
Abstract
Background
Colon adenocarcinoma (COAD) is one of the most common gastrointestinal cancers globally. Molecular aberrations of tumor suppressors and/or oncogenes are the main contributors to tumorigenesis. However, the exact underlying mechanisms of COAD pathogenesis are clearly not known yet. In this regard, there is an urgent need to indicate promising potential diagnostic and prognostic biomarkers in COAD patients.
Methods
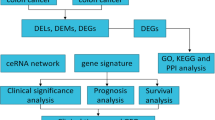
In the current study, level 3 RNA-Seq and miR-Seq data and corresponding clinical data of colon adenocarcinoma (COAD) were retrieved from the TCGA database. The “limma” package in R software was utilized to indicate the differentially expressed genes. For in silico functional analysis, GO and KEGG signaling pathways were conducted. PPI network was constructed based on the STRING online database by Cytoscape 3.7.2. A ceRNA network was also constructed by “GDCRNATools” package in R software. Kaplan-Meier survival analysis (log-rank test) and ROC curve analysis were used to indicate the diagnostic and prognostic values of the biomarkers.
Results
The differential expression data demonstrated that 2995 mRNAs, 205 lncRNAs, and 345 miRNAs were differentially expressed in COAD. The GO and KEGG pathway analysis indicated that the differentially expressed mRNAs were primarily enriched in canonical processes in cancer. The PPI network showed that the CDKN2A, CCND1, MYC, E2F, CDK4, BRCA2, CDC25B, and CDKN1A proteins were the critical hubs. In addition, the Kaplan-Meier analysis revealed that 215 mRNAs, 14 lncRNAs, and 39 miRNAs were associated with overall survival time in the patients. Also, the ceRNA network data demonstrated that three lncRNAs including MIR17HG, H19, SNHG1, KCNQ1OT1, MALAT1, GAS5, SNHG20, OR2A1-AS1, and MAGI2-AS3 genes were involved in the development of COAD.
Conclusions
Our data suggested several promising lncRNAs in the diagnosis and prognosis of patients with COAD.
Similar content being viewed by others


Background
Colon adenocarcinoma (COAD) is one of the most common gastrointestinal (GI) cancers and is the second leading cause of cancer-related death, globally [1, 2]. It is demonstrated that COAD occurs in approximately 5% of overall population at any given time in the world [3]. Despite the current screenings and therapies such as endoscopic resection and radical surgery, nearly half of the patients are diagnosed as advanced cases of COAD, experiencing tumor recurrence and relapse. COAD tumorigenesis has complicated multi-step processes including colon epithelial cell proliferation, aberration in differentiation, apoptosis resistance, survival, and invasion mechanisms [4]. Molecular aberrations of tumor suppressors and/or oncogenes are also one of the main contributors in different types of tumors especially COAD tumorigenesis [5]. However, due to complicacy of the underlying molecular pathways, the exact pathogenic contributors of COAD have not yet been clarified. Hence, there is an urgent need to indicate promising diagnostic and prognostic biomarkers for COAD. Recent investigations have highlighted the role of non-coding RNAs in the tumorigenesis of various malignancies. Among different kinds of non-coding RNAs, long non-coding RNA (lncRNA) is a putative class of non-coding RNA with more than 200 nucleotides in length, without any open-reading-frame (ORF) to encode proteins [5, 6]. Interestingly, a large body of evidence indicates that lncRNAs plays critical roles in a variety of biological processes including cell proliferation, cellular development, differentiation, carcinogenesis, and metastasis through modulating gene expression at the transcriptional and posttranscriptional levels directly or by recruiting chromatin remodeling factors [6,7,8]. Aberrant expression of lncRNAs has been well-documented in different sorts of cancers [9]. Dysregulation of lncRNA HOTAIR, H19, MALAT1, SNHG7, GAS8-AS, and NEAT1 were extensively well-studied and have been demonstrated to contribute in tumorigenesis and poor prognosis [5, 9,10,11,12,13]. Numerous investigations have shown that the lncRNAs can exert their function by competing endogenous RNA (ceRNA) crosstalk. For instance, it has been shown that lncRNA SCARNA2 was overexpressed in COAD tissues and it remarkably correlated with chemoresistance. Mechanistically, SCARNA2 via targeting miR-342-3p, upregulates EGFR and BCL2 expression in COAD cells [14]. Furthermore, overexpression of lncRNA SNHG1 has been shown to promote epithelial-mesenchymal transition (EMT) by binding to miR-497/miR-195-5p in COAD cells [15]. Also, lncRNA BDNF-AS was downregulated in COAD patients and served as a tumor suppressor gene. Unsurprisingly, ectopic expression of BDNF-AS suppressed cell proliferation and migration via epigenetically downregulating GSK-3β expression through EZH2 [16]. Moreover, several investigations have considered lncRNAs as therapeutic opportunities in COAD. For instance, it has been demonstrated that overexpression of LINC00152 can promote Fascin actin-bundling protein 1 (FSCN1) expression via sponging miR-632 and miR-185-3p, which consequently leads to proliferation and metastasis in COAD [17]. A recent study has demonstrated that targeting lncRNA FLANC by 1,2-dioleoyl-sn-glycero-3-phosphatidylcholine nanoparticles loaded with a specific small interfering RNA, decreased metastasis without any significant toxicity. They proposed that FLANC may act as a novel therapeutic strategy in COAD [18].
Additionally, many researches have suggested the potency of lncRNAs as biomarkers in the blood and serum. They suggested microvesicles and exosomes as carriers, being protected and stabilized in circulation [19]. In the current study, we comprehensively investigate lncRNAs, miRNAs, and mRNAs expressions from a public database, “Cancer Genome Atlas (TCGA)” and we constructed a ceRNA network in COAD. Also, we proposed novel potential biomarkers for COAD.
Methods
Sample and data collection
Clinical data of COAD were retrieved from the TCGA database (https://portal.gdc.cancer.gov/repository). The inclusion criteria were: (1) the histopathological diagnosis was COAD; (2) having complete demographic data including age, vital status, race, ethnicity, pathological stage, TNM classification, and overall survival time. Totally, 459 COAD were enrolled in this study. Two hundred and thirty participants had age > 68 years and 229 patients had age ≤ 68 and 243 and 216 patients were male and female, respectively. Among 459 patients, only 4 patients were Hispanic or Latino and 271 were non-Hispanic or non-Latino. Two hundred and fourteen patients were white, 29 patients were Black or African American, 11 were Asian and 1 American Indian/Alaska native. Pathological stages of I, II, III, and IV were 76, 178, 129 and 65, respectively. The clinical characteristics are summarized in Table 1.
RNA-Seq and miR-Seq data analysis
RNA-Seq and miR-Seq Level 3 data were collected from the TCGA database. The raw count of the reads of RNA-Seq and miR-Seq data was normalized by Voom and TMM normalization methods. All the analyses were conducted in R software. The “limma” package in R software was utilized to indicate the differentially expressed mRNAs (DEmRNAs), lncRNAs (DElncRNAs), and miRNAs (DEmiRNAs) between normal solid tissues and primary tumors. The concluded data were filtered based on the |log2 fold change (FC)| > 1 for DEmRNA, DElncRNA, and DEmiRNA. P-value < 0.05 and false discovery rate (FDR) < 0.05 were considered as significant thresholds.
Functional enrichment analysis and protein-protein interaction (PPI) network
For in silico functional enrichment analysis, gene ontology (GO) in three domains including biological processes, cellular components, and molecular functions, in addition to Kyoto Encyclopedia of Genes and Genomes (KEGG) signaling pathways were conducted. The GO and KEGG outputs were visualized by R software (ggplot2 package). The PPI network was constructed based on the STRING online database by Cytoscape 3.7.2. Molecular Complex Detection (MCODE) was used to analyze and predict the interactions (score value > 0.4).
LncRNA-miRNA-mRNA ceRNA network construction
LncRNA-miRNA-mRNA ceRNA network was constructed by “GDCRNATools” (http://bioconductor.org/packages/devel/ bioc/html/GDCRNATools.html) package in R software based on starbase database [14]. The nodes and edges were virtualized by Cytoscape 3.7.2.
Statistical analysis
All the differentially expressed data were analyzed by using R software (3.5.2) through the “GDCRNATools” package. Kaplan-Meier survival analysis (log-rank test) was used to indicate the relation between over or downregulation of RNA, based on median expression with patient’s survival time. ROC curve analysis, univariate, and multivariate Cox regression analysis were conducted by SPSS v21. P-value < 0.05 was considered as a significant threshold.
Results
Differentially expressed genes
Our data demonstrated that 2995 mRNAs (1094 up-regulated and 1901 down-regulated) were differentially expressed in COAD. Moreover, 205 lncRNAs (128 up-regulated and 77 down-regulated) were identified that were deferentially expressed in patients. Three hundred and forty-five miRNAs containing 183 up-regulated and 162 down-regulated have been found with differential expression in the COAD samples. The data are presented in Figs. 1, 2 and Tables 2, 3.

Bar graph of the differentially expressed genes in the COAD samples. TEC: To be Experimentally Confirmed; TR: T cell receptor; IG: Immunoglobulin

Volcano plot of the differentially expressed genes and miRNAs. a Volcano plot of differentially expressed lncRNAs and mRNAs. Overexpressed genes are demonstrated in red and down-regulated genes are demonstrated in green. b Volcano plot of differentially expressed miRNA. Overexpressed and down-regulated genes are demonstrated in red and green, respectively
GO enrichment and KEGG pathway analysis
GO enrichment analysis demonstrated that the differentially expressed mRNAs were enriched in different biological processes such as leukocyte migration, extracellular matrix organization, T cell activation, mitotic nuclear division, and adaptive immune response. Furthermore, GO analysis in cellular component revealed that the differentially expressed mRNAs predominantly contributed to collagen-containing extracellular matrix, basement membrane, microvillus, apical part of cell, and external side of plasma membrane. GO molecular function domain indicated that the genes were mainly enriched in glycosaminoglycan binding, heparin binding, sulfur compound binding, extracellular matrix structural constituent, and cytokine activity. GO outputs are presented in Fig. 3. In addition, KEGG pathway analysis indicated that the differentially expressed genes in the COAD patients remarkably participated in pathways involving in cancer, cell cycle, PPAR signaling pathway, PI3K-Akt signaling pathway, Wnt signaling pathway, and p53 signaling pathway (Fig. 4).

GO enrichment analysis of the differentially expressed mRNAs in COAD (Top 10 GO enrichment are presented)

KEGG signaling pathway analysis of the differentially expressed mRNAs in COAD (Top 20 KEGG terms are presented)
PPI network construction
The PPI network was constructed based on the STRING database to better understand the roles of the differentially expressed mRNAs. The data demonstrated that CDKN2A, CCND1, MYC, E2F, CDK4, BRCA2, CDC25B, and CDKN1A were the protein-protein interaction (PPI) critical hubs (Fig. 5).

Protein-protein interaction (PPI) network of the differentially mRNAs in COAD (score > 0.4) with Node:118, eadge:1745, MCADE score: 29.82
Kaplan-Meier survival analysis of differentially expressed genes
Kaplan-Meier survival analysis was used to indicate the association of differentially expressed mRNAs, lncRNAs, miRNA, and prognosis of COAD patients. The data showed that 215 mRNAs, 14 lncRNAs, and 39 miRNAs were associated with overall survival time in the patients. The top 10 hits of each group are presented in Table 4.
Diagnostic analysis of differentially expressed lncRNAs
AUC analysis was conducted to demonstrate the diagnostic value of each lncRNAs in the COAD samples. All 205 differentially expressed lncRNAs indicated significant diagnostic values. The top 50 hits of the lncRNAs are summarized in Table 5.
Novel lncRNA biomarkers
After merging the overall survival, and the diagnostic value data, we found that 14 lncRNAs had high ranks in prognostic and diagnostic areas which can be considered as COAD biomarkers. The data are presented in Table 6.
Kaplan-Meier and ROC curve analysis were conducted for the top three lncRNAs (AC087388.1, SLC16A1-AS1, and ELFN1-AS1) from aforementioned analysis shown in Fig. 6. Moreover, univariate and multivariate analysis were conducted to demonstrate the power of the lncRNAs and to diminish the covariate effects. Univariate and multivariate analysis are summarized in Table 7.

Kaplan-Meier and ROC curve analysis of the AC087388.1, SLC16A1-AS1, and ELFN1-AS1). a Kaplan-Meier curve of AC087388.1. b Kaplan-Meier curve of SLC16A1-AS. c Kaplan-Meier curve of ELFN1-AS1. d ROC curve of the lncRNAs
LncRNA-miRNA-mRNA ceRNA network construction
According to ceRNA hypothesis, which implicates that lncRNAs regulate mRNA expression level by competing the shared miRNAs in cells, a ceRNA network was built based on lncRNAs, mRNAs, and miRNAs expression in the samples based on starbase online tool in R software. The nodes and edges were drawn by Cytoscape 3.7.2. The ceRNA network data demonstrated important lncRNAs including MIR17HG, H19, SNHG1, KCNQ1OT1, MALAT1, GAS5, SNHG20, OR2A1-AS1, and MAGI2-AS3, which have implied in the development of COAD (Fig. 7).

LncRNA-miRNA-mRNA ceRNA network construction of the differentially expressed genes in COAD (Red: LncRNA, Yellow: miRNA, and Green: mRNA)
Discussion
LncRNAs regulate critical and canonical biological functions in different types of normal human cells and in a variety of tumor cells [20]. An escalating number of investigations have reported the function of lncRNAs in tumor proliferation, cell invasion and migration, chemotherapy resistance, and stemness capability in tumorigenesis and progression of COAD [21,22,23]. However, the exact underlying mechanisms of lncRNAs in progression of COAD are still unclear. So far, several different biological regulatory functions have been proposed for lncRNAs. Some previous studies have demonstrated that lncRNAs regulate mRNA expression via binding and sponging miRNA known as competing endogenous RNA theory, which generates a new aspect in the lncRNA regulatory mechanism [24, 25]. To the best of our knowledge, only a few investigations have displayed ceRNA networks between lncRNAs and miRNAs in COAD. Thus, a clear image of lncRNAs-miRNAs links still remains uncharacterized. In the current study, we studied the differentially expressed genes including lncRNAs, miRNAs, and mRNAs in the COAD patients based on TCGA database. Gene set enrichment by GO and KEGG signaling pathway identified the differentially expressed genes which were significantly enriched in cell proliferation, differentiation, protein phosphorylation, and signaling pathways. Furthermore, KEGG signaling pathway analysis demonstrated several canonical signaling pathways including Wnt, PI3K/Akt and PPAR signaling pathways that have been shown to contribute in tumor progression [26, 27]. A mounting of evidence has emphasized on Wnt/ β-catenin signaling pathway, promoting tumor growth, invasion and metastasis, and chemoresistance in COAD [28, 29]. For instance, it has been demonstrated that lncRNA H19 overexpression induces the EMT of colorectal cancer (CRC) cells by sponging miR-29b-3p to directly upregulate PGRN and activate Wnt axis [30]. Moreover, the up-regulation of lncRNA colorectal cancer-associated lncRNA (CCAL) promotes CRC progression through suppressing the activator protein 2α (AP-2α) to initiate Wnt/β-catenin signaling pathway [31]. In the present study, the KEGG analysis indicated that the peroxisome proliferator-activated receptor (PPAR) pathway contributes in Wnt signaling. It has been shown that the PPAR signaling pathway reduces cell proliferation and inhibits tumorigenesis in different types of cancers. Down-regulation of PPAR-α has been correlated with poor clinicopathological features of CRC that was remarkably higher in well to moderately differentiated adenocarcinoma than in mucinous adenocarcinoma [32]. In addition, lncRNA TINCR modulates PPAR signaling pathway through binding to miR-107 to up-regulate CD36 in CRC [33]. Recently, the PPAR aberration expression and its prime roles in gastrointestinal tract has been extensively reviewed [34].
It has been shown that PI3K/Akt signaling pathway had prominent roles in carcinogenesis of a variety of cancers particularly COAD. LncRNA AB073614 can take under control CRC growth and invasion by PI3K/Akt signaling pathway [35]. In addition, lncRNA SNHG7 elevated GALNT7 level and induced PI3K/Akt/mTOR pathway by sponging miR-34a in CRC cells [36]. Our ceRNA network data demonstrated important lncRNAs including MIR17HG, H19, SNHG1, KCNQ1OT1, MALAT1, GAS5, SNHG20, OR2A1-AS1, and MAGI2-AS3 which previously have been highlighted in the development of COAD. LncRNA MAGI2-AS3 have been discovered to play a crucial role as a tumor suppressor in breast cancer by targeting Fas/FasL in tumor cells [37]. Moreover, MAGI2-AS3 hampers hepatocellular carcinoma cell growth and its invasion through sponging miR-374b-5p to up-regulate SMG1 axis [38]. On the other hand, overexpression of MAGI2-AS3 has been explained to promote tumor progression by absorbing miR-141/200a and consequently, up-regulating ZEB1 which is an EMT promoting transcription factor, in gastric cancer cells [39]. MAGI2-AS3 up-regulation has also been shown to induce CRC proliferation and migration by modulating miR-3163 through upregulating TMEM106B [40].
LncRNA SNHG1 is a prominent lncRNA that is involved in a variety of cancers. SNHG1 expression is associated with unfavorable overall survival and tumor recurrence in patients with COAD. Moreover, SNHG1 promote cell growth and cell migration via upregulating EZH2 and miR-154a-5p in COAD [41]. LncRNA KCNQ1OT1 can promote EMT by decreasing miRNA-217 expression to upregulate ZEB1 axis in COAD [42]. Furthermore, KCNQ1OT1 has been demonstrated to promote chemoresistance of oxaliplatin by iR-34a/ATG4B pathway and it is associated with poor prognosis in COAD [43]. A previous study showed that lncRNA MALAT1 was remarkably upregulated in COAD cells. MALAT1 can promote metastasis of COAD via RUNX2 as a survival factor in tumor cells [44]. MALAT1 evokes EMT and angiogenesis via sponging miR-1265p to upregulate VEGFA, SLUG, and TWIST [45]. Several investigations demonstrated that lncRNA GAS5 can act as a tumor suppressor gene by different actions. It has been illustrated that GAS5 inhibited angiogenesis and metastasis via regulating Wnt signaling pathway in COAD cells [46]. Finally, lncRNA SNHG20 has been reported overexpressed prominently in CRC tissues in comparison to normal ones. Overexpression of SNHG20 was correlated with poor prognosis in the patients [47]. Although, there are several similar studies, the novelties of the current study include; an extensive exploration of lncRNA, mRNA and miRNA signatures, revealing the diagnostic and prognostic value of lncRNA, and constructing a COAD lncRNA-miRNA-mRNA ceRNA network.
Hence, our data elucidated that, the suggested lncRNAs can be considered as potential promising biomarkers, which could drive tumorigenesis through hijacking canonical signaling pathways in COAD.
Conclusions
Our data highlighted the importance of lncRNA regulatory networks that might provide a promising therapeutic approach for clinical application by considering lncRNA hubs as potential efficient biomarkers.
Availability of data and materials
The miRNA-Seq and RNA-Seq genotype data analyzed in the current study are available through the open access datasets retrieved from TCGA [Project ID: TCGA-COAD, Project Name: Colon Adenocarcinoma (dbGaP Study Accession: phs000178)].
Abbreviations
- COAD:
-
Colon adenocarcinoma
- GI:
-
Gastrointestinal
- LncRNA:
-
Long non-coding RNA
- ORF:
-
Open-reading-frame
- ceRNA:
-
Competing endogenous RNA
- EMT:
-
Epithelial-mesenchymal transition
- DEmRNAs:
-
Differentially expressed mRNAs
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- CAMs:
-
Cell adhesion molecules
- CRC:
-
Colorectal cancer
- CCAL:
-
Colorectal cancer-associated lncRNA
- AP-2α:
-
Activator protein 2α
- PPAR:
-
Peroxisome proliferator-activated receptor
References
Luo K, Geng J, Zhang Q, Xu Y, Zhou X, Huang Z, Shi K-Q, Pan C, Wu J. LncRNA CASC9 interacts with CPSF3 to regulate TGF-β signaling in colorectal cancer. J Exp Clin Cancer Res. 2019;38(1):249.
Kerachian MA, Poudineh A, Thiery JP. Cell free circulating tumor nucleic acids, a revolution in personalized Cancer medicine. Crit Rev Oncol Hematol. 2019;102827.
Soltani G, Poursheikhani A, Yassi M, Hayatbakhsh A, Kerachian M, Kerachian MA. Obesity, diabetes and the risk of colorectal adenoma and cancer. BMC Endocr Disord. 2019;19(1):113.
Yu HM, Wang C, Yuan Z, Chen GL, Ye T, Yang BW. LncRNA NEAT1 promotes the tumorigenesis of colorectal cancer by sponging miR-193a-3p. Cell Prolif. 2019;52(1):e12526.
Zhao Y, Chu Y, Sun J, Song R, Li Y, Xu F. LncRNA GAS8-AS inhibits colorectal cancer (CRC) cell proliferation by downregulating lncRNA AFAP1-AS1. Gene. 2019;710:140–4.
Yousefi H, Maheronnaghsh M, Molaei F, Mashouri L, Aref AR, Momeny M, Alahari SK. Long noncoding RNAs and exosomal lncRNAs: classification, and mechanisms in breast cancer metastasis and drug resistance. Oncogene. 2019:1–22.
Bian Z, Jin L, Zhang J, Yin Y, Quan C, Hu Y, Feng Y, Liu H, Fei B, Mao Y. LncRNA—UCA1 enhances cell proliferation and 5-fluorouracil resistance in colorectal cancer by inhibiting miR-204-5p. Sci Rep. 2016;6:23892.
Xu M, Chen X, Lin K, Zeng K, Liu X, Xu X, Pan B, Xu T, Sun L, He B. lncRNA SNHG6 regulates EZH2 expression by sponging miR-26a/b and miR-214 in colorectal cancer. J Hematol Oncol. 2019;12(1):3.
Zhang PF, Wu J, Wu Y, Huang W, Liu M, Dong ZR, Xu BY, Jin Y, Wang F, Zhang XM. The lncRNA SCARNA2 mediates colorectal cancer chemoresistance through a conserved microRNA-342-3p target sequence. J Cell Physiol. 2019;234(7):10157–65.
C-f L, Y-c L, Wang Y, Sun L-B. the effect of LncRNA H19/miR-194-5p axis on the epithelial-mesenchymal transition of colorectal adenocarcinoma. Cell Physiol Biochem. 2018;50(1):196–213.
Xue D, Lu H, Xu HY, Zhou CX, He XZ. Long noncoding RNA MALAT 1 enhances the docetaxel resistance of prostate cancer cells via miR-145-5p-mediated regulation of AKAP 12. J Cell Mol Med. 2018;22(6):3223–37.
Shan Y, Ma J, Pan Y, Hu J, Liu B, Jia L. LncRNA SNHG7 sponges miR-216b to promote proliferation and liver metastasis of colorectal cancer through upregulating GALNT1. Cell Death Dis. 2018;9(7):722.
Zhang M, Weng W, Zhang Q, Wu Y, Ni S, Tan C, Xu M, Sun H, Liu C, Wei P. The lncRNA NEAT1 activates Wnt/β-catenin signaling and promotes colorectal cancer progression via interacting with DDX5. J Hematol Oncol. 2018;11(1):113.
Zhu Y, Bian Y, Zhang Q, Hu J, Li L, Yang M, Qian H, Yu L, Liu B, Qian X. Construction and analysis of dysregulated lncRNA-associated ceRNA network in colorectal cancer. J Cell Biochem. 2019;120(6):9250–63.
Bai J, Xu J, Zhao J, Zhang R. lncRNA SNHG1 cooperated with miR-497/miR-195-5p to modify epithelial–mesenchymal transition underlying colorectal cancer exacerbation. J Cell Physiol. 2019.
Zhi H, Lian J. LncRNA BDNF-AS suppresses colorectal cancer cell proliferation and migration by epigenetically repressing GSK-3β expression. Cell Biochem Funct. 2019.
Ou C, Sun Z, He X, Li X, Fan S, Zheng X, Peng Q, Li G, Li X, Ma J. Targeting YAP1/LINC00152/FSCN1 signaling axis prevents the progression of colorectal cancer. Adv Sci. 2020;7(3):1901380.
Pichler M, Rodriguez-Aguayo C, Nam SY, Dragomir MP, Bayraktar R, Anfossi S, Knutsen E, Ivan C, Fuentes-Mattei E, Lee SK: Therapeutic potential of FLANC, a novel primate-specific long non-coding RNA in colorectal cancer. Gut 2020.
Mashouri L, Yousefi H, Aref AR, Mohammad Ahadi A, Molaei F, Alahari SK. Exosomes: composition, biogenesis, and mechanisms in cancer metastasis and drug resistance. Mol Cancer. 2019;18(1):75.
Chen X, Zeng K, Xu M, Hu X, Liu X, Xu T, He B, Pan Y, Sun H, Wang S. SP1-induced lncRNA-ZFAS1 contributes to colorectal cancer progression via the miR-150-5p/VEGFA axis. Cell Death Dis. 2018;9(10):982.
Xiao Z, Qu Z, Chen Z, Fang Z, Zhou K, Huang Z, Guo X, Zhang Y. LncRNA HOTAIR is a prognostic biomarker for the proliferation and chemoresistance of colorectal cancer via MiR-203a-3p-mediated Wnt/ss-catenin signaling pathway. Cell Physiol Biochem. 2018;46(3):1275–85.
Sun J, Hu J, Wang G, Yang Z, Zhao C, Zhang X, Wang J. LncRNA TUG1 promoted KIAA1199 expression via miR-600 to accelerate cell metastasis and epithelial-mesenchymal transition in colorectal cancer. J Exp Clin Cancer Res. 2018;37(1):106.
Ren J, Ding L, Zhang D, Shi G, Xu Q, Shen S, Wang Y, Wang T, Hou Y. Carcinoma-associated fibroblasts promote the stemness and chemoresistance of colorectal cancer by transferring exosomal lncRNA H19. Theranostics. 2018;8(14):3932.
Jia Z, Peng J, Yang Z, Chen J, Liu L, Luo D, He P. Long non-coding RNA TP73-AS1 promotes colorectal cancer proliferation by acting AS a ceRNA for miR-103 to regulate PTEN expression. Gene. 2019;685:222–9.
Ma S, Yang D, Liu Y, Wang Y, Lin T, Li Y, Yang S, Zhang W, Zhang R. LncRNA BANCR promotes tumorigenesis and enhances adriamycin resistance in colorectal cancer. Aging (Albany NY). 2018;10(8):2062.
Ouyang S, Zheng X, Zhou X, Chen Z, Yang X, Xie M. LncRNA BCAR4 promotes colon cancer progression via activating Wnt/β-catenin signaling. Oncotarget. 2017;8(54):92815.
Tong JL, Zhang CP, Nie F, Xu XT, Zhu MM, Xiao SD, Ran ZH. MicroRNA 506 regulates expression of PPAR alpha in hydroxycamptothecin-resistant human colon cancer cells. FEBS Lett. 2011;585(22):3560–8.
Zhang Z, Zhou C, Chang Y, Zhang Z, Hu Y, Zhang F, Lu Y, Zheng L, Zhang W, Li X. Long non-coding RNA CASC11 interacts with hnRNP-K and activates the WNT/β-catenin pathway to promote growth and metastasis in colorectal cancer. Cancer Lett. 2016;376(1):62–73.
Han P, Li J-W, Zhang B-M, Lv J-C, Li Y-M, Gu X-Y, Yu Z-W, Jia Y-H, Bai X-F, Li L. The lncRNA CRNDE promotes colorectal cancer cell proliferation and chemoresistance via miR-181a-5p-mediated regulation of Wnt/β-catenin signaling. Mol Cancer. 2017;16(1):9.
Ding D, Li C, Zhao T, Li D, Yang L, Zhang B. LncRNA H19/miR-29b-3p/PGRN axis promoted epithelial-mesenchymal transition of colorectal cancer cells by acting on Wnt signaling. Mol Cells. 2018;41(5):423.
Ma Y, Yang Y, Wang F, Moyer M-P, Wei Q, Zhang P, Yang Z, Liu W, Zhang H, Chen N. Long non-coding RNA CCAL regulates colorectal cancer progression by activating Wnt/β-catenin signalling pathway via suppression of activator protein 2α. Gut. 2016;65(9):1494–504.
Morinishi T, Tokuhara Y, Ohsaki H, Ibuki E, Kadota K, Hirakawa E. Activation and expression of peroxisome proliferator-activated receptor alpha are associated with tumorigenesis in colorectal carcinoma. PPAR Res. 2019;2019.
Zhang X, Yao J, Shi H, Gao B, Zhang L. LncRNA TINCR/microRNA-107/CD36 regulates cell proliferation and apoptosis in colorectal cancer via PPAR signaling pathway based on bioinformatics analysis. Biol Chem. 2019;400(5):663–75.
Fucci A, Colangelo T, Votino C, Pancione M, Sabatino L, Colantuoni V. The role of peroxisome proliferator-activated receptors in the esophageal, gastric, and colorectal cancer. PPAR Res. 2012;2012.
Wang Y, Kuang H, Xue J, Liao L, Yin F, Zhou X. LncRNA AB073614 regulates proliferation and metastasis of colorectal cancer cells via the PI3K/AKT signaling pathway. Biomed Pharmacother. 2017;93:1230–7.
Li Y, Zeng C, Hu J, Pan Y, Shan Y, Liu B, Jia L. Long non-coding RNA-SNHG7 acts as a target of miR-34a to increase GALNT7 level and regulate PI3K/Akt/mTOR pathway in colorectal cancer progression. J Hematol Oncol. 2018;11(1):89.
Yang Y, Yang H, Xu M, Zhang H, Sun M, Mu P, Dong T, Du S, Liu K. Long non-coding RNA (lncRNA) MAGI2-AS3 inhibits breast cancer cell growth by targeting the Fas/FasL signalling pathway. Hum Cell. 2018;31(3):232–41.
Yin Z, Ma T, Yan J, Shi N, Zhang C, Lu X, Hou B, Jian Z. LncRNA MAGI2-AS3 inhibits hepatocellular carcinoma cell proliferation and migration by targeting the miR-374b-5p/SMG1 signaling pathway. J Cell Physiol. 2019.
Li D, Wang J, Zhang M, Hu X, She J, Qiu X, Zhang X, Xu L, Liu Y, Qin S. LncRNA MAGI2-AS3, regulated by BRD4, is an independent prognostic marker and promotes gastric cancer progression via maintaining ZEB1 overexpression. Mol Ther Nucleic Acids. 2019.
Ren H, Li Z, Tang Z, Li J, Lang X. Long noncoding MAGI2-AS3 promotes colorectal cancer progression through regulating miR-3163/TMEM106B axis. J Cell Physiol. 2019.
Xu M, Chen X, Lin K, Zeng K, Liu X, Pan B, Xu X, Xu T, Hu X, Sun L. The long noncoding RNA SNHG1 regulates colorectal cancer cell growth through interactions with EZH2 and miR-154-5p. Mol Cancer. 2018;17(1):1–16.
Bian Y, Gao G, Zhang Q, Qian H, Yu L, Yao N, Qian J, Liu B, Qian X. KCNQ1OT1/miR-217/ZEB1 feedback loop facilitates cell migration and epithelial-mesenchymal transition in colorectal cancer. Cancer Biol Ther. 2019;20(6):886–96.
Xu M, Xu X, Pan B, Chen X, Lin K, Zeng K, Liu X, Xu T, Sun L, Qin J. LncRNA SATB2-AS1 inhibits tumor metastasis and affects the tumor immune cell microenvironment in colorectal cancer by regulating SATB2. Mol Cancer. 2019;18(1):1–16.
Ji Q, Cai G, Liu X, Zhang Y, Wang Y, Zhou L, Sui H, Li Q. MALAT1 regulates the transcriptional and translational levels of proto-oncogene RUNX2 in colorectal cancer metastasis. Cell Death Dis. 2019;10(6):1–17.
Sun Z, Ou C, Liu J, Chen C, Zhou Q, Yang S, Li G, Wang G, Song J, Li Z. YAP1-induced MALAT1 promotes epithelial–mesenchymal transition and angiogenesis by sponging miR-126-5p in colorectal cancer. Oncogene. 2019;38(14):2627–44.
Song J, Shu H, Zhang L, Xiong J. Long noncoding RNA GAS5 inhibits angiogenesis and metastasis of colorectal cancer through the Wnt/β-catenin signaling pathway. J Cell Biochem. 2019;120(5):6937–51.
Li C, Zhou L, He J, Fang X-Q, Zhu S-W, Xiong M-M. Increased long noncoding RNA SNHG20 predicts poor prognosis in colorectal cancer. BMC Cancer. 2016;16(1):655.
Acknowledgments
This study was part of a Ph.D. dissertation (AP). We would like to thank Hassan Yousefi for the English editing of the manuscript.
Funding
This study was funded by the Mashhad University of Medical Sciences, Mashhad, Iran with the grant number: 981287. The funding body did not have any role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
AP, MRA, NN and MAK were all participated in study design, data analysis, and preparation of the drafted manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Poursheikhani, A., Abbaszadegan, M.R., Nokhandani, N. et al. Integration analysis of long non-coding RNA (lncRNA) role in tumorigenesis of colon adenocarcinoma. BMC Med Genomics 13, 108 (2020). https://doi.org/10.1186/s12920-020-00757-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-020-00757-2