
Abstract
Background
Inflammatory response plays a key role in the development of insulin resistance (IR) in obesity. Oxidative stress triggers the replication of the mitochondrial genome and division of the organelle. The purpose of this study was to identify the relationship of chemerin and TNF-α with mitochondrial DNA (mtDNA) copy number in subcutaneous adipose tissue (SAT) and visceral adipose tissue (mesentery of the small intestine (Mes), greater omentum (GO) and blood mononuclear cells (MNCs)) in patients with obesity with/without type 2 diabetes mellitus (T2DM).
Methods
The study included 142 obese patients and 34 healthy donors. The samples used for the study were peripheral venous blood (MNCs) and ATs (SAT, Mes and GO). The measurement of mtDNA copy number was done by droplet digital PCR. Quantitative determination of insulin, adiponectin, TNF-α and chemerin in serum/plasma was performed by flow-through fluorometry and commercial ELISA kit. Statistical analysis and graphs were obtained in R Statistical Software (version 3.3.1).
Results
The increase in body mass index (BMI) was accompanied by an increase in TNF-α production, an increase in mtDNA copy number in SAT and a decrease in mtDNA copy number in MNCs. The negative association between plasma chemerin and mtDNA copy number in the Mes, as well as between mtDNA copy number and chemerin expression in the Mes, in the group with BMI > 40 kg/m2 without T2DM demonstrates the protective effect of chemerin against the development of IR via the regulation of mtDNA copy number in adipose tissues.
Conclusions
We thus speculated the existence of a compensatory mechanism in which leads to the increased number of mtDNA copies under the influence of proinflammatory factors. Based on the obtained data, we propose that reducing mtDNA copy number in cases of morbid obesity without T2DM has a positive effect on carbohydrate metabolism, which may help maintain glucose levels within reference values.
Obesity, type 2 diabetes, mtDNA, cytokines, TNF-a, chemerin.
Similar content being viewed by others


Background
Inflammatory response plays a key role in the development of insulin resistance (IR) in obesity [1, 2]. The initial pathogenetic factors in the development of IR in obesity are not fully understood. Mitochondria are involved in the process of lipolysis in adipocytes and are the main source of ATP in cells [3]. The malfunctioning of mitochondria leads to an energy crisis in insulin-dependent tissues and forms the foundation for the formation of IR [4, 5]. The high content of free fatty acids induces mitochondrial DNA (mtDNA) damage and causes the production of endoplasmic reticulum stress markers, protein degradation and apoptosis, contributing to increased oxidative stress in skeletal muscle cells and adipose tissues (ATs) [6]. Oxidative stress triggers the replication of the mitochondrial genome and division of the organelle [7, 8].
Some authors have studied the role of chemerin in regulating the function of mitochondria [8]. Сhemerin participates in processes of lipogenesis, metabolism, angiogenesis, inflammation, and proliferation and differentiation of adipocytes by regulating the expression of respective genes [9]. The mechanism underlying the effect chemerin has on mitochondrial function is mostly unknown. Xie Q. et al. (2015) have shown that chemerin-induced mitochondrial dysfunction contributes to a reduction in mitochondrial biogenesis and an increase in mitochondrial autophagy. The dual role of chemerin in inflammatory processes and metabolism may provide a link between chronic inflammation and energy metabolism in obesity, as well as the complications associated with this condition.
The purpose of this study was to define the relationship of chemerin and TNF-α with mtDNA copy number in subcutaneous adipose tissue (SAT) and visceral adipose tissue (VAT; mesentery of the small intestine (Mes), greater omentum (GO) and blood mononuclear cells (MNCs)) in obese patients with/without type 2 diabetes mellitus (T2DM).
Materials and methods
Study subjects
The study included 142 obese patients treated at the Regional Clinical Hospital of the Kaliningrad Region (Table 1). The control group consisted of 34 healthy donors with normal anthropometric and biochemical parameters of carbohydrate and lipid metabolism.
Patients with abdominal obesity were ranked by the degree of obesity into the following groups: 2 - BMI = 24.9–29.9 kg/m2; 3 and 4 - BMI = 30.0–34.9 kg/m2; 5 and 6 - BMI = 35–39.9 kg/m2; and 7 and 8 - BMI > 40 kg/m2; patients were ranked by the state of carbohydrate metabolism as follows: patients with T2DM (groups 4, 6, and 8) and patients without T2DM (groups 2, 3, 5, and 7).
-
Patients with pre-obesity were included in the second group.
-
The third and fourth groups comprised patients with grade I obesity stratified by the state of carbohydrate metabolism.
-
The fifth and sixth groups comprised patients with grade II obesity stratified by the state of carbohydrate metabolism.
-
Patients with BMI > 40 kg/m2 were in the seventh and eighth groups with grade III obesity and stratified by the state of carbohydrate metabolism.
All groups were comparable age- and sex-wise.
The samples used for the study were peripheral venous blood (MNCs) and ATs (SAT, Mes and GO).
The presence of obesity and T2DM was established on the basis of a detailed clinical and instrumental examination in a specialized hospital, guided by the World Health Organization (1999–2013) criteria for diagnosing diabetes and other types of hyperglycemia [10]. Informed consent was signed by all patients. Verification of the diagnosis and recruitment of patients into the study groups was carried out at the Department of Reconstructive and Plastic Surgery on the basis of regional clinical hospital in Kaliningrad.
Permission to conduct the study was obtained from the local ethics committee (Minute No. 4 of the meeting of the Local Ethics Committee at the Innovation Park of the Immanuel Kant Baltic Federal University, dated October 23, 2013).
Droplet digital PCR
DNA extraction from MNCs and ATs (SAT, Mes and GO) was carried out using commercial QIAamp DNA Mini Kit according to the manufacturer’s protocol (Qiagen, USA).
The DNA concentration in the samples was measured with an Implen NanoPhotometer spectrophotometer at a wavelength of 260 nm. The quality of extracted DNA was determined from the absorption ratio at 260 and 280 nm (A260/280 > 1.8).
mtDNA copy number was determined by ddPCR. For this, the extracted DNA samples were pretreated with ApaI restriction endonuclease (New England BioLabs, Ipswich, MA, USA) (1 μl (10 U) ApaI, 5 μl 10x CutSmart® buffer, and ~ 30 ng DNA sample), and then the reaction mixture was incubated for 3–4 h at 37 °C. The components of the ddPCR itself included a commercial PCR mixture, selected fluorescent TaqMan probes, and oligonucleotide primers (Table 2). Oligonucleotide primers and TaqMan probes labeled with fluorescent HEX and FAM were selected to amplify the corresponding portions of the mitochondrial gene ND1 (encoding subunit 1 of the NADH dehydrogenase complex) and the single-copy nuclear gene RPP30 (encoding the P/MRP subunit P30 subunit); the sequences are indicated in Table 2. The composition of the multiplex mixture of ddPCR included 10 μl of commercial 2x ddPCR Master Mix (Bio-Rad, Pleasanton, USA), 1 μl of each primer (final concentration 450 nM), FAM- and HEX-labeled TaqMan probes (the final concentration of each probe in the mixture was 125 nM) and 8 μl of DNA sample. The prepared reaction mixture was loaded into an automatic droplet generator (Bio-Rad, Pleasanton, USA), where it was emulsified into approximately 15–20 thousand drops, each approximately one nanoliter in volume.
In the next step, the emulsified ddPCR mixture was transferred to standard 96-well plates and amplified in a thermocycler (Bio-Rad T100 thermal cycler) according to the following temperature protocol: 95 °C for 10 min, 40 cycles at 94 °C for 30 s and 53 °C for 60 s and a final cycle at 98 °C for 10 min. After the amplification reaction, strength of fluorescence was measured using the QX200 Droplet Reader (Bio-Rad, Pleasanton, CA) which operates similarly to a flow cytometer. The data were analyzed in the QuantaSoft program suite (version 1.7.4.0917). The absolute number of copies of mtDNA per cell was calculated using methods outlined below.
Quantitative PCR
Total RNA from homogenized AT biopsies was isolated using ExtractRNA kit (Eurogen, Russia). The resulting RNA was dissolved in 30 μl of RNAse- and DNAse-free water. The purity and concentration of isolated RNA were determined using a spectrophotometer (Nanovue Plus, GE Healthcare Bio-Sciences, Sweden). The quality of total RNA was determined by the RNA Integrity Number (RIN). Reverse transcription was performed using (dT) 23 (Beagle, Russia) and LV-revertase (Eurogen, Russia). To determine the levels of relative gene expression qPCR was performed using the qPCR mix-HS SYBR reagents (Eurogen, Russia). Approximately 4 μl cDNA was used as template, and B2M was used as the reference gene. The primer sequences are shown in Table 2.
Blood chemistry
Parameters of carbohydrate and lipid metabolism were studied on an automatic biochemical analyzer CA-180 (Furuno Electric Co., Ltd., Japan).
Enzyme-linked immunosorbent assay
The concentration of molecules (TNF-α and chemerin) was measured in serum/plasma using a sandwich enzyme-linked immunosorbent assay (ELISA) (Vector-Best kits, Russia The range of measured concentrations is 0–1000 pg / ml, sensitivity - 2 pg / ml; BIO Vendor, Czech Republic, the range of measured concentrations is 0.25–8 ng / ml, sensitivity 0.1 ng / ml).
Flow cytometry
Quantitative determination of insulin and adiponectin in plasma was performed by flow-through fluorometry using commercial test systems (Bio-Rad, USA) on a two-beam laser automated analyzer (BioPlex® 200 Systems, Bio-Rad, USA) and BioPlex Manager (Bio-Rad, USA).
Statistical analysis
Verification of the normality of quantative indicator distribution was carried out using the Kolmogorov-Smirnov and the Shapiro-Wilk tests. Because the investigated samples fit under normal distribution, the hypothesis of the equality of the mean sample values was verified using Student’s t-tests. To assess the significance of differences between independent quantitative samples that do not follow a normal distribution law, non-parametric Kruskal-Wallis test was used. For detecting statistically significant differences between groups, a pairwise analysis was performed using the Mann-Whitney test. Differences were considered significant at the level of p < 0.05.
Correlations between the studied indices were determined using the Spearman correlation analysis and linear regression. For the analysis of an adequate linear regression model, the following regression residues were considered: the lack of autocorrelation of residues (Durbin-Watson test, p-values> 0.05) and its normal distribution and the consistency of the dispersion residues (heteroscedasticity test, p-values< 0.05 were considered significant). Statistical analysis and graphs were obtained in R Statistical Software (version 3.3.1) and Statistica 10.
Results
Plasma level of components of carbohydrate metabolism, proinflammatory mediators (TNF-α and CRP) and adipokines (chemerin and adiponectin)
The level of glucose and the HOMA-IR was higher in obese patients with T2DM (with grade I, II, and III obesity) than in patients without T2DM and control individuals (p < 0.05) (Table 3). The plasma level of proinflammatory molecules (C-reactive protein (CRP) and TNF-α) increased along with BMI (Table 3).
The serum level of CRP was 8.95 (5.04–14.34) mmol/l in the group with T2DM (BMI > 40 kg/m2) and 13.4 (9.9–15.99) mmol/l in the group without T2DM (BMI > 40 kg/m2), exceeding the values in the control group of 4.41 (3.12–5.38) mmol/l (p < 0.05).
The plasma level of TNF-α in obese patient with T2DM exceeded that in patients without T2DM and control individuals (p < 0.05).
Correlation (r = 0.76, p < 0.001) and regression (r2 = 70, p < 0.001) analyses revealed an association between the concentration of TNF-α and BMI in all groups (p < 0.05) (Fig. 1a, b).

Correlation and regression relationships between studied metabolites.Note: Correlation - a, c, d, e, f, g, h, i, j, k, l, m, n, o, p, s, t, u, v, w, x; Regression – b, q, r, y
Plasma level of chemerin increased in patients without T2DM (with grade I, II and III obesity); however, the plasma level of chemerin in patients with T2DM did not differ from that in control individuals (with grade II and III obesity).
The plasma level of chemerin in patients without T2DM (with grade I, II and III obesity) and in patients with T2DM (with grade I obesity) exceeded the control values and values in pre-obese individuals (p < 0.05). In pre-obese individuals, the plasma level of chemerin was positively correlated with BMI (r = 0.71, p < 0.05) (Fig. 1c).
Positive correlations between plasma level of chemerin and CRP were observed in patients with T2DM (r = 0.76, p < 0.01) (Fig. 1d).
Furthermore, the plasma level of adiponectin in patients without T2DM (III degree of obesity) was higher than in pre-obese patients (4.44 (2.87–5.21) μg/ml vs. 1.74 (1.29–4.16) μg/ml).
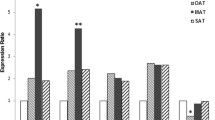
Expression of mediators (TNF-α, RARRES2, and ADIPOQ) in different ATs
The expression of the TNF-α gene was increased in all fat stores relative to that in control. The highest level of TNF-α was detected VAT (GO and Mes) (Fig. 2a). The expression of the RARRES2 gene (encoding chemerin) increased in SAT in patients with T2DM with respect to that in control individuals. In patients with T2DM (with grade II and III obesity), the expression of the RARRES2 gene increased in the GO and SAT.

Changes in the level of relative expression in adipose tissue of different locations in obese patients associated with and without type 2 diabetes
Interestingly, the expression of TNF-α and RARRES2 genes in the GO and SAT in patients with T2DM was higher than in patients without T2DM (Fig. 2b).
The plasma level of TNF-α was positively correlated with the expression of the RARRES2 gene in the Mes in all groups with BMI < 40 kg/m2 (Fig. 1e). The expression level of the TNF-α gene was positively correlated with the expression level of the RARRES2 gene in the GO in patients with T2DM (with grade I and II obesity) (r = 0.66, p < 0.05 and r = 0.67, p < 0.05, respectively) (Fig. 1f, g).
Positive correlations were also observed between the expression of the TNF-α gene in the GO (r = 0.34, p < 0.05), Mes (r = 0.34, p < 0.05) and SAT (r = 0.37, p < 0.05) and the plasma level of chemerin in all groups with BMI < 40 kg/m2 (Fig. 1h, i, j).
In general, the expression of the ADIPOQ gene in the Mes tended to increase in patients without T2DM and tended to decrease in patients with T2DM. The ADIPOQ gene is expressed predominantly in the SAT in patients with T2DM and in the GO in patients without T2DM (Fig. 2c). Under physiological conditions, chemerin may have anti-inflammatory properties within the visceral fluid, and this was confirmed by positive correlations between the expression of ADIPOQ and RARRES2 genes in the GO (r = 0.7, p < 0.05) in the control group (Fig. 1k).
mtDNA copy number in different ATs and MNCs
In general, there was an increase in mtDNA copy number in different ATs with an increase in BMI (Table 4).
mtDNA copy number in the GO was 1619 (963–2228) in patients with T2DM (with grade II obesity), which was higher than the reference value and higher than the value in patients with T2DM (with grade I obesity) (590 (541–633) (p < 0,05). mtDNA copy number in the GO was 2-times higher (1080 (769–1663)) in patients with T2DM (with grade III obesity) than in patients with grade I obesity 590 (541–633) (p < 0,05). Furthermore, mtDNA copy number in the GO in patients with T2DM (with grade III obesity) was higher than that in patients without T2DM (with grade III obesity) (1080 (769–1663) vs. 733 (465–834)) (p < 0,05).
A positive correlation was observed between mtDNA copy number in the GO with the expression of the TNF-α gene in the GO in pre-obese individuals (r = 0.81, p < 0.05), and a negative correlation was observed between these indices and the plasma level of CRP (r = − 0.8, r = − 0.79, p < 0.05) (Fig. 1l, m, n).
The number of copies of mtDNA in the Mes (1630 (1360–1835)) in patients with T2DM (with grade II obesity) was higher than in pre-obese individuals 841 (549–11,160)) (p < 0,05). mtDNA copy number in the Mes in patients with grade III obesity was lower than that in patients with grade II obesity regardless of the presence/absence of T2DM. mtDNA copy number in the Mes was negatively related to the plasma level of chemerin in patients without T2DM (with grade III obesity) (r = − 0.72, p < 0.05) (Fig. 1o).
Moreover, mtDNA copy number in the SAT was higher in patients with T2DM than in patients without T2DM. mtDNA copy number in the SAT was 1059 (635–2484) in patients with T2DM (with grade III obesity, respectively) and exceeded the control value of 462 (316–820). Furthermore, mtDNA copy number in SAT (1059 (635–2484)) in patients with T2DM was higher than that in patients without T2DM (with grade III obesity) (Table 4).
Positive correlation and regression were observed between mtDNA copy number in the SAT and the plasma level of TNF-α (r = 0.34, r2 = 24.7, p < 0.05) and mtDNA copy number in the GO (r2 = 0.27, p < 0.05) in all study groups (Fig. 1p, q, r).
Positive correlations were also observed between mtDNA copy number and the expression level of the ADIPOQ gene (r = 1.0, p < 0.01) in SAT in patients without T2DM (with grade I obesity) (Fig. 1s).
mtDNA copy number in the GO and SAT was positively correlated with serum glucose level (r = 0.39, r = 0.38, p < 0.05) in patients with T2DM (BMI > 40 kg/m2) (Fig. 1t, u). In contrast, mtDNA copy number in the Mes was not correlated with blood glucose level (r = − 0.9, p < 0.05) in patients without T2DM (with grade I obesity) (Fig. 1v).
mtDNA copy number in the MNCs was higher in the pre-obese group (323 (180–414)) than in all other study groups (p < 0.05). However, mtDNA copy number in MNCs was lower in patients with T2DM (with grade II obesity) than in patients without T2DM (with grade II obesity) and in pre-obese and control individuals (Table 4). mtDNA copy number in MNCs was also negatively correlated with the plasma level of chemerin in pre-obese individuals (r = − 0.77, p < 0.05) (Fig. 1w). However, mtDNA copy number in the SAT was negatively associated with the expression of the RARRES2 gene in patients without T2DM (with grade III obesity) (r = − 0.84, p < 0.05; r2 = − 0.69, p < 0.05) (Fig. 1x, y).
Discussion
Hypertrophy of adipocytes contributes to the development of hypoxia in AT during obesity and the release of free fatty acids during lipolysis [11]. In ATs, immune cells and macrophages are recruited by inflammatory mediators, such as HIF-1 and MCP-1 [12]. Macrophages ingest necrotic adipocytes and produce chemokines and proinflammatory mediators, including chemerin and TNF-α [12, 13]. Thus, a positive inflammatory feedback loop is formed.
In all obese patients, with or without T2DM, the expression level of the TNF-α gene in the GO, Mes, and SAT and the plasma level of TNF-α increase along with the BMI. Correlation and regression analyses revealed an association between the BMI and the plasma level of TNF-α in all study groups (Fig. 1a, b).
In our study, the change in metabolic parameters at the earliest stage of obesity in pre-obese patients was particularly interesting (BMI = 25–29.9 kg/m2). Positive correlation was observed between mtDNA copy number with the expression of the TNF-α gene in the GO in pre-obese individuals (r = 0.81, p < 0.05), and a negative correlation was observed between these indices and the plasma level of CRP (Fig. 1l, m, n). CRP characterizes a systemic inflammatory response [14]. The associations revealed by us indicate the local course of inflammation in the AT at early stages of obesity.
Increasing BMI is accompanied by an increase in TNF-α production, which ultimately leads to an increase in mtDNA copy number [1, 8]. Mitochondria generate superoxide, hydrogen peroxide and hydroxyl radicals, contributing to damage protein, DNA, RNA and lipids. Oxidative stress promotes nuclease activity against mtDNA, and this leads to the accumulation of mtDNA fragments [13, 15]. The disruption of the native structure of mtDNA in IR is reflected by a decrease in the efficiency of oxidative phosphorylation and ATP synthesis [4].
We speculate the existence of a compensatory mechanism in which mtDNA copy number increases under the influence of proinflammatory factors. This hypothesis was confirmed by positive correlations of the plasma level of TNF-α with mtDNA copy number in the SAT and GO in all study groups (Fig. 1p, q, r).
mtDNA copy number in the VAT (GO and MES), regardless of the state of carbohydrate metabolism, increased in patients with grade II and III obesity in relation to that in patients with grade I obesity. However, in patients with morbid obesity (BMI > 40 kg/m2) with and without T2DM, mtDNA copy number in the Mes was reduced by a factor of 2 compared to that in patients with grade II obesity and was comparable to that in control individuals. This correlation was not observed in other tissues (GO, SAT or MNCs).
A reduction in the number of copies of mtDNA in the GO in patients with morbid obesity in comparison with that in patients with grade I and II obesity indicates a change in the energy metabolism in this fat depot. When the TNF signaling pathway is activated, in addition to the initiation of mitochondrial division by DNM1L, the activation of caspases takes place, initiating the cell death programs apoptosis and necrosis [8, 16, 17]. Studies in animal models with obesity have shown that a change in mitochondrial biogenesis is a possible response to prolonged exposure to reactive oxygen species (ROS) – in the first months the amount of mtDNA increases to compensate for the damage induced by oxidation, and under further stress, intracellular depletion occurs beyond the recovery capacity of the cell, and the amount mtDNA drops sharply [18].
mtDNA dysfunction is less associated with damage to single copies of mtDNA but more so with a dysregulation of mtDNA copy number in the cell [19]. Recently, Ye J. (2013) proposed a concept regarding the relationship between energy metabolism in mitochondria and IR [20]. The decrease in mtDNA copy number detected by us can be considered an adaptive mechanism for the “soft-separation” of the electron transport chain, which also protects cells from the vicious circle of ROS production. Reducing the level of ROS by suppressing its production or stimulating utilization is a promising approach in the treatment of IR [20].
Mitochondria are involved in the regulation of carbohydrate metabolism and contribute to the pathogenesis of T2DM in obesity [21]. We detected high plasma TNF-α level and mtDNA copy numbers in different tissues in patients with T2DM with respect to those in obese patients without T2DM. mtDNA copy numbers in the GO (grade II and grade III) and SAT (grade III) in obese patients with T2DM significantly exceeded those in patients without T2DM. A positive correlation of mtDNA copy numbers in the GO and SAT with serum glucose level was found in T2DM patients with morbid obesity (BMI > 40 kg/m2), and a negative correlation of mtDNA copy numbers in the GO with glucose was found in patients without T2DM (with grade I obesity), which, in our opinion, helps maintain normoglycemia (Fig. 1t, u, v). Based on these findings, we postulate a positive effect of reducing mtDNA in AT on carbohydrate metabolism in morbid obesity, which may help maintain glucose levels within reference values.
An increase in the mass of VATs is associated with an increased inflammatory background and development of IR [22], and the role of SATs is not completely determined. The change in SAT metabolism in obesity may be protective in nature with respect to IR and its complications [8, 23]. We observed positive correlations between the amount of mtDNA and expression level of the ADIPOQ gene (Fig. 1s) in the SAT in patients without T2DM (with grade I obesity), which was confirmed in the study data. In this case, an increase in the production of adiponectin can suppress the production of TNF-α in this type of fat depot [23].
With respect to the homeostasis of glucose, we have demonstrated the dual role of chemerin in obesity: positive - with obesity and no disruptions in carbohydrate metabolism and negative - with obesity associated with T2DM. The location of chemerin synthesis plays a key role in metabolism [24]. Chemerin is an adipokine regulating the processes of lipogenesis, metabolism and inflammation through the activation of chemokine-like receptor-1 (CMKLR1) [8]. Other receptors for the ligand are CMKLR1 or Chem23, GPR1 and CCRL2 [25]. Some studies indicate the proinflammatory properties of chemerin [26,27,28]. Сhemerin affects the production of proinflammatory mediators (TNF-α, IL-1β, and IL-6), which leads to the activation of the MAPK pathway and suppression of insulin signal transmission in human skeletal muscle cells [29, 30]. In patients with BMI < 40 kg/m2, we observed positive correlations between plasma chemerin level and TNF-α gene expression in the GO, Mes and SAT (Fig. 1h, i, j).
TNF-α can also affect the production of chemerin in differentiated adipocytes of the 3 T3-L1 line [31, 32]. In patients with BMI < 40 kg/m2, the plasma level of TNF-α was positively correlated with the expression of the RARRES2 gene in the Mes (Fig. 1e), suggesting that these mediators are mutually regulated. In the patients without T2DM (with grade I and II obesity), the relative expression levels of TNF-α and RARRES2 genes in the GO were correlated (Fig. 1f, g).
Chemerin increases the secretion of proinflammatory and prodiabetic adipokines, with negative systemic effects on proinflammatory adipokines [26, 27]. Subclinical inflammation in AT is systemic in morbid obesity, which can be confirmed by positive associations of the plasma level of chemerin with CRP in patients with T2DM (r = 0.76, p < 0.01) (Fig. 1d).
In contrast, chemerin can prevent TNF/TNFA activation by inhibiting the NF-kB and CRK/p38 signaling pathways [8] by binding to the CMKLR1 receptor [8]. Under physiological conditions, chemerin may play anti-inflammatory roles within the visceral fluid. This was confirmed by the positive correlations between the expression of ADIPOQ and RARRES2 genes in the GO in the control group (Fig. 1k).
The level of chemerin expression in the GO and SAT increased in patients with T2DM and did not change in patients without T2DM relative to that in control individuals. In contrast, the plasma level of chemerin increased mainly in obese patients without T2DM. Given the presence of several isoforms of chemerin with both pro- and anti-inflammatory activity, it is logical to assume that the production of proinflammatory forms is mainly prevalent in obese patients with T2DM, whereas anti-inflammatory forms are likely synthesized in other locations.
In turn, the activation of CMKLR1 exerts negative effects on mitochondrial biogenesis and division [8]. The plasma level of chemerin was negatively correlated with mtDNA copy number in MNCs (Fig. 1w) and positively correlated with BMI in pre-obese individuals (Fig. 1c). A reciprocal link between mtDNA duplication and chimeric plasma level was also observed. The plasma level of chemerin was negatively correlated with mtDNA copy number in the Mes in patients without T2DM (with grade III obesity) (Fig. 1o). In addition, in all obese patients without T2DM, an increase in the plasma level of chemerin and its expression in biopsy samples of the Mes was recorded.
Therefore, it can be speculated that chemerin regulates local mtDNA copy number in the GO and Mes in cases of morbid obesity. In biopsy specimens of the prostate, mtDNA copy number was negatively associated with the expression of the RARRES2 gene in patients with morbid obesity without T2DM (Fig. 1x, y). A negative relationship was observed between mtDNA copy number in biopsy specimens of the GO and plasma chemerin and chemerin expression in the GO. Thus, in patients with morbid obesity without T2DM, chemerin may exert a protective effect against the development of IR by regulating the number of copies of mtDNA.
The dual role of chemerin in inflammatory processes and metabolism provides a link between chronic inflammation and energy metabolism in obesity, as well as the complications associated with these processes.
It should be noted that antioxidant protective enzymes in different tissues function with different efficacy [33, 34] and may display different responses to genotoxic agents during obesity. In this regard, it is necessary to consider the heterogeneity of the response of different tissues of the body to cytotoxic damage. Therefore, it can be assumed that the amount of mtDNA in various types of insulin-dependent tissues, such as AT and blood cells, is an important indicator of various metabolic disorders. The quantification of mtDNA in various biological samples can thus be a valid tool for predicting and evaluating the effectiveness of treatment of IR.
The obtained data on changes in copy number of mtDNA can be used to diagnose early disorders of carbohydrate metabolism in obese patients. In addition, further studies of the effect of different isomers of chimerin on mitochondrial biogenesis may contribute to the development of targeted therapy for type 2 diabetes.
Conclusions
1. We assume the existence of a compensatory mechanism represented by an increase in mtDNA copy number under the influence of proinflammatory factors.
2. Based on the data obtained, we postulate a positive effect of reducing mtDNA copy number in morbid obesity in the absence of T2DM on carbohydrate metabolism, which can help maintain glucose levels within reference values.
3. The negative association between plasma chemerin and mtDNA copy number in the Mes, as well as between mtDNA copy number and chemerin expression in the Mes, in the group with BMI > 40 kg/m2 without T2DM demonstrates the protective effect of chemerin against the development of IR via the regulation of the number of copies of mtDNA in adipose tissues.
Abbreviations
- AT:
-
Adipose tissue
- ATP:
-
Adenosine triphosphate
- BMI:
-
Body mass index
- ddPCR:
-
Droplet Digital PCR
- GO:
-
Greater omentum
- IR:
-
Insulin resistance
- Mes:
-
Mesentery of the small intestine
- MNC:
-
Blood mononuclear cells
- mtDNA:
-
Mitochondrial DNA
- SAT:
-
Subcutaneous adipose tissue
- T2DM:
-
Type 2 diabetes mellitus
References
Yu T, Gao M, Yang P, Pei Q, Liu D, Wang D, Zhang X, Liu Y. Topical insulin accelerates cutaneous wound healing in insulin-resistant diabetic rats. Am J Transl Res. 2017;9(10):4682–93.
Zhang Z, Liu H, Liu J. Akt activation: a potential strategy to ameliorate insulin resistance. Diabetes Res Clin Pract. 2017. https://doi.org/10.1016/j.diabres.2017.10.004.
Serra D, Mera P, Malandrino MI, Mir JF, Herrero L. Mitochondrial fatty acid oxidation in obesity. Antioxid Redox Signal. 2013. https://doi.org/10.1089/ars.2012.4875.
Pagel-Langenickel I, Bao J, Pang L, Sack MN. The role of mitochondria in the pathophysiology of skeletal muscle insulin resistance. Endocr Rev. 2010;31(1):25–51.
Maechler P, Wollheim CB. Mitochondrial function in normal and diabetic beta-cells. Nature. 2001;414(6865):807–12.
Yuzefovych LV, Musiyenko SI, Wilson GL, Rachek LI. Mitochondrial DNA damage and dysfunction, and oxidative stress are associated with endoplasmic reticulum stress, protein degradation and apoptosis in high fat diet-induced insulin resistance mice. PLoS One. 2013;8(1):e54059.
Zhang B, Davidson MM, Zhou H, Wang C, Walker WF, Hei TK. Cytoplasmic irradiation results in mitochondrial dysfunction and DRP1-dependent mitochondrial fission. Cancer Res. 2013;73(22):6700–10.
Xie Q, Deng Y, Huang C, Liu P, Yang Y, Shen W, Gao P. Сhemerine-induced mitochondrial dysfunction in skeletal muscle. J Cell Mol Med. 2015;19(5):986–95.
Laudes M. Role of WNT signalling in the determination of human mesenchymal stem cells into preadipocytes. J Mol Endocrinol. 2011;46(2):65–72.
American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2014. https://doi.org/10.2337/dc14-S081.
Snodgrass RG, Boß M, Zezina E, Weigert A, Dehne N, Fleming I, Brüne B, Namgaladze D. Hypoxia potentiates palmitate-induced pro-inflammatory activation of primary human macrophages. J Biol Chem. 2016;291(1):413–24.
Bergmann K, Sypniewska G. Diabetes as a complication of adipose tissue dysfunction. Is there a role for potential new biomarkers? Clin Chem Lab Med. 2013;51(1):177–85.
Aliev G, Li Y, Palacios HH, Obrenovich ME. Oxidative stress induced mitochondrial DNA deletion as a hallmark for the drug development in the context of the cerebrovascular diseases. Recent Pat Cardiovasc Drug Discov. 2011;6(3):222–41.
Lemieux I, Pascot A, Prud'homme D, Alméras N, Bogaty P, Nadeau A, Després JP BJ. Elevated C-reactive protein: another component of the atherothrombotic profile of abdominal obesity. Arterioscler Thromb Vasc Biol. 2001;21(6):961–7.
Peng TI, Yu PR, Chen JY, Wang HL, Wu HY, Wei YH, Jou MJ. Visualizing common deletion of mitochondrial DNA-augmented mitochondrial reactive oxygen species generation and apoptosis upon oxidative stress. Biochim Biophys Acta. 2006;1762(2):241–55.
TNF-signaling pathway KEGG PATHWAY. https://www.kegg.jp/kegg-bin/show_pathway?ko04668+K17065, Accessed 27 Mar 2018.
Ježek J, Cooper KF, Strich R. Reactive Oxygen Species and Mitochondrial Dynamics: The Yin and Yang of Mitochondrial Dysfunction and Cancer Progression. Antioxidants (Basel). 2018;7(1). https://doi.org/10.3390/antiox7010013.
Wang PW, Kuo HM, Huang HT, Chang AY, Weng SW, Tai MH, Chuang JH, Chen IY, Huang SC, Lin TK, Liou CW. Biphasic response of mitochondrial biogenesis to oxidative stress in visceral fat of diet-induced obesity mice. Antioxid Redox Signal. 2014;20(16):2572–88.
Marín-García J. Mitochondrial DNA repair: a novel therapeutic target for heart failure. Heart Fail Rev. 2016;21(5):475–87.
Ye J. Mechanisms of insulin resistance in obesity. Frontiers Med. 2013;7(1):14–24.
Peti-Peterdi J. High glucose and renin release: the role of succinate and GPR91. Kidney Int. 2010. https://doi.org/10.1038/ki.2010.333.
Hagman DK, Larson I, Kuzma JN, Cromer G, Makar K, Rubinow KB, Foster-Schubert KE, van Yserloo B, Billing PS, Landerholm RW, Crouthamel M, Flum DR, Cummings DE, Kratz M. The short-term and long-term effects of bariatric/metabolic surgery on subcutaneous adipose tissue inflammation in humans. Metabolism. 2017. https://doi.org/10.1016/j.metabol.2017.01.030.
Pivovarova O, Gögebakan Ö, Sucher S, Groth J, Murahovschi V, Kessler K, Osterhoff M, Rudovich N, Kramer A, Pfeiffer AF. Regulation of the clock gene expression in human adipose tissue by weight loss. Int J Obes. 2016. https://doi.org/10.1038/ijo.2016.34.
Zabel BA, Kwitniewski M, Banas M, Zabieglo K, Murzyn K, Cichy J. Сhemerine regulation and role in host defense. Am J Clin Exp Immunol. 2014;3(1):1–19.
Zylla S, Pietzner M, Kühn JP, Völzke H, Dörr M, Nauck M, Friedrich N. Serum сhemerine is associated with inflammatory and metabolic parameters-results of a population-based study. Obesity (Silver Spring). 2017. https://doi.org/10.1002/oby.21735.
Sell H, Laurencikiene J, Taube A, Eckardt K, Cramer A, Horrighs A, Arner P, Eckel J. Сhemerine is a novel adipocyte-derived factor inducing insulin resistance in primary human skeletal muscle cells. Diabetes. 2009;58(12):2731–40.
Fülöp P, Seres I, Lőrincz H, Harangi M, Somodi S, Paragh G. Association of сhemerine with oxidative stress, inflammation and classical adipokines in non-diabetic obese patients. J Cell Mol Med. 2014;18(7):1313–20.
Rourke JL, Dranse HJ, Sinal CJ. CMKLR1 and GPR1 mediate сhemerine signaling through the RhoA/ROCK pathway. Mol Cell Endocrinol. 2015;417:36–51.
Kaur J, Adya R, Tan BK, Chen J, Randeva HS. Identification of сhemerine receptor (ChemR23) in human endothelial cells: сhemerine-induced endothelial angiogenesis. Biochem Biophys Res Commun. 2010. https://doi.org/10.1016/j.bbrc.2009.12.150.
Neves KB, Nguyen Dinh Cat A, Lopes RA, Rios FJ, Anagnostopoulou A, Lobato NS, de Oliveira AM, Tostes RC, Montezano AC, Touyz RM. Сhemerine Regulates Crosstalk Between Adipocytes and Vascular Cells Through Nox. Hypertension. 2015. https://doi.org/10.1161/HYPERTENSIONAHA.115.05616.
Parlee SD, Ernst MC, Muruganandan S, Sinal CJ, Goralski KB. Serum сhemerine levels vary with time of day and are modified by obesity and tumor necrosis factor-{alpha}. Endocrinology. 2010. https://doi.org/10.1210/en.2009-0794.
Parlee SD, McNeil JO, Muruganandan S, Sinal CJ, Goralski KB. Elastase and tryptase govern TNFα-mediated production of active сhemerine by adipocytes. PLoS One. 2012. https://doi.org/10.1371/journal.pone.0051072.
Sivitz WI, Yorek MA. Mitochondrial dysfunction in diabetes: from molecular mechanisms to functional significance and therapeutic opportunities. Antioxid Redox Signal. 2010;12(4):537–77.
Gruzdeva OV, Akbasheva OE, Dyleva YA, Antonova LV, Matveeva VG, Uchasova EG, Fanaskova EV, Karetnikova VN, Ivanov SV, Barbarash OL. Adipokine and cytokine profiles of Epicardial and subcutaneous adipose tissue in patients with coronary heart disease. Bull Exp Biol Med. 2017 Sep;163(5):608–11. https://doi.org/10.1007/s10517-017-3860-5.
Acknowledgments
No acknowledgments.
Funding
Publication of this article was supported by Russian Foundation for Basic Research 18–015-00084-a and Organization of Scientific Research 20.4986.2018/VU.
Availability of data and materials
The data that support the findings of this study are available on request from the corresponding author Skuratovskaia D.A. The data are not publicly available due to containing information that could compromise research participant privacy/consent.
About this supplement
This article has been published as part of BMC Medical Genomics Volume 12 Supplement 2, 2019: Selected articles from BGRS\SB-2018: medical genomics. The full contents of the supplement are available online at https://bmcmedgenomics.biomedcentral.com/articles/supplements/volume-12-supplement-2.
Author information
Authors and Affiliations
Contributions
PZ received the samples of adipose tissue. DS, MV performed the experiments and molecular analyses and contributed to the interpretation and discussion of the results related to these experiments. LL, IM and DS designed and planned the study and drafted and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Authors’ information
Daria Skutratovskaia – PhD student, Biologist of Laboratory immunology and cell biotechnology, Immanuel Kant Baltic federal university.
Pavel Zatolokin – PhD, head of Department of Reconstructive and endoscopic surgery, Kaliningrad Regional Hospital.
Maria Vasilenko – PhD, Researcher of Laboratory of Immunology and Cell Biotechnologies Immanuel Kant Baltic Federal University.
Mazunin Ilia – PhD, Head of Laboratory of molecular genetics technologies, Immanuel Kant Baltic federal university.
Larisa Litvinova – MD, PhD, Head of Laboratory immunology and cell biotechnology, Immanuel Kant Baltic federal university.
Ethics approval and consent to participate
The study was carried out in accordance with World Medical Association (WMA) Declaration of Helsinki (2000) and the Protocol to the Convention on Human Rights and Biomedicine (1999). The study protocol was approved by the Local Ethical Committee of the Innovation Park of the Immanuel Kant Baltic Federal University (Protocol No. 4 from October 23, 2013). All study participants signed informed consent to take part in a research study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Skuratovskaia, D., Zatolokin, P., Vulf, M. et al. Interrelation of chemerin and TNF-α with mtDNA copy number in adipose tissues and blood cells in obese patients with and without type 2 diabetes. BMC Med Genomics 12 (Suppl 2), 40 (2019). https://doi.org/10.1186/s12920-019-0485-8
Published:
DOI: https://doi.org/10.1186/s12920-019-0485-8