, NY: Guilford Press; 1998.
Puig-Antich J, Ryan N. Kiddie schedule for affective disorders and schizophrenia. Pittsburgh, PA: Western Psychiatric Institute; 1986.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–40.
McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40(10):4288–97.
Sun J, Nishiyama T, Shimizu K, Kadota K. TCC: an R package for comparing tag count data with robust normalization strategies. BMC Bioinform. 2013;14:219.
Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10):R106.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
Law CW, Chen Y, Shi W, Smyth GK. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15(2):R29.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47.
Hardcastle TJ, Kelly KA. Empirical Bayesian analysis of paired high-throughput sequencing data with a beta-binomial distribution. BMC Bioinform. 2013;14:135.
Fernandes AD, Macklaim JM, Linn TG, Reid G, Gloor GB. ANOVA-like differential expression (ALDEx) analysis for mixed population RNA-Seq. PLoS ONE. 2013;8(7):e67019.
Lutz NW, Banerjee P, Wilson BJ, Ma J, Cozzone PJ, Frank MH. Expression of Cell-Surface marker ABCB5 causes characteristic modifications of glucose, amino acid and phospholipid metabolism in the G3361 melanoma-initiating cell line. PLoS ONE. 2016;11(8):e0161803.
Glessner JT, Bradfield JP, Wang K, Takahashi N, Zhang H, Sleiman PM, Mentch FD, Kim CE, Hou C, Thomas KA, et al. A genome-wide study reveals copy number variants exclusive to childhood obesity cases. Am J Hum Genet. 2010;87(5):661–6.
Nigg JT, Johnstone JM, Musser ED, Long HG, Willoughby MT, Shannon J. Attention-deficit/hyperactivity disorder (ADHD) and being overweight/obesity: New data and meta-analysis. Clin Psychol Rev. 2016;43:67–79.
Xu M, Xie YA, Abouzeid H, Gordon CT, Fiorentino A, Sun Z, Lehman A, Osman IS, Dharmat R, Riveiro-Alvarez R, et al. Mutations in the spliceosome component CWC27 cause retinal degeneration with or without additional developmental anomalies. Am J Hum Genet. 2017;100(4):592–604.
Xiahou Z, Wang X, Shen J, Zhu X, Xu F, Hu R, Guo D, Li H, Tian Y, Liu Y, et al. NMI and IFP35 serve as proinflammatory DAMPs during cellular infection and injury. Nat Commun. 2017;8(1):950.
Dunn GA, Nigg JT, Sullivan EL. Neuroinflammation as a risk factor for attention deficit hyperactivity disorder. Pharmacol Biochem Behav. 2019;182:22–34.
Han MR, Han KM, Kim A, Kang W, Kang Y, Kang J, Won E, Tae WS, Cho Y, Ham BJ. Whole-exome sequencing identifies variants associated with structural MRI markers in patients with bipolar disorders. J Affect Disord. 2019;249:159–68.
Jin J, Bhatti DL, Lee KW, Medrihan L, Cheng J, Wei J, Zhong P, Yan Z, Kooiker C, Song C, et al. Ahnak scaffolds p11/Anxa2 complex and L-type voltage-gated calcium channel and modulates depressive behavior. Mol Psychiatry. 2019;25:1039–49.
Haase H, Alvarez J, Petzhold D, Doller A, Behlke J, Erdmann J, Hetzer R, Regitz-Zagrosek V, Vassort G, Morano I. Ahnak is critical for cardiac Ca(V)1.2 calcium channel function and its beta-adrenergic regulation. FASEB J. 2005;19(14):1969–77.
Sha S, Xu J, Lu ZH, Hong J, Qu WJ, Zhou JW, Chen L. Lack of JWA enhances neurogenesis and long-term potentiation in hippocampal dentate gyrus leading to spatial cognitive potentiation. Mol Neurobiol. 2016;53(1):355–68.
Miao SH, Sun HB, Ye Y, Yang JJ, Shi YW, Lu M, Hu G, Zhou JW. Astrocytic JWA expression is essential to dopaminergic neuron survival in the pathogenesis of Parkinson’s disease. CNS Neurosci Ther. 2014;20(8):754–62.
Ikemoto MJ, Inoue K, Akiduki S, Osugi T, Imamura T, Ishida N, Ohtomi M. Identification of addicsin/GTRAP3-18 as a chronic morphine-augmented gene in amygdala. NeuroReport. 2002;13(16):2079–84.
Wu Y, Chen R, Zhao X, Li A, Li G, Zhou J. JWA regulates chronic morphine dependence via the delta opioid receptor. Biochem Biophys Res Commun. 2011;409(3):520–5.
Goncalves TM, Southey BR, Rodriguez-Zas SL. Interplay between amphetamine and activity level in gene networks of the mouse striatum. Bioinform Biol Insights. 2018;12:1177932218815152.
Cheng Y, Quinn JF, Weiss LA. An eQTL mapping approach reveals that rare variants in the SEMA5A regulatory network impact autism risk. Hum Mol Genet. 2013;22(14):2960–72.
Hamnett R, Crosby P, Chesham JE, Hastings MH. Vasoactive intestinal peptide controls the suprachiasmatic circadian clock network via ERK1/2 and DUSP4 signalling. Nat Commun. 2019;10(1):542.
Park GY, Jang DH, Lee DW, Jang JH, Joo J. Hereditary sensory and autonomic neuropathy 2B caused by a novel RETREG1 mutation (c.765dupT) and paternal uniparental isodisomy of chromosome 5. Front Genet. 2019;10:1085.
Forman OP, Hitti RJ, Pettitt L, Jenkins CA, O’Brien DP, Shelton GD, De Risio L, Quintana RG, Beltran E, Mellersh C. An inversion disrupting FAM134B is associated with sensory neuropathy in the border collie dog breed. G3 (Bethesda). 2016;6(9):2687–92.
Islam F, Gopalan V, Lam AK. RETREG1 (FAM134B): A new player in human diseases: 15 years after the discovery in cancer. J Cell Physiol. 2018;233(6):4479–89.
Polimanti R, Zhao H, Farrer LA, Kranzler HR, Gelernter J. Ancestry-specific and sex-specific risk alleles identified in a genome-wide gene-by-alcohol dependence interaction study of risky sexual behaviors. Am J Med Genet B Neuropsychiatr Genet. 2017;174(8):846–53.
Hechtman L, Swanson JM, Sibley MH, Stehli A, Owens EB, Mitchell JT, Arnold LE, Molina BS, Hinshaw SP, Jensen PS et al. Functional adult outcomes 16 years after childhood diagnosis of attention-deficit/hyperactivity disorder: MTA results. J Am Acad Child Adolesc Psychiatry 2016, 55(11):945–952 e942.
Mathew T, Avati A, D’Souza D, Therambil M. Expanding spectrum of RARS2 gene disorders: Myoclonic epilepsy, mental retardation, spasticity, and extrapyramidal features. Epilepsia Open. 2018;3(2):270–5.
Zhang J, Zhang Z, Zhang Y, Wu Y. Distinct magnetic resonance imaging features in a patient with novel RARS2 mutations: A case report and review of the literature. Exp Ther Med. 2018;15(1):1099–104.
Chen H, Huang S, Han X, Zhang J, Shan C, Tsang YH, Ma HT, Poon RY. Salt-inducible kinase 3 is a novel mitotic regulator and a target for enhancing antimitotic therapeutic-mediated cell death. Cell Death Dis. 2014;5:e1177.
Jantscher F, Pirker C, Mayer CE, Berger W, Sutterluety H. Overexpression of Aurora-A in primary cells interferes with S-phase entry by diminishing Cyclin D1 dependent activities. Mol Cancer. 2011;10:28.
Nudel R, Benros ME, Krebs MD, Allesoe RL, Lemvigh CK, Bybjerg-Grauholm J, Borglum AD, Daly MJ, Nordentoft M, Mors O, et al. Immunity and mental illness: findings from a Danish population-based immunogenetic study of seven psychiatric and neurodevelopmental disorders. Eur J Hum Genet. 2019;27:1445–55.
Wang CJ, Chidiac P. RGS2 promotes the translation of stress-associated proteins ATF4 and CHOP via its eIF2B-inhibitory domain. Cell Signal. 2019;59:163–70.
Asselmann E, Hertel J, Schmidt CO, Homuth G, Nauck M, Beesdo-Baum K, Grabe HJ, Pane-Farre CA. Interplay between RGS2 and childhood adversities in predicting anxiety and depressive disorders: Findings from a general population sample. Depress Anxiety. 2018;35(11):1104–13.
Rorabaugh BR, Chakravarti B, Mabe NW, Seeley SL, Bui AD, Yang J, Watts SW, Neubig RR, Fisher RA. Regulator of G Protein Signaling 6 Protects the Heart from Ischemic Injury. J Pharmacol Exp Ther. 2017;360(3):409–16.
Rorabaugh BR, Sprague L, Norman H, Seeley SL, D’Souza MS. Regulator of G protein signaling 2 differentially regulates nicotine-induced anxiolytic- and antidepressant-like effects in mice. Eur J Neurosci. 2018;48(5):2110–7.
Okimoto N, Bosch OJ, Slattery DA, Pflaum K, Matsushita H, Wei FY, Ohmori M, Nishiki T, Ohmori I, Hiramatsu Y, et al. RGS2 mediates the anxiolytic effect of oxytocin. Brain Res. 2012;1453:26–33.
Oliveira-Dos-Santos AJ, Matsumoto G, Snow BE, Bai D, Houston FP, Whishaw IQ, Mariathasan S, Sasaki T, Wakeham A, Ohashi PS, et al. Regulation of T cell activation, anxiety, and male aggression by RGS2. Proc Natl Acad Sci USA. 2000;97(22):12272–7.
Raab A, Popp S, Lesch KP, Lohse MJ, Fischer M, Deckert J, Hommers L. Increased fear learning, spatial learning as well as neophobia in Rgs2(-/-) mice. Genes Brain Behav. 2018;17(4):e12420.
Luessen DJ, Hinshaw TP, Sun H, Howlett AC, Marrs G, McCool BA, Chen R. RGS2 modulates the activity and internalization of dopamine D2 receptors in neuroblastoma N2A cells. Neuropharmacology. 2016;110(Pt A):297–307.
Calipari ES, Sun H, Eldeeb K, Luessen DJ, Feng X, Howlett AC, Jones SR, Chen R. Amphetamine self-administration attenuates dopamine D2 autoreceptor function. Neuropsychopharmacology. 2014;39(8):1833–42.
van Duijn CM, Dekker MC, Bonifati V, Galjaard RJ, Houwing-Duistermaat JJ, Snijders PJ, Testers L, Breedveld GJ, Horstink M, Sandkuijl LA, et al. Park7, a novel locus for autosomal recessive early-onset parkinsonism, on chromosome 1p36. Am J Hum Genet. 2001;69(3):629–34.
Kehagia AA, Housden CR, Regenthal R, Barker RA, Muller U, Rowe J, Sahakian BJ, Robbins TW. Targeting impulsivity in Parkinson’s disease using atomoxetine. Brain. 2014;137(Pt 7):1986–97.
Lee CJ, Wu CC, Chou WJ, Lee MJ, Chou MC, Lee SY, Wang LJ. Mitochondrial-associated protein biomarkers in patients with attention-deficit/hyperactivity disorder. Mitochondrion. 2019;49:83–8.
Hurt JA, Obar RA, Zhai B, Farny NG, Gygi SP, Silver PA. A conserved CCCH-type zinc finger protein regulates mRNA nuclear adenylation and export. J Cell Biol. 2009;185(2):265–77.
Fallahi P, Katz R, Toma I, Li R, Reiner J, VanHouten K, Carpio L, Marshall L, Lian Y, Bupp S, et al. Aspirin Insensitive Thrombophilia: Transcript profiling of blood identifies platelet abnormalities and HLA restriction. Gene. 2013;520(2):131–8.
Chawla LS, Toma I, Davison D, Vaziri K, Lee J, Lucas R, Seneff MG, Nyhan A, McCaffrey TA. Acute appendicitis: transcript profiling of blood identifies promising biomarkers and potential underlying processes. BMC Med Genomics. 2016;9(1):40.
Ashtari N, Jiao X, Rahimi-Balaei M, Amiri S, Mehr SE, Yeganeh B, Marzban H. Lysosomal Acid phosphatase biosynthesis and dysfunction: a mini review focused on lysosomal enzyme dysfunction in brain. Curr Mol Med. 2016;16(5):439–46.
Sittig LJ, Carbonetto P, Engel KA, Krauss KS, Palmer AA. Integration of genome-wide association and extant brain expression QTL identifies candidate genes influencing prepulse inhibition in inbred F1 mice. Genes Brain Behav. 2016;15(2):260–70.
Rubio RM, Depledge DP, Bianco C, Thompson L, Mohr I. RNA m(6) A modification enzymes shape innate responses to DNA by regulating interferon beta. Genes Dev. 2018;32(23–24):1472–84.
Du K, Zhang L, Lee T, Sun T. m(6)A RNA Methylation Controls Neural Development and Is Involved in Human Diseases. Mol Neurobiol. 2019;56(3):1596–606.
Stojkovic V, Fujimori DG. Mutations in RNA methylating enzymes in disease. Curr Opin Chem Biol. 2017;41:20–7.
Habtemichael EN, Alcazar-Roman A, Rubin BR, Grossi LR, Belman JP, Julca O, Loffler MG, Li H, Chi NW, Samuel VT, et al. Coordinated regulation of vasopressin inactivation and glucose uptake by action of TUG protein in muscle. J Biol Chem. 2015;290(23):14454–61.
Sharma SR, Gonda X, Tarazi FI. Autism Spectrum Disorder: Classification, diagnosis and therapy. Pharmacol Ther. 2018;190:91–104.
Strittmatter L, Li Y, Nakatsuka NJ, Calvo SE, Grabarek Z, Mootha VK. CLYBL is a polymorphic human enzyme with malate synthase and beta-methylmalate synthase activity. Hum Mol Genet. 2014;23(9):2313–23.
Reus GZ, Scaini G, Furlanetto CB, Morais MO, Jeremias IC, Mello-Santos LM, Freitas KV, Quevedo J, Streck EL. Methylphenidate treatment leads to abnormalities on krebs cycle enzymes in the brain of young and adult rats. Neurotox Res. 2013;24(2):251–7.
Baltussen LL, Rosianu F, Ultanir SK. Kinases in synaptic development and neurological diseases. Prog Neuropsychopharmacol Biol Psychiatry. 2018;84(Pt B):343–52.
Witoelar A, Jansen IE, Wang Y, Desikan RS, Gibbs JR, Blauwendraat C, Thompson WK, Hernandez DG, Djurovic S, Schork AJ, et al. Genome-wide pleiotropy between parkinson disease and autoimmune diseases. JAMA Neurol. 2017;74(7):780–92.
Won H, Mah W, Kim E, Kim JW, Hahm EK, Kim MH, Cho S, Kim J, Jang H, Cho SC, et al. GIT1 is associated with ADHD in humans and ADHD-like behaviors in mice. Nat Med. 2011;17(5):566–72.
Klein M, van der Voet M, Harich B, van Hulzen KJ, Onnink AM, Hoogman M, Guadalupe T, Zwiers M, Groothuismink JM, Verberkt A, et al. Converging evidence does not support GIT1 as an ADHD risk gene. Am J Med Genet B Neuropsychiatr Genet. 2015;168(6):492–507.
Kim H, Kim JI, Kim H, Kim JW, Kim BN. Interaction effects of GIT1 and DRD4 gene variants on continuous performance test variables in patients with ADHD. Brain Behav. 2017;7(9):e00785.
Li YS, Qin LX, Liu J, Xia WL, Li JP, Shen HL, Gao WQ. GIT1 enhances neurite outgrowth by stimulating microtubule assembly. Neural Regen Res. 2016;11(3):427–34.
Zhang H, Webb DJ, Asmussen H, Horwitz AF. Synapse formation is regulated by the signaling adaptor GIT1. J Cell Biol. 2003;161(1):131–42.
Claing A, Perry SJ, Achiriloaie M, Walker JK, Albanesi JP, Lefkowitz RJ, Premont RT. Multiple endocytic pathways of G protein-coupled receptors delineated by GIT1 sensitivity. Proc Natl Acad Sci USA. 2000;97(3):1119–24.
Kapuralin K, Curlin M, Mitrecic D, Kosi N, Schwarzer C, Glavan G, Gajovic S. STAM2, a member of the endosome-associated complex ESCRT-0 is highly expressed in neurons. Mol Cell Neurosci. 2015;67:104–15.
Endo K, Takeshita T, Kasai H, Sasaki Y, Tanaka N, Asao H, Kikuchi K, Yamada M, Chenb M, O’Shea JJ, et al. STAM2, a new member of the STAM family, binding to the Janus kinases. FEBS Lett. 2000;477(1–2):55–61.
Felder B, Radlwimmer B, Benner A, Mincheva A, Todt G, Beyer KS, Schuster C, Bolte S, Schmotzer G, Klauck SM, et al. FARP2, HDLBP and PASK are downregulated in a patient with autism and 2q37.3 deletion syndrome. Am J Med Genet A. 2009;149A(5):952–9.
Chistiakov DA, Kuzenkova LM, Savost’anov KV, Gevorkyan AK, Pushkov AA, Nikitin AG, Vashakmadze ND, Zhurkova NV, Podkletnova TV, Namazova-Baranova LS, et al. Genetic analysis of 17 children with Hunter syndrome: identification and functional characterization of four novel mutations in the iduronate-2-sulfatase gene. J Genet Genomics. 2014;41(4):197–203.
Wang X, Herr RA, Rabelink M, Hoeben RC, Wiertz EJ, Hansen TH. Ube2j2 ubiquitinates hydroxylated amino acids on ER-associated degradation substrates. J Cell Biol. 2009;187(5):655–68.
Marcus DK, Barry TD. Does attention-deficit/hyperactivity disorder have a dimensional latent structure? A taxometric analysis. J Abnorm Psychol. 2011;120(2):427–42.
Haslam N, Williams B, Prior M, Haslam R, Graetz B, Sawyer M. The latent structure of attention-deficit/hyperactivity disorder: a taxometric analysis. Aust N Z J Psychiatry. 2006;40(8):639–47.
Levy F, Hay DA, McStephen M, Wood C, Waldman I. Attention-deficit hyperactivity disorder: a category or a continuum? Genetic analysis of a large-scale twin study. J Am Acad Child Adolesc Psychiatry. 1997;36(6):737–44.
Wang J, Samuels DC, Zhao S, Xiang Y, Zhao YY, Guo Y. Current research on non-coding ribonucleic acid (RNA). Genes (Basel). 2017;8(12):366.
Kremerskothen J, Zopf D, Walter P, Cheng JG, Nettermann M, Niewerth U, Maraia RJ, Brosius J. Heterodimer SRP9/14 is an integral part of the neural BC200 RNP in primate brain. Neurosci Lett. 1998;245(3):123–6.
Musa S, Eyaid W, Kamer K, Ali R, Al-Mureikhi M, Shahbeck N, Al Mesaifri F, Makhseed N, Mohamed Z, AlShehhi WA, et al. A middle eastern founder mutation expands the genotypic and phenotypic spectrum of mitochondrial MICU1 deficiency: a report of 13 patients. JIMD Rep. 2019;43:79–83.
Edfors F, Danielsson F, Hallstrom BM, Kall L, Lundberg E, Ponten F, Forsstrom B, Uhlen M. Gene-specific correlation of RNA and protein levels in human cells and tissues. Mol Syst Biol. 2016;12(10):883.
Liao C, Laporte AD, Spiegelman D, Akcimen F, Joober R, Dion PA, Rouleau GA. Transcriptome-wide association study of attention deficit hyperactivity disorder identifies associated genes and phenotypes. Nat Commun. 2019;10(1):4450.
Chen J, Zhao X, Cui L, He G, Wang X, Wang F, Duan S, He L, Li Q, Yu X, et al. Genetic regulatory subnetworks and key regulating genes in rat hippocampus perturbed by prenatal malnutrition: implications for major brain disorders. Aging (Albany NY). 2020;12(9):8434–58.
Li H, Wang X, Lu X, Zhu H, Li S, Duan S, Zhao X, Zhang F, Alterovitz G, Wang F, et al. Co-expression network analysis identified hub genes critical to triglyceride and free fatty acid metabolism as key regulators of age-related vascular dysfunction in mice. Aging (Albany NY). 2019;11(18):7620–38.
Morrow JD, Qiu W, Chhabra D, Rennard SI, Belloni P, Belousov A, Pillai SG, Hersh CP. Identifying a gene expression signature of frequent COPD exacerbations in peripheral blood using network methods. BMC Med Genomics. 2015;8:1.
Lee BC, Lee J. Cellular and molecular players in adipose tissue inflammation in the development of obesity-induced insulin resistance. Biochim Biophys Acta. 2014;1842(3):446–62.
Dall’Olio F. Glycobiology of aging. Subcell Biochem. 2018;90:505–26.
Ruocco LA, Viggiano D, Pignatelli M, Iannaccone T, Rimoli MG, Melisi D, Curcio A, De Lucia S, Carboni E, Gironi Carnevale UA, et al. Galactosilated dopamine increases attention without reducing activity in C57BL/6 mice. Behav Brain Res. 2008;187(2):449–54.
Ruocco LA, Viggiano D, Viggiano A, Abignente E, Rimoli MG, Melisi D, Curcio A, Nieddu M, Boatto G, Carboni E, et al. Galactosylated dopamine enters into the brain, blocks the mesocorticolimbic system and modulates activity and scanning time in Naples high excitability rats. Neuroscience. 2008;152(1):234–44.
Viggiano D, Vallone D, Sadile A. Dysfunctions in dopamine systems and ADHD: evidence from animals and modeling. Neural Plast. 2004;11(1–2):97–114.
Acknowledgements
The authors are grateful to The Michigan State University Twins Registry, and the Co-Directors Dr. Kelly Klump and Dr. S. Alexandra Burt, for providing blood samples and clinical data for these studies, and to the OHSU Genomics Core, especially Beth Wilmot, for RNA sample processing. The authors are also very grateful to Debra Dederich of SeqLL for her dedicated work in sample processing for RNAseq data generation. The authors are grateful to Richard Wargowsky for preparation of color figures, and to Maxim Ri for help with bioinformatic analysis.
Funding
The authors are grateful to The Abracadabra Foundation, and the St. Laurent Family, through The St. Laurent Institute, for their generous philanthropic support of this work. The funding organizations were apprised of, but had no role in the design, analysis, or interpretation of the data, nor were they involved in the writing of the manuscript.
Author information
Authors and Affiliations
Contributions
JN, GSL, and TM were responsible for the overall conception, design, and execution of the studies. GSL, DS, and DJ conducted the RNA sequencing, alignment, and DEG analysis. JN and EB conducted clinical assessments of ADHD, and analyzed resulting data for comparison to RNA expression. YV and DA developed a custom bioinformatics pipeline and conducted bioinformatics analysis. TM and JN analyzed DEGs for pathways and patterns relevant to ADHD. The manuscript was principally drafted by TM and JN, with input from all the authors. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All human subjects gave written informed consent under protocols approved by either the Oregon Health and Science University (OHSU) Institutional Review Board (approval # IRB10543) and the Michigan State University Twin Registry (MSUTR IRB). For subjects under 16 years old, written informed consent was obtained from the parents or legal guardians. Additionally, for subjects under age 16, written informed assent of the subject was obtained, and for subjects over age 16, written informed consent was obtained. All research complied with applicable ethical and human participant guidelines.
Consent for publication
Not applicable, no individually identifiable information is provided.
Competing interests
TM holds an equity interest in True Bearing Diagnostics, Inc., which is developing blood RNA-based biomarkers for several disorders, but not specifically in the area of ADHD at this time. DJ is employed by and holds an equity interest in SeqLL, Inc. All other authors do not report competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1
. Enrollment details.
Additional file 2
. Transcript IDs by 9 DEG methods for case/control study.
Additional file 3
. Transcript IDs by 9 DEG methods for discordant twin study.
Additional file 4
. Case control T-test 524 transcripts.
Additional file 5
. Discordant twins paired T-test 505 transcripts.
Additional file 6
. Discordance DEG correlation.
Additional file 7
. Detailed methods.
Additional file 8
. DEG Concordance between methods.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
McCaffrey, T.A., St. Laurent, G., Shtokalo, D. et al. Biomarker discovery in attention deficit hyperactivity disorder: RNA sequencing of whole blood in discordant twin and case-controlled cohorts. BMC Med Genomics 13, 160 (2020). https://doi.org/10.1186/s12920-020-00808-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-020-00808-8