
Abstract
Background
The microdeletion of chromosome 13 has been rarely reported. Here, we report a 14-year old Asian female with a de novo microdeletion on 13q12.3.
Case presentation
The child suffered mainly from two types of epileptic seizures: partial onset seizures and myoclonic seizures, accompanied with intellectual disability, developmental delay and minor dysmorphic features. The electroencephalogram disclosed slow waves in bilateral temporal, together with generalized spike-and-slow waves, multiple-spike-and-slow waves and slow waves in bilateral occipitotemporal regions. The exome sequencing showed no pathogenic genetic variation in the patient’s DNA sample. While the single nucleotide polymorphism (SNP) array analysis revealed a de novo microdeletion spanning 2.324 Mb, within the cytogenetic band 13q12.3.
Conclusions
The epilepsy may be associated with the mutation of KATNAL1 gene or the deletion unmasking a recessive mutation on the other allele, and our findings could provide a phenotypic expansion.
Similar content being viewed by others


Background
Epilepsy refers to a chronic neurologic disorder, and it leads to impairments of cognitive and behavioral function. Although the mechanism of epilepsy is still unclear, it is believed that genetic cause is strongly associated with epilepsy of infancy and childhood. Moreover, the genetic cause of epilepsy was initially demonstrated in 2001, with a finding that all seven children in a study of Dravet syndrome had a de novo SCN1A mutation. With the development of molecular techniques, more discoveries between genetics and epilepsy have been revealed [1,2,3]. Besides, chromosome 13 owns one of the lowest gene densities among human chromosomes and structural and functional variations of it may lead to 13q-syndrome, but interstitial deletion of 13q12.3 has only rarely been reported [4].
M. Drummond-Borg et al. [5] have reported a complex chromosome rearrangement involving chromosome 2,13, and 20 in the normal mother of a girl with mild clinical features, developmental delay and an interstitial deletion of 13q12.1-q14.1. Also, another 14 cases of de novo 13q partial deletions (seven terminal and seven interstitial) ranging from 4.2 to 75.7 Mb showed varying degrees of intellectual disability and specific clinical features, among them, 8 had central nervous system anomalies, 6 had eyes abnormalities, 9 had facial dysmorphisms and 10 had hand or feet anomalies [6]. Der Kaloustian et al. [7] have revealed a patient with an interstitial deletion of 2.1 Mb at 13q12.11 who had mild developmental delay, craniofacial dysmorphism, a pectus excavatum, narrow shoulders, malformed toes café-au-lait spots. Furthermore, a child with approximate 12 Mb deletion involving chromosome bands 13q12.3-13q14.11 showed immunodeficiency with elevated IgM levels, mild and transient cerebellar ataxia, and developmental delay [8].
However, currently, there is no report illustrating the correction between microdeletion of 13q12.3 and epilepsy. Here, we present an Asian (Chinese) patient with a microdeletion of 2.324 Mb on 13q12.3, who displayed the recurrent unconsciousness with convulsion for 7 years.
Case presentation
The proband is a 14-year-old Asian female. She is the first child of nonconsanguineous parents who have another healthy child. The family history was unremarkable. She was delivered by forceps-assisted vaginal delivery at gestational age 40 weeks, with 20 h of labor. Her birth weight was 4250 g. She was not able to suckle for several days after she was born. A developmental delay was observed since her first few months of life. The child had the capability of independent walking at 17 months. At the age of 20 months, she was able to speak. The child occurred hyperpyretic convulsion twice at age of four and a half, and five respectively.
At the age of 7 years, she initially appeared absence, unconsciousness, right skew of head and eyes, head back, tumble, limb convulsion, lips cyanosis, rustle in the throat, sustaining for 2 to 4 min, coupled with headache, emesis and impaired consciousness. This situation occurred 3 to 4 times per year. Electroencephalogram (EEG) was obtained at 8 years old, which showed frequent epileptic discharge in right medial temporal lobe at awake, significantly paroxysmal epileptic discharge in bilateral lobe during sleep, and sporadic epileptic discharge in central and superior lobe during sleep. Then the same attack occurred twice at the ages of 9 and 10 respectively. At the age of 11, the frequency of attack increased to four times per year, and even more regular at age of 12. Therefore, a second EEG was performed at her age of 12 and revealed epileptic discharge in occipital lobe, suspecting her of absence seizure. Meanwhile, brain magnetic resonance imaging (MRI) manifested signal enhancement in bilateral hippocampus on FLAIR sequence. Between 13 and 14 years of age inclusive, epileptic seizures took place about every 7 to 10 days, with rapid vibration of lower limbs occurring dozens of times per day. Thus, a third EEG was performed to the child that illustrated slow waves in bilateral temporal regions, significant in left, with generalized spike-and-slow waves, multiple-spike-and-slow waves and slow waves in bilateral occipitotemporal, significant in left.
After admitting, the physical examination revealed specific facial features with ocular hypertelorism, low insertion of the columella, malar flattening, bend of index and ring fingers and transverse lines in both palms. Cognitive performance was unable to be assessed. Routine metabolic assays were performed the day after admission. Serum sodium valproate: 94.53μg/ml (reference value: 50.0–100.0μg/ml), thyrotropin: 8.30uIU/ml (0.34–5.6uIU/ml, mildly high), fibrinogen: 4.07 g/l (2.0–4.0 g/l, mildly high), alanine aminotransferase: 57iu/L (5.0–40.0iu/L, mildly high), indirect bilirubin: 2.99umol/L (3.42–15.1umol/L, mildly low), γ – glutamyltranspeptidase: 57iu/L (7.0–50.0iu/L, mildly high), and blood ammonia: 70μg/dL (0.0–100.0μg/dL). Moreover, her abdominal ultrasound showed no obvious abnormality in her gall bladder, spleen, pancreas and kidney, while low echo was found in left lobe of liver, which indicated non-uniform fatty liver.
The patient was diagnosed with two types of epileptic seizures: the first type of seizure was partial onset seizures characterized by recurrent absence, unconsciousness, convulsion of four extremities, and aconuresis. The partial onset seizures could last 2 to 4 min and occurred about every 7 to 10 days. The second type of seizure was myoclonic seizures, which manifested as rapid myoclonic jerks of bilateral lower limbs. The frequency of this type of seizure was dozens of times per day. She was provided levetiracetam (43.5 mg/kg/day), lamotrigine (3.0 mg/kg/day), and sodium valproate (14.5 mg/kg/day).
Genetic study
Methods
Exome sequencing was performed. The whole area of exons and adjacent area of introns (50 bp) were captured by SeqCap EZ MedExome Kit (Roche Nimblegen) from segmented, spliced, amplified, and purified DNA sample obtained from peripheral blood of the patient. Then, the captured DNA was eluted, amplified and purified, later sequenced by Illumina. Moreover, comparison and identification of genetic variation was used Nextgene V2.3.4 software and UCSC hg19 human reference genomic sequence which collected the coverage of targeted region and average sequencing depth at the same time. The average sequencing depth of targeted area of whole exome sequencing was 100.05X, among which the sequencing depth was more than 20X in 95.35% targeted sequence. Besides, Sanger sequencing was applied to verify the genetic variation reported by abovementioned approach.
SNP array was conducted by Infinium Global Screening Array. Hence, chromosome abnormalities such as the heteroploidy, deletion, repetition, and uniparental disomy of chromosome fragment, of autosomes and sex chromosomes were detected.
Results
The exome sequencing was normal that no pathogenic genetic variation was detected in the patient’s or her mother’s DNA sample (Supplementary Table 1).
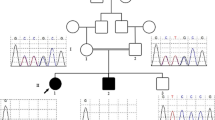
The SNP array (details in Fig. 1) revealed a microdeletion with an approximate size of 2.324 Mb on the 13q12.3 region (arr [hg19] 13q12.3(29,376,209-31,700,395) × 1), which included 9 unique genes: MTUS2, SLC7A1, UBL3, KATNAL1, HMGB1, USPL1, ALOX5AP, MEDAG3, and TEX26. However, the mutation was not found in her parents, the pedigree of the proband is presented in Fig. 2.

The SNP array of the patient. A: Array-CGH on chromosome 13. B: The ideogram of chromosome 13. C: The genes involved in the 13q12.3 deletion

The pedigree of the proband.  normal male,
normal male,  normal female,
normal female,  proband
proband
Discussion and conclusions
Here, we report a 14-year-old girl being the carrier of a 2.324 Mb microdeletion on 13q12.3 with a novel phenotype: epilepsy. The deletion of chromosome 13q12.3 has been reported only rarely and we merely find three related studies (details shown on Table 1). The clinical phenotypes were complex with facial dysmorphism, hand or feet anomalies, intellectual disability, development delay, and other anomalies. And most of them had poor prognoses. Moreover, an identical mutation site with our case has been found in a study [4] that three probands mainly presented intellectual disability, postnatal microcephaly, and eczema/atopic dermatitis, however, none of them showed epileptic seizures.
Nevertheless, none of the genes contained in the deleted region are associated with a known monogenic phenotype. MTUS2 regulates a protein involved in cardiac hypertrophy and neural differentiation, which has been confirmed in mouse and chicken models [9]. SLC7A1 is related to the function of endothelium and the decrease of nitric oxide (NO) level, it also presents a genetic susceptibility to spontaneous hypertension [10, 11]. USPL1 represents a third type of SUMO protease, and its expression increases in breast tumor tissue [17, 18]. ALOX5AP has been linked to modulate the leukotriene biosynthesis pathway and increase the risk for myocardial infarction, stroke and restenosis [19, 20]. The functions of UBL3, MEDAG3, and TEX26 were not clear. However, currently no evidence has demonstrated the association between these genes and epilepsy (refer to Table 2).
We speculate genes that are highly expressed in central nervous system (CNS) are dosage sensitive, which may lead to the occurrence of epilepsy. HMGB1 and KATNAL1 are highly expressed in CNS. HMGB1 is a possible dosage-sensitive gene encoding a ubiquitous nonhistone chromosomal protein expressed in the brain, which regulates inflammatory responses leading to neuronal excitability and seizures [14]. However, it presents a gain-of-function rather than loss-of-function effect. Both animal and human studies have demonstrated that overexpression of HMGB1 induces epilepsy via regulating TLR4/NF-κB and p38MAPK signaling pathways [15, 16]. Moreover, KATNAL1 was initially identified with a circadian deficit, it has a similar domain structure as KATNA1 and the amino acid sequence is 80% identical. Studies has confirmed that loss-of-function effect of KATNA1 performs reduction of neuronal migration, axonal elongation and axonal branching [12, 21]. A recent study has found its association with the development of neuronal function and behavior. As well as multiple morphological abnormalities and defects in neuronal migration and morphology were detected in KATNAL1 mutant mice [13]. Therefore, we assume that the mutation of KATNAL1 is one of the reasons why the proband presents epilepsy. On the other hand, it is rather usual to have an epilepsy in children with chromosomal abnormalities, a possibility that this manifestation might be in fact due to the deletion unmasking a recessive mutation on the other allele also cannot be excluded.
We describe a novel phenotype of 13q12.3 microdeletion, characterized by spontaneously recurrent epileptic seizures, as absence, unconscientiousness, convulsion, rapid vibration of lower limbs with facial and volar dysmorphism, intellectual disability, and developmental delay. Our findings could be a phenotypic expansion (a set of observed phenotypic features extended beyond those previously reported in association with a particular locus) of 13q12.3 microdeletion. However, the underlying mechanism between 13q12.3 microdeletion and epilepsy remains unclear, which requires further in vitro and/or in vivo studies.
Availability of data and materials
The original data analysis reports (in Chinese) provided by the Beijing Zhongtong Lanbo Clinical Research Institute are available at DOI: https://doi.org/10.6084/m9.figshare.12948311 and https://doi.org/10.6084/m9.figshare.12948302. The summarized analysis reports in English (translated from the original reports) can be found in supplementary Table 1 and supplementary Table 2. The hg19 human reference genomic sequence dataset used in our study was from USCS Genome Brower repository, direct web link: http://genome.ucsc.edu/cgi-bin/hgTracks?db=hg19&lastVirtModeType=default&lastVirtModeExtraState=&virtModeType=default&virtMode=0&nonVirtPosition=&position=chrX%3A15578261%2D15621068&hgsid=900320981_ExrDPx8GaC7hku1X7VbmrfpRld4I. Any clarifications regarding this study can be directed to the corresponding author.
Abbreviations
- SNP:
-
Single nucleotide polymorphism
- EEG:
-
Electroencephalogram
- MRI:
-
Magnetic resonance imaging
- CNS:
-
Central nervous system
- N/A:
-
Information not available
- CT:
-
Computerized tomography
References
McTague A, Howell KB, Cross JH, Kurian MA, Scheffer IE. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. 2016;15(3):304–16.
Hussain SA. Epileptic Encephalopathies. Continuum (Minneap Minn). 2018;24(1, Child Neurology):171–85.
Guerreiro CA. Epilepsy: is there hope? Indian J Med Res. 2016;144(5):657–60.
Bartholdi D, Stray-Pedersen A, Azzarello-Burri S, Kibaek M, Kirchhoff M, Oneda B, et al. A newly recognized 13q12.3 microdeletion syndrome characterized by intellectual disability, microcephaly, and eczema/atopic dermatitis encompassing the HMGB1 and KATNAL1 genes. Am J Med Genet A. 2014;164A(5):1277–83.
Drummond-Borg M, Kulharya AS, Tonk V, Garcia-Heras J. Maternal complex chromosome rearrangement ascertained through a del (13)(q12.1q14.1) detected in her mildly affected daughter. Am J Med Genet. 2002;107(1):61–3.
Ballarati L, Rossi E, Bonati MT, Gimelli S, Maraschio P, Finelli P, et al. 13q deletion and central nervous system anomalies: further insights from karyotype-phenotype analyses of 14 patients. J Med Genet. 2007;44(1):e60.
Der Kaloustian VM, Russell L, Aradhya S, Richard G, Rosenblatt B, Melançon S. A de novo 2.1-Mb deletion of 13q12.11 in a child with developmental delay and minor dysmorphic features. Am J Med Genet A. 2011;155A(10):2538–42.
Cirillo E, Romano R, Romano A, Giardino G, Durandy A, Nitsch L, et al. De novo 13q12.3-q14.11 deletion involving BRCA2 gene in a patient with developmental delay, elevated IgM levels, transient ataxia, and cerebellar hypoplasia, mimicking an A-T like phenotype. Am J Med Genet A. 2012;158A(10):2571–6.
Du Puy L, Beqqali A, Monshouwer-Kloots J, Haagsman HP, Roelen BAJ, Passier R. CAZIP, a novel protein expressed in the developing heart and nervous system. Dev Dyn. 2009;238(11):2903–11.
Yang Z, Kaye DM. Mechanistic insights into the link between a polymorphism of the 3'UTR of the SLC7A1 gene and hypertension. Hum Mutat. 2009;30(3):328–33.
Wang Y. Jin L: miRNA-145 is associated with spontaneous hypertension by targeting SLC7A1. Exp Ther Med. 2018;15(1):548–52.
Hatakeyama E, Hayashi K. KATNAL1 is a more active and stable isoform of katanin, and is expressed dominantly in neurons. Biochem Biophys Res Commun. 2018;507(1–4):389–94.
Banks G, Lassi G, Hoerder-Suabedissen A, Tinarelli F, Simon MM, Wilcox A, et al. A missense mutation in Katnal1 underlies behavioural, neurological and ciliary anomalies. Mol Psychiatry. 2018;23(3):713–22.
Maroso M, Balosso S, Ravizza T, Liu J, Aronica E, Iyer AM, et al. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat Med. 2010;16(4):413–9.
Shi Y, Zhang L, Teng J, Miao W. HMGB1 mediates microglia activation via the TLR4/NF-κB pathway in coriaria lactone induced epilepsy. Mol Med Rep. 2018;17(4):5125–31.
Yang W, Li J, Shang Y, Zhao L, Wang M, Shi J, et al. HMGB1-TLR4 Axis plays a regulatory role in the pathogenesis of mesial temporal lobe epilepsy in immature rat model and children via the p38MAPK signaling pathway. Neurochem Res. 2017;42(4):1179–90.
Bermejo JL, Kabisch M, Dünnebier T, Schnaidt S, Melchior F, Fischer HP, et al. Exploring the association between genetic variation in the SUMO isopeptidase gene USPL1 and breast cancer through integration of data from the population-based GENICA study and external genetic databases. Int J Cancer. 2013;133(2):362–72.
Schulz S, Chachami G, Kozaczkiewicz L, Winter U, Stankovic-Valentin N, Haas P, et al. Ubiquitin-specific protease-like 1 (USPL1) is a SUMO isopeptidase with essential, non-catalytic functions. EMBO Rep. 2012;13(10):930–8.
Evans JF, Ferguson AD, Mosley RT, Hutchinson JH. What's all the FLAP about?: 5-lipoxygenase-activating protein inhibitors for inflammatory diseases. Trends Pharmacol Sci. 2008;29(2):72–8.
Al-Shemari H, Bossé Y, Hudson TJ, Cabaluna M, Duval M, Lemire M, et al. Influence of leukotriene gene polymorphisms on chronic rhinosinusitis. BMC Med Genet. 2008;9:21.
Toyo-Oka K, Sasaki S, Yano Y, Mori D, Kobayashi T, Toyoshima YY, et al. Recruitment of katanin p60 by phosphorylated NDEL1, an LIS1 interacting protein, is essential for mitotic cell division and neuronal migration. Hum Mol Genet. 2005;14(21):3113–28.
Acknowledgements
We would like to appreciate the support from the family for this publication.
Funding
This work was supported by Beijing Municipal Administration of Hospitals’ Youth Program, Grant No. QML 20150904. This funding body had no influence on case treatment, study design, data analysis, interpretation of data and writing the manuscript.
Author information
Authors and Affiliations
Contributions
Conceptualization, Y.F; resources, Y.F; writing—original draft preparation, M.W. and B.L.; illustrating, proofreading and editing, Z.L., Y.J. and Y.F; supervision, Y.F; project administration, Y.F; funding acquisition, Y.F. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the human research ethic committees of Beijing Hospital of Traditional Chinese Medicine. Written informed consent was obtained from the parents/guardians.
Consent for publication
Written informed consent was obtained from the parents/guardians for the publication of this study.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1 Supplementary Table 1
: The results of exome sequencing of the proband and her mother.
Additional file 2 Supplementary Table 2:
The results of SNP array of the proband.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, M., Li, B., Liao, Z. et al. A novel phenotype of 13q12.3 microdeletion characterized by epilepsy in an Asian child: a case report. BMC Med Genomics 13, 144 (2020). https://doi.org/10.1186/s12920-020-00801-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-020-00801-1