
Abstract
Background
A plethora of cases are reported in the literature with iso- and ring-chromosome 18. However, co-occurrence of these two abnormalities in an individual along with a third cell line and absence of numerical anomaly is extremely rare.
Case presentation
A 7-year-old female was referred for diagnosis due to gross facial dysmorphism and severe developmental delay. She presented with dysmorphic features, hypo/hyper pigmentation of the skin, intellectual disability and craniosynostosis. G-banding chromosome analysis suggested mos 46,XX,psu idic(18)(p11.2)[25]/46,XX,r(?18)[30]. Additional analysis by molecular karyotyping suggested pure partial deletion of 15 Mb on 18p (18p11.32p11.21). Lastly, multiple rearrangements and detection of a third cell line (ring chr18 and interstitial deletion) of chr18 was observed by multi-color banding.
Conclusion
The current study presents a novel case of chromosomal abnormalities pertaining to chromosome 18 across 3 cell lines, which were delineated with a combinatorial approach of diagnostic methods.
Similar content being viewed by others


Background
Structural anomalies involving chromosome 18 (chr18) are relatively frequently observed with an incidence at birth of ~ 1/40,000 [1,2,3,4]. These anomalies are grouped into 18q-, 18p-, ring 18 and tetrasomy 18p [5]. Ring chromosome 18 (r18) has been reported in several cases where a normal maternal or paternal chromosome is replaced by r18. Majority of the patients are female and present with a milder clinical phenotype compared to patients with chromosome 18 long-arm deletions [6]. In most cases, the inherent instability of ring chromosomes leads to loss of the ring, double ring, and/or multiple copies of the ring chromosome in a variable percentage of cells [7]. r18 and isochromosome anomaly individually are well documented in the literature. However, their occurrence together as mosaic cell lines have been reported only in a few cases, yet [8–10]. Isochromosome 18q (92% cells) along with r18 (8% cells) was reported by Souraty et al. [8] in a girl with congenital anomalies. Madan et al. [9] reported a baby girl with mosaicism consisting of cells of an iso-pseudo dicentric chromosome 18 (psu idic(18)(p11). Another case (a baby girl) with triple mosaicism involving chromosome 18 [46,XX,del (18)(p11.23)/46,XX,idic(18)(p11.23)/46,XX,r (18)] was reported by Bocian et al. [10]. Similarly, co-occurrence of r18 and an acentric fragment are also detected rarely [11]. It is rare to observe multiple structural rearrangements replacing chr18 in the absence of numerical alterations. We report here a novel case with gross dysmorphism and developmental delay in which we observed a mosaic karyotype consisting of cells with an psu idic(18)(p11.2) together with a r18 and a derivative chr18 with interstitial deletion replacing the respective chromosome 18 in a mosaic pattern.
Case presentation
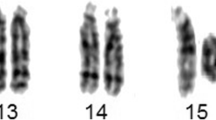
A female child was referred for diagnosis at the age of 7 years due to dysmorphism and severe developmental delay. She was the first child born to a non-consanguineous young couple [father age: 28 years, mother age 26 years at birth of the proband] with an uneventful natural pregnancy. She was born full term but with delayed crying i.e. breathing [APGAR: n.a.] and a birth weight of 2.5 kg. Her development timeline showed her ability to stand at 8 months, walk unaided at 2.5 years and speak only bi-syllable words at the time of referral. Her height was 103 cm, weight of 15 kg and head circumference of 49 cm. She presented with several dysmorphic features, most notably: mild asymmetry of face, squint eyes, large ears, depressed nasal bridge, flat occiput, as well as webbed neck, broad trunk, wide nipple, pectus excavatum, scoliosis and hypo/hyper pigmentation on skin. Intellectual disability was also observed as she was unable to understand the verbal commands as per her age. Craniosynostosis was detected on the MRI. Peripheral blood sample collection and written informed consent was obtained in accordance with the Helsinki declaration. Chromosome analysis of the patient and parents was performed with 72 h lymphocyte culture and standard GTG-banding, which revealed a mosaicism involving two cell lines. The karyotype was interpreted according to the International System for Human Cytogenomic Nomenclature (ISCN 2016) [12] as mos 46,XX,psu idic(18)(p11.2)[25]/46,XX,r(?18)[30]. The presence of pseudoisodicentric chr18 was confirmed by both C-banding and fluorescence in situ hybridization (FISH) using a centromere 18 specific probe (data not shown). Karyotype of the parents was normal, confirming a de novo origin of the rearranged chromosome 18 (Fig. 1 a and b).

Partial G-banded karyotype showing two cell lines as mos 46,XX,iso(18)(p11.2)[25]/46,XX,r(?18)[30]dn. a Pseudo isodicentric chr18 and (b) ring chromosome of various sizes
Further molecular cytogenetic studies were carried out to in order to clarify the structural rearrangements. Assessment of breakpoints on chromosome 18 was carried out using Affymetrix CytoScan™ 750 K SNP array on peripheral blood derived genomic DNA. Data was analyzed using Chromosome Analysis Suite (ChAS) that revealed multiple CNV calls on #18: arr [GRCh38] 18p11.32p11.21(136226_15181209)× 1,18q11.2q21.2(24160227_51024296)×2~3,18q22.2q23(69,349,902-80,255,845)×1 ~ 2,18q22.3q23(73,278,051_79,324,294)×1,18q23(80,132,201_80,255,845)×1. This identified a partial terminal 15 Mb deletion at chromosome 18 from 18p11.32p11.21, which was previously undetected by karyotyping (Fig. 2). Additionally, multiple CNVs showing mosaic gain and loss along with terminal deletion on q- arm were observed. Our hypothesis was that these may be due to various structural rearrangements observed on chr18 via karyotyping.

Chromosomal microarray showed 15 Mb deletion at 18p11.32p11.21 and a 6.1 Mb deletion at 18q22.3q23 i.e. arr[GRCh38] 18p11.32p11.21(136226_15181209)×1,18q22.3q23(73278051_80,255,845)×1. The figure shows, predicted deletion segments by CytoScan software, weighted log2 ratio, copy number state estimated by CytoScan software and B allele frequency
However, to further characterize these complex structural anomalies and assess the distribution of different cell lines, we carried out multi-color banding (MCB). We utilized MCB probes encompassing whole chromosome 18, as described previously [13], and twenty-eight metaphases were evaluated. MCB enabled detection of a third cell line in addition to the two cell lines identified by the G-banded karyotype (pseudo isodicentric chr18 and r18) and further resolved the chromosome architecture as mos 46,XX,der (18)(qter➔p11.21::p11.21➔qter)[13]/46,XX,r (18)(::p11.21➔q22.2::)[12]/ 46,XX,del(18)(q11.2q22)[3] (Figs. 3 and 4). The del(18)(q11.2q22) can also be described as der (18)(pter➔q11.2::q22➔qter). No further evidence of rearrangement was seen with this study.

Multicolour banding (MCB) FISH study showed clone 1 containing pseudo isodicentric chromosome of chromosome 18p11.21 [psu idic (18)(qter➔p11.21::p11.21➔qter)], clone 2 containing ring chromosome 18 [r (18)(::p11.21➔q22.2::)], and clone 3 containing intertsitial deletion of 18q [del (18)(q11.2q22)] i.e. mos 46,XX,der (18)(qter➔p11.21::p11.21➔qter) [13]/46,XX,r(18)(::p11.21➔q22.2::)[12]/46,XX,del(18)(q11.2q22)[3]. Due to break and fusion in derivative del(18)(q11.2q22) pseudocolours change and the underlaying alteration can only be followed up correctly in fluorochrome profiles (result not shown)

Diagrammatic representation of mosaic chromosomal abberations observed in the patient. Clone 1 consists of a pseudo isodicentric 18, clone 2 consists of a ring chromosome and clone 3 consits of deleted chromosome 18
Discussion and conclusions
The severity of the phenotypic manifestation depends on the nature and extent of the chromosomal abnormality and degree of mosaicism. In addition, the proportion of cell lines with different structural chromosomal rearrangements is also critical in the expression of a given phenotype (Table 1) [10]. In the present case, partial deletion of chr18p (p11.2➔pter; pure monosomy) was detected in two of the three cell-lines. One of the most frequent autosomal deletions is the monosomy of short arm of chr18. Growth failure, intellectual disability of varying degree, hypertelorism and nystagmus, short nose with anteverted nostrils, wide mouth, prominent upper lip, micrognathia, and large, protruding, low-set ears, depressed nasal bridge, short neck, wide mouth and widely spaced nipples are features usually seen in the 18p deletion, as seen in our case as well [14, 15]. Rarer or only occasionally observed malformations include premature craniosynostosis, as also seen in our case [16]. The mean age at diagnosis for craniosynostosis (oxycephaly in our case) is around 6 years. Elevated intracranial pressure is also a common clinical feature (affecting more than 60% of the patients). Many of the children have intellectual deficits and ophthalmic complications like papilledema, however, ophthalmic complications were not observed in our case [17].
The association between holoprosencephaly and deletion of chr18p was first described by Johnson and Bachman 1976 [18] and 18p11.3 is defined as one of the critical regions for holoprosencephaly (HPE) [19]. The case reported here showed deletion on 18p11.2 however HPE was not observed. Abnormal skin pigmentation as a phenotypic feature in chromosomal mosaicism involving chr18 has been previously reported confirming hypomelanosis of Ito [10], however, our current case portrayed only mosaic skin pigmentation as previously reported [20, 21]. Neurocutaneous syndromes have nonspecific neuroradiologic abnormalities with variable frequency, like hypoplasia of corpus callosum, periventricular white matter damages, cerebral atrophy, porencephalic cysts, neuronal heterotopias etc., which this child didn’t have [22].
Three different structural rearrangements were seen in the present case. The major two cell lines- pseudo isodicentric chr18 (qter➔p11.21::p11.21➔qter) and r18 (::p11.21➔q22.2::) were seen almost with same proportion. Pseudo isodicentric chr18 was confirmed by both karyotyping and MCB in our case. We also demonstrated that the isochromosome was dicentric with two genetically identical arms which is observed rarely as compared to monocentric isochromosome. Formation of dicentric isochromosomes demands breakage and subsequent U-type reunion of sister chromatids conjoined with a partial loss of p arm. Such an event can be hypothesized to occur as a part of breakage of a chromatid followed by failed or improper DNA repair. The current patient demonstrated an isochromosome along with two alike chromatids. Therefore, we propose that the event could have happened during the second meiotic division; with no recombination occurring during the first meiotic division. Such a mechanism has been described previously [23]. In the proband r18 was confirmed solely using MCB-FISH. Its formation might have occurred simultaneously as an independent event with second break on the q arm leading to the formation of ring chromosome. Thus, two independent events causing three strand breaks might have resulted in to isodicentric chromosome along with the ring as suggested by Madan et al. [9]. Coexistence of r18, partial 18p and 18q deletion further strengthens the proposed mechanistic hypothesis. In addition, absence of cell line with normal chr18 indicate that the rearrangements occurred before conception in the germ cells. Only two cases have been published so far with a multiple rearrangement to the best of our knowledge [9, 11].
The correlation of phenotype with genotype in ring chromosome carriers is difficult to conduct due to the size of deletion and the number of genes involved at each end of the chromosome. Therefore, it becomes imperative to determine primary deletions and secondary loss/gain associated with ring chromosome formation that may occur due to the instability of the ring as observed in the present case. The occurrence of r18 together with a duplication of a chr18 segment in acentric form is occasional [2, 9, 11]. Small percentage of the cells observed with interstitial deletion chr18 was comprehensively characterized solely by MCB technique though the fragment looked bigger than r18 under microscope (Fig. 3).
In conclusion, the patient reported herein presents a novel set of chromosomal abnormalities that have never been reported before to occur concurrently. Mosaicism of two or more cell lines with unbalanced structural aberrations is an extremely rare phenomenon. However, it involves chr18 in majority of the cases [24]. The combinatorial use of molecular cytogenetics modalities invariably serves a great value in the comprehensive characterization of the structurally altered chromosomes. In addition, it helps to elucidate genotype-phenotype correlation facilitating the search for candidate genes.
Availability of data and materials
Data generated and analysed from the Affymetrix CytoScan™ 750 K SNP chip during this study is available as Supplementary File 1 in this published article.
Abbreviations
- CDH7:
-
Cadherin 7
- CMA:
-
Chromosomal microarray
- CNV:
-
Copy Number Variation
- CTDP1:
-
Carboxy-Terminal domain phosphatase 1
- FISH:
-
Fluorescence in situ hybridization
- MCB:
-
Multicolor banding
- ISCN:
-
International System for Human Cytogenomic Nomenclature
- r(18):
-
Ring chromosome 18
- SNP:
-
Single nucleotide polymorphism
- HPE:
-
Holoprosencephaly
References
Cody JD, Ghidoni PD, DuPont BR, Hale DE, Hilsenbeck SG, Stratton RF, et al. Congenital anomalies and anthropometry of 42 individuals with deletions of chromosome 18q. Am J Med Genet. 1999;85:455–62.
Stankiewicz P, Brozek I, Helias-Rodzewicz Z, Wierzba J, Pilch J, Bocian E, et al. Clinical and molecular cytogenetic studies in seven patients with ring chromosome 18. Am J Med Genet. 2001;101:226–39.
Baumer A, Giovannucci Uzielli ML, Guarducci S, Lapi E, Rothlisberger B, Schinzel A. Meiotic origin of two ring chromosomes 18 in a girl with developmental delay. Am J Med Genet. 2002;113:101–4.
Miller K, Pabst B, Ritter H, Nurnberg P, Siebert R, Schmidtke J, et al. Chromosome 18 replaced by two ring chromosomes of chromosome 18 origin. Hum Genet. 2003;112:343–7.
Cody JD, Hasi-Zogaj M, Heard P, Hill A, Rupert D, Sebold C, et al. The chromosome 18 clinical re source center. Mol Genet Genomic Med. 2018;6:416–21. https://doi.org/10.1002/mgg3.385.
Schinzel A. Catalogue of unbalanced chromosome aberrations in man. 2nd ed. New York Berlin: de Gruyter; 2001.
Wyandt HE. Ring autosomes: identification, familial transmission, causes of phenotypic effects and in vitro mosaicism. In: Daniel A, editor. The cytogenetics of mammalian autosomal rearrangements. New York: Liss; 1988. p. 667–95.
Souraty N, Sanlaville D, Chedid R, Le Lorch M, Maurin ML, Ghanem L, et al. Cytogenetic investigation of a child with a mosaic isochromosome 18q and ring 18q. Eur J Med Genet. 2007;50:379–85.
Madan K, Vlasveld L, Barth PG. Ring-18 and isopseudodicentric-18in the same child: a hypothesis to account for common origin. Ann Genet. 1981;1:12–6.
Bocian E, Mazurczak T, Bulawa E, Stanczak H, Rowicka G. Triple structural mosaicism of chromosome 18 in a child with MR/MCA syndrome and abnormal skin pigmentation. J Med Genet. 1993;30:614–5.
Carreira IM, Mascarenhas A, Matoso E, Couceiro AB, Ramos L, Dufke A, et al. Three unusual but cytogenetically similar cases with up to five different cell lines involving structural and numerical abnormalities of chromosome 18. J Histochem Cytochem. 2007;55(11):1123–8.
McGowan-Jordan J, Simons A, Schmid M. An international system for human cytogenomic nomenclature. Basal: S. Kager; 2016.
Weise A, Mrasek K, Fickelscher I, Claussen U, Cheung SW, Cai WW, et al. Molecular definition of high-resolution multicolor banding probes: first within the human DNA sequence anchored FISH banding probe set. J Histochem Cytochem. 2008;56(5):487–93. https://doi.org/10.1369/jhc.2008.950550.
Wester U, Bondeson ML, Edeby C, Anneren G. Clinical and molecular characterization of individuals with 18p deletion: a genotype-phenotype correlation. Am J Med Genet. 2006;140A:1164e1171.
Portnoï MF, Gruchy N, Marlin S, Finkel L, Denoyelle F, Dubourg C, et al. Midline defects in deletion 18p syndrome: clinical and molecular characterization of three patients. Clin Dysmorphol. 2007;16(4):247–52.
Faust J, Habedank M, Nieuwenhuijsen C. The 18 p- syndrome. Report of four cases. Eur J Pediatr. 1976;123:59–66. https://doi.org/10.1007/BF00497681.
Khanna PC, Thapa MM, Iyer RS, Prasad SS. Pictorial essay: the many faces of Craniosynostosis. Ind J Radiol Imag. 2011;21(1):49–56. https://doi.org/10.4103/0971-3026.76055.
Johnson G, Bachman R. A 46,XY,del (18)(pter->p1.100:) cebocephalic child from a 46,XX,t (12;18)(18pter->18p1100::12qter->12pter) normal parent. Hum Genet. 1976;34:103–6.
Gripp KW, Wotton D, Edwards MC, Roessler E, Ades L, Meinecke P, et al. Mutations in TGIF cause holoprosencephaly and link NODAL signalling to human neural axis determination. Nat Genet. 2000;25:205–8.
Thomas IT, Frias JL, Cantu ES, Lafer CZ, Flannery DB, Graham JG. Association of pigmentary anomalies with chromosomal and genetic mosaicism and chimerism. Am J Hum Genet. 1989;45:193–205.
Cantu E, Thomas I, Frias J. Unusual cytogenetic mosaicism involving chromosome 14 abnormalities in a child with an MR/MCA syndrome and abnormal pigmentation. Clin Genet. 1989;36:189–95.
Bodemer C. Incontinentia pigmenti and hypomelanosis of Ito. Handb Clin Neurol. 2013;111:341e7.
Bugge M, Brandt C, Petersen M. DNA studies of mono- and pseudodicentric isochromosomes 18q. Am J Med Genet. 2004;127A:230e233.
de Pater JM, Smeets DF, Scheres JM. Unique mosaicism of structural chromosomal rearrangement: is chromosome 18 preferentially involved? Am J Med Genet. 2003;119A:26e31.
Acknowledgements
We are grateful to the family of the patient for their kind cooperation and permission.
Funding
No funding was received for the said work.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: FS and HS. Patient recruitment and clinical analysis: KP, DJ, FS and HS. Cytogenetic and molecular analysis: FS, HS and TL. Wrote the first draft of the manuscript: HS, FS and ST. Made critical revisions and approved final version: HS, TL, DJ, KP, ST, JS, FS. The authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Present case under submission has been approved by the institutional ethics committee [FRIGE’s Institute of Human Genetics] wide approval number FRIGE/IEC/15/2016 dated 16th March, 2016. This process is in accordance with the Helsinki declaration.
A written informed consent was obtained from the parents before enrolling for the investigations [This was in accordance with the requirement of the institutional ethics committee].
Consent for publication
Written informed consent was obtained from the parents for publication of identifying images and clinical details since the patient was under the age of 18 years. A copy of the written consent is available for review.
Competing interests
The authors declare that they have no conflict of interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sheth, H., Trivedi, S., Liehr, T. et al. Mosaic chromosome 18 anomaly delineated in a child with dysmorphism using a three-pronged cytogenetic techniques approach: a case report. BMC Med Genomics 13, 141 (2020). https://doi.org/10.1186/s12920-020-00796-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-020-00796-9