
Abstract
Background
With the increasing amount of high-throughput genomic sequencing data, there is a growing demand for a robust and flexible tool to perform interaction analysis. The identification of SNP-SNP, SNP-CpG, and higher order interactions helps explain the genetic etiology of human diseases, yet genome-wide analysis for interactions has been very challenging, due to the computational burden and a lack of statistical power in most datasets.
Results
The wtest R package performs association testing for main effects, pairwise and high order interactions in genome-wide association study data, and cis-regulation of SNP and CpG sites in genome-wide and epigenome-wide data. The software includes a number of post-test diagnostic and analysis functions and offers an integrated toolset for genetic epistasis testing.
Conclusions
The wtest is an efficient and powerful statistical tool for integrated genetic epistasis testing. The package is available in CRAN: https://CRAN.R-project.org/package=wtest.
Similar content being viewed by others


Background
The etiology of complex disorder involves an interplay of polygenic biomarkers, lifestyle and environmental factors [1]. Robust and efficient statistical tools are needed to perform interaction analysis in high volume genome data. Besides SNP-SNP interactions, the analysis of interactions of SNPs and cytosine-phosphate-guanine (CpG) sites might provide novel insight into the regulatory mechanism DNA methylation and gene expression underlying complex diseases.
Here we introduce a software that provides estimations for different types of genetic associations, including the main effect, second or higher order interaction, and gene-methylation interaction. This package is built upon the W-test [2] to perform epistasis testing. The statistic compares distributional differences of a set of biomarkers in cases and controls and follows a chi-squared distribution with data-set adaptive degrees of freedom. The method has the advantage of correcting p-value bias caused by complicated genetic architectures. Flexible implementation options are provided. The package can calculate SNP-CpG epistasis for biomarkers located in physical proximity of the input genome and epigenome. A number of post-test diagnostic, visualization and statistical genetic analysis functions are provided for model diagnosis. This is the first statistical software providing functions for direct gene-methylation interaction and high-order interaction evaluations in genome and epigenome dataset.
Implementation
Design
The wtest package is based on the W-test [2] to measure the association between binary phenotype and categorical genetic data. To test the association of a subset marker, a k by 2 contingency table can be formed, where k is the number of non-empty category combination formed by the SNP-set, and 2 is the binary phenotype. The statistic tests for the existence of distributional difference of a subset in the case group from a comparison control group, and it takes the following form,
where n1i and n0i are the number of cases and controls in the ith cell of the contingency table; N1 and N0 are the total cell counts of cases and controls; \(\hat {p}_{1i} = n_{1i}/N_{1}\) and \(\hat {p}_{0i} = n_{0i}/N_{0}\) are the conditional cell probabilities of the ith cell of the contingency table; and SEi is the standard error of the ith log odds ratio. The W-test follows a chi-squared distribution of f degrees of freedom. The scalar h and degree of freedom f take forms of covariance matrices of the log odds ratios and are estimated from bootstrapped samples under the null hypothesis by the large sample theory. The W-test inherits a data-set adaptive degree of freedom that absorbs the genetic variation not attribute to phenotypes, therefore robust to complicated genetic architectures. In this software, we further extend it to evaluate high-order interaction effect and gene-methylation interaction effect. For gene-methylation interaction, methylation data are clustered into two categories according to high and low methylation levels by two-mean clustering algorithm. We also use a novel triangular network diagram to display interaction effects up to the third order. Extensive simulation studies testing the power and type I error of the W-test can be found in Wang, Sun et al. (2016) [2] and Sun et al. (2017) [3].
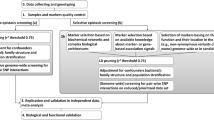
Implementation
Figure 1 demonstrates the major functions in the package and illustrates the implementation step by step using example data in the package. The implementation is performed in two steps: (1) Estimation of parameters h and f; (2) Testing by the W-test. Step 1. Estimation of parameters h and f. In genotype data, the hf() function is called, and in genotype and methylation data, the function hf.snps.meth() is called. Parameter h is the scaler in Eq. (1) and f is the degrees of freedom of a chi-squared distribution of the W-test. The two parameters are estimated using bootstrap samples with permutated phenotypes (null hypothesis) for B times. Simulations suggest that the estimation converges at B>400 when the number of variables is 1000 and the number of subjects is 1000 (Additional file 1). If step 1 is not performed, the p-value of W-test will be calculated by default h and f: h=k/(k−1) and f=k−1. In this case, k is the integer categorical combinations formed by the marker set. When k=2, the W-test is equivalent to the odds ratio test for a 2-by-2 table.

Integrated genetic epistasis testing and functions
Step 2. Testing by the W-test. The wtest() evaluates main and second order interaction and wtest.high() evaluates third or higher order interaction in genotype data. The wtest.snps.meth() calculates SNP-CpG interactions for genome and epigenome data. Oftentimes users are interested to explore the interactions among biomarkers with a certain level of main effect signals. The input.pval option in the function can be used to screen candidate SNPs according to their p-values to form interaction sets. While the output.pval option allows the convenient output of interaction sets reaching a p-value threshold. In function wtest.snps.meth(), positions of the biomarkers are input alongside the genome and epigenome data sets, and the window size to calculate cis-regulation relationship can be specified. The methylation.recode() function transforms the methylation data into high and low methylated levels. For high order interaction calculation, a simple check for sample size can be done by estimating the average number of cell counts formed by a set, and a high order is feasible if the number is at least two. A reference table could be found in Additional file 2 with suggested sample sizes for various order of interactions.
Diagnostic checking for test statistic distribution can be performed by w.diagnosis(), which plots the W-test statistics histograms from the observed data and the curve of the chi-squared distribution using estimated parameters, indexed by the number of categorical combinations k. Close overlaying of the densities indicates the goodness of fit of estimation. An example is shown in the real data application section. The w.qqplot() function assists the diagnostic of probability distribution and degree of population stratification.
Results
Real data example
The software is applied to a number of real data analysis with novel biomarker findings and interesting implications [2–9]. Here we demonstrate its usage by two data sets: a genotypic dataset for bipolar disorder from the Genetic Association Information Network (GAIN) project, and a gene-methylation data for the lipid control treatment.
Application I. GAIN bipolar disorder dataset
This data contains 653 bipolar disorder patients and 1767 healthy controls, and 46,181 SNPs of chromosome 6 [10]. The result of h and f estimation can be found in Additional file 3. At second order interaction (order=2), setting input.pval=0.001 and output.pval=0.001, the function would output second order epistasis marker pairs with p-value <0.001. Figure 2 is the diagnostic plot for this estimation using w.diagnosis() function. The estimated red color chi-square curve follows closely with the histogram of the test statistics calculated from the observed data, showing a good estimation of the parameters.

Diagnostic plot by w.diagnostics. At each combination size k, the estimated red color chi-square curve follows closely with the histogram of the W-test statistics calculated from the observed data, showing a good estimation of the parameters
Data analysis identified one SNP with significant main effect: rs2495982 near GRM4, p-value =2.06×10−7. GRM4 is a major excitatory neurotransmitter in central nervous system and it is a susceptible gene for bipolar disorder and schizophrenia [11, 12]. For interaction effects, a number of SNP sets surpassed the Bonferroni corrected significance level. The top SNPs identified from different orders of interaction are listed in Additional file 4, and the interaction network up to the third order is plotted in a triangular network in Fig. 3. Each colored triangle in the network indicates a significant third order interaction, and the bold edge shows a significant second order interaction. It could be seen from the plot that the strongest interaction is formed by the gene set (SYNE1, BTBD9, RPL12P2) in the middle of the plot, in which BTBD9 plays a key role and extends to form significant combinations with FGD2 and CDKAL1. The BTBD9 is reported to be associated with neuropsychiatric disorders such as restless legs syndrome in Schizophrenia and the Tourette Syndrome [13, 14]. The gene encodes the BTB/POZ domain-containing protein that involved in protein-protein interactions [15], and is highly expressed in brain tissues [16]. It is very encouraging to discover this gene with known physical protein interaction function from pure computational and statistical perspective.

Triangular network for third order genetic interactions
Application II. gene-methylation interaction analysis for lipid control data
This application was originally reported in Sun et al 2018 [3]. The data set contains 476 diabetic patients undergone lipid control treatments, and 150,000 candidate SNP-CpG pairs within 10kb genome distance (window.size=10,000). The phenotype is whether or not a subject responded to the treatment, calculated by comparing the before and after treatment triglyceride levels [3]. The h and f are estimated by hf.snps.meth(), and the gene-methylation interactions are calculated by wtest.snps.meth(). Table 1 summarized the top 5 markers identified by gene-methylation interaction associations. The cluster of genes is found to be involved in neuronal and retinal functions, including MPPED2 [17] and GUCY2E [18].
Performance
The speed of the wtest package is evaluated on a laptop computer of 1.6GHz Intel Core i5 processor and 4GB RAM. Simulation data are used to compare the speed of different methods. On a data set consists of 5000 subjects and 100 SNPs, when B=200, n.sample=1000, the time elapsed for estimating h and f is 40.5s. After h and f calculation or assuming default values, the time used to evaluate main effects is 0.04s, and took 1.69s for second order interaction. In the same environment, the running time for existing tests for interaction yields 36.41s by chi-squared test and 130.56s by logistic regression. In the real data set, the genome-wide main effect calculation on 5000 subjects and 500,000 SNPs took around 5 min; and second order interaction calculation on 8000 SNPs used around 3.5 h.
Conclusions
Genetic epistasis testing is important to fathom the massive genomic data, and it also provides a way to explore the relationship between diseases and various types of biomarkers. This package offers an integrated toolset to analyse the association of genetic signals at all levels: from main effects, high order interactions, to gene-methylation interactions. The software is available in CRAN from https://CRAN.R-project.org/package=wtest under the GPL-2.0 license.
Availability and requirements
Project name: wtest
Project home page: https://CRAN.R-project.org/package=wtest
Operation systems: Platform independent
Programming language: R (>= 3.1), C++
License: GPL (>= 2)
Restrictions to use by non-academics: None
Availability of data and materials
The data mentioned in figure 1 are provided in the wtest package at https://CRAN.R-project.org/package=wtest. Raw sequence data for application 1 and 2 are available via the referenced manuscripts.
Abbreviations
- CpG:
-
cytosine-phosphate-guanine
- GAIN:
-
association information network
- SNP:
-
Single-nucleotide polymorphism
References
Hunter DJ. Gene-environment interactions in human diseases. Nat Rev Genet. 2005; 6(4):287–98.
Wang MH, Sun R, Guo J, Weng H, Lee J, Hu I, et al.A fast and powerful W-test for pairwise epistasis testing. Nucleic Acids Res. 2016; 44(12):e115.
Sun R, Weng H, Men R, Xia X, Chong KC, Wu WKK, et al.Gene-methylation epistatic analyses via the W-test identifies enriched signals of neuronal genes in patients undergoing lipid-control treatment. BMC Proc. 2018; 12(Suppl 9):53.
Wang YM, Ma L, Lu SY, Chan TCY, Yam JCS, Tang SM, et al.Analysis of multiple genetic loci reveals MPDZ-NF1B rs1324183 as a putative genetic marker for keratoconus. Br J Ophthalmol. 2018; 102(12):1736–41.
Wu WKK, Sun R, Zuo T, Tian Y, Zeng Z, Ho J, et al.A novel susceptibility locus in MST1 and gene-gene interaction network for Crohn’s disease in the Chinese population. J Cell Mol Med. 2018; 22(4):2368–77.
Wang MH, Chang B, Sun R, Hu IC, Xia XX, Wu WKK, et al.Stratified polygenic risk prediction model with application to CAGI bipolar disorder sequencing data. Hum Mutat. 2017; 38(9):1235–9.
Sun R, Weng HY, Hu IC, Guo JF, Wu WKK, Zee BCY, et al.A W-test collapsing method for rare-variant association testing in exome sequencing data. Genet Epidemiol. 2016; 40(7):591–6.
Wang MH, Weng H, Sun R, Lee J, Wu WKK, Chong KC, et al.A Zoom-Focus algorithm (ZFA) to locate the optimal testing region for rare variant association tests. Bioinformatics. 2017; 33(15):2330–6.
Uppu S, Krishna A. A deep hybrid model to detect multi-locus interacting SNPs in the presence of noise. Int J Med Inform. 2018; 119:134–51.
McInnis MG, Dick DM, Willour VL, Avramopoulos D, MacKinnon DF, Simpson SG, et al.Genome-wide scan and conditional analysis in bipolar disorder: Evidence for genomic interaction in the National Institute of Mental Health Genetics Initiative bipolar pedigrees. Biol Psychiat. 2003; 54(11):1265–73.
Fallin MD, Lasseter VK, Avramopoulos D, Nicodemus KK, Wolyniec PS, McGrath JA, et al.Bipolar I disorder and schizophrenia: A 440-single-nucleotide polymorphism screen of 64 candidate genes among Ashkenazi Jewish case-parent trios. Am J Hum Genet. 2005; 77(6):918–36.
Kato T. Molecular genetics of bipolar disorder and depression. Psychiatr Clin Neurosci. 2007; 61(1):3–19.
Janik P, Berdynski M, Safranow K, Zekanowski C. The BTBD9 gene polymorphisms in Polish patients with Gilles de la Tourette syndrome. Acta Neurobiol Exp (Wars). 2014; 74(2):218–26.
Guo Y, Su L, Zhang J, Lei J, Deng X, Xu H, et al.Analysis of the BTBD9 and HTR2C variants in Chinese Han patients with Tourette syndrome. Psychiatr Genet. 2012; 22(6):300–3.
Gene N. BTBD9 BTB domain containing 9 [Homo sapiens (human)]. 2018. https://www.ncbi.nlm.nih.gov/gene/114781#gene-expression. Accessed 1 Nov 2018.
Fagerberg L, Hallstrom BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, et al.Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics. 2014; 13(2):397–406.
Gormley P, Anttila V, Winsvold BS, Palta P, Esko T, Pers TH, et al.Meta-analysis of 375,000 individuals identifies 38 susceptibility loci for migraine. Nat Genet. 2016; 48(10):1296.
Perrault I, Rozet JM, Calvas P, Gerber S, Camuzat A, Dollfus H, et al.Retinal-specific guanylate cyclase gene mutations in Leber’s congenital amaurosis. Nat Genet. 1996; 14(4):461–4.
Acknowledgements
This research was partly conducted using the resources of the High-Performance Cluster Computing Centre, Hong Kong Baptist University, which receives funding from Research Grant Council, University Grant Committee of the HKSAR and Hong Kong Baptist University.
About this supplement
This article has been published as part of BMC Medical Genomics Volume 12 Supplement 9, 2019: Proceedings of the Joint International GIW & ABACBS-2019 Conference: medical genomics. The full contents of the supplement are available online at https://bmcmedgenomics.biomedcentral.com/articles/supplements/volume-12-supplement-9.
Funding
Publication costs are funded by NSFC [31871340]. The study sponsor has no role in the study design and in the collection, analysis, and interpretation of data.
Author information
Authors and Affiliations
Contributions
RS created the package and draft the manuscript. MHW supervised the project, tested the package and revised the manuscript. All authors conceived the project, read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
Maggie H. Wang is a shareholder of Beth Bioinformatics Co., Ltd; Benny CY Zee is a shareholder of Beth Bioinformatics Co., Ltd and Health View Bioanalytics Ltd.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1
Convergency simulation study. The coefficient of variance of h at different B for pairwise interactions. Simulated dataset contains 1000 subjects and 1000 SNPs. A convergent h and f estimation is reached at B>400.
Additional file 2
Reference table of sample size estimation. When the averaged MAF is 0.3 and the sample size is greater than the estimated sample size, no more than 25% cells have averaged cell count less than 2 in the contingency tables.
Additional file 3
h and f estimation for main effects, second order interaction, and third order interaction analysis.
Additional file 4
Top three identified sNPs at different levels of interaction orders. Note: Bonferroni corrected significant thresholds: main effect p-value <1.1×10−6, second order interaction p-value <4.69×10−11, and third order interaction p-value <3.05×10−15. Gene: the gene located within 35kb of the identified SNPs.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Sun, R., Xia, X., Chong, K.C. et al. wtest: an integrated R package for genetic epistasis testing. BMC Med Genomics 12 (Suppl 9), 180 (2019). https://doi.org/10.1186/s12920-019-0638-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-019-0638-9