
Abstract
Background
Corynebacterium pseudotuberculosis (Cp) is a gram-positive bacterium that is classified into equi and ovis serovars. The serovar ovis is the etiological agent of caseous lymphadenitis, a chronic infection affecting sheep and goats, causing economic losses due to carcass condemnation and decreased production of meat, wool, and milk. Current diagnosis or treatment protocols are not fully effective and, thus, require further research of Cp pathogenesis.
Results
Here, we mapped known protein-protein interactions (PPI) from various species to nine Cp strains to reconstruct parts of the potential Cp interactome and to identify potentially essential proteins serving as putative drug targets. On average, we predict 16,669 interactions for each of the nine strains (with 15,495 interactions shared among all strains). An in silico sanity check suggests that the potential networks were not formed by spurious interactions but have a strong biological bias. With the inferred Cp networks we identify 181 essential proteins, among which 41 are non-host homologous.
Conclusions
The list of candidate interactions of the Cp strains lay the basis for developing novel hypotheses and designing according wet-lab studies. The non-host homologous essential proteins are attractive targets for therapeutic and diagnostic proposes. They allow for searching of small molecule inhibitors of binding interactions enabling modern drug discovery. Overall, the predicted Cp PPI networks form a valuable and versatile tool for researchers interested in Corynebacterium pseudotuberculosis.
Similar content being viewed by others


Background
Corynebacterium pseudotuberculosis (Cp) belongs to the supra generic CMNR group (Corynebacterium, Mycobacterium, Nocardia, Rhodococcus) of bacteria [1]. It is an intracellular Gram-positive pathogenic bacterium that is fimbriated, non-motile and non-capsulated [2] and is present in two serovars: ovis and equi [3]. The serovar equi infects mainly horses and cattle while the serovar ovis is the etiological agent of caseous lymphadenitis (CLA), a chronic infectious disease affecting mainly sheep and goat populations. It can also infect humans upon occupational exposure [4, 5]. CLA is prevalent in several countries around the world [6–21] and causes significant economic losses due to low carcass quality, a decrease in the production of meat, wool and milk [22, 23], while also causing animal mortality due to suppurative meningoencephalitis [24]. The available methods for CLA diagnosis or treatment are not effective enough and require further research to tackle the threats posed by C. pseudotuberculosis. Hence, it becomes important to know how the genes, transcripts, proteins and other molecules inside the bacterial cells interact with each other and with the outer environment to perform their biological functions [25–29]. From this perspective, the study of proteins and their interactions allows for a better understanding of the molecular mechanism of cells at a system level [30, 31]. The protein-protein interactions (PPI) form a complex network represented as a graph, where the nodes represent proteins and undirected edges connecting these nodes represent the interactions between the proteins [32, 33]. Generally, PPI networks have shown to be a great vehicle for developing new hypotheses and designing novel laboratory experiments [34, 35]. Furthermore, essential proteins can be identified by topological analysis. An essential protein is defined as a gene which demonstrates to be lethal for the organism when subject to a knock-out [36]. Therefore, essential proteins are potential drug targets [37–41], enabling the development of new drugs against pathogenic microorganisms [42–45].
Generally, in silico reconstruction of biological networks is a long standing problem and is applied to various different types of networks. As prominent example may serve the reconstruction of the regulatory network of various different Corynebacteria which has become a widely used resource [46, 47].
In this manuscript, we predicted the potential PPI networks of nine strains of Cp serovar ovis using the interolog mapping method. The interolog mapping method was already successfully applied in several other studies, for example to predict the interactions in Mycobacterium tuberculosis [48], Leishmania spp. [49], mouse [50] and Bacillus licheniformis [51].
While Yu et al. [57] used an identity > 80 % in their “generalized interolog mapping” to transfer interactions, we have refined this cut-off in one of our previous studies by means of an exhaustive in silico evaluation [52]. We used the experimentally validated and manual curated small-scale interactions from the DIP database (Database of Interacting Proteins) [53] as the gold standard and further collected the interactions from three different and independent PPI databases (STRING (search tool for recurring instances of neighbouring genes) [54], IntAct [55] and PSIbase (database of Protein Structural Interactome map) [56]) as the input for the network transfer and aimed to reconstruct the interactions in the DIP database. In this setting we archived a specificity of 0.95, sensitivity of 0.83 and a precision of 0.99 when we compared our predictions with the gold standard [52].
In a different study, Yu et al. archived an accuracy of 54 % when employing a similar method for transferring the interactome from C. elegans to S. cerevisiae [57]; two evolutionarily rather different organisms. In this study, we are convinced that our predictions are more reliable as with C. glutamicum we have an exhaustively studied model organism at hand which is evolutionary very close to Cp [46, 47, 58].
Due to this exhaustive previous work, we only perform a brief in silico sanity check of the derived networks before identifying essential proteins which might be promising targets for further wet-lab experiments. It is important to note that the reported PPI networks are a mere list of potential interactions and should serve as a basis for further research. The experimental validation for the predicted potential interactome is out of the scope of this study.
Results and discussion
Prediction of C. pseudotuberculosis PPI network
For all nine strains of Cp, we predicted a total of 150,019 potential protein-protein interactions involving 10,370 of the in total 18,890 proteins (Table 1).
The analysis of the prediction origin shows that the vast majority of interactions were mapped from phylogenetically close organisms, belonging to the genus Corynebacterium (in ~99 % of the cases) but also reveals some predictions from more distant organisms (Additional file 1: Figure S1).
Validation of the network properties
As described above, in order to check the credibility of our network predictions, we performed statistical sanity checks on the network topology. We were able to show that the node degree distribution approximately follows a power-law distribution and in combination with shortest-path analysis, suggest that the predicted networks have a scale-free topology, both prevalent and relevant characteristics pertaining to biological networks. The clustering coefficient, correlation and regression analysis using the R-Squared values from predicted Cp interaction networks and the Shapiro-Wilk [59] normality test demonstrated that the degree distribution of predicted interaction networks do not follow a normal distribution (p-value < 2.2e-16) (Additional file 2). All analyses suggest that the networks were not formed by spurious interactions but originated due to a biological growth process. Moreover, the high Clustering Coefficient of the predicted networks suggest the existence of self-organization inside the biological cell motivated by the interactions [60]. Furthermore, we were able to confirm the existence of several clusters of our networks by means of literature research, increasing the confidence in the methodology and the predictions (Additional file 3). Please note, that these test comprise mere sanity checks of the potential networks and should not be misinterpreted as exhaustive proof for correctness of the potential interactions.
Not surprisingly, due to the extremely clonal life-style [61], almost all predicted interactions are found in all Cp ovis strains (i.e., core-interactome). Strain specific interactions or the accessory interactions are also of great interest as they might explain the biological specifics of a strain. However, here we focused on exploring the core-interactome of the nine Cp ovis strains aiming to better understand the serovar ovis in general and derive potential viable targets for further wet-lab research.
Essential proteins
Essential proteins are proteins which have a lethal effect when removed from the organism. It was shown that the node degree of a protein (i.e., the number of interactions of that protein) is correlated with the lethality [62, 63]. Thus, potential essential genes may be identified by identifying hub nodes in the network, i.e., nodes with a very high node degree (refer to the Methods section for details).
In our predicted networks, we identified 181 hub proteins each having 68 or more interactions. In the set of hub proteins, we find proteins involved in biological processes related to carbon metabolism, cell envelope and cell wall, DNA metabolism, nucleotides biosynthesis, folding, translocation, ribosomal translation factors, tRNA synthetase, RNA metabolism and respiratory pathways, among others. Aiming to verify the essentiality of these Cp hubs, we searched for homologous proteins in the database of essential genes (DEG) [64, 65]. Among the 181 hub proteins, 180 had homologous counterparts already stored as essential in DEG, showing the effectiveness of our method for identifying the essential proteins (Additional file 4).
The DNA repair protein (RecN), was the only essential protein not found in DEG, apparently being exclusive to Cp. RecN is responsible for maintaining DNA integrity when exposed to various stress conditions. Despite the conserved mechanism, both metabolic pathways and proteins can differ in each species [66]. This indicates the essentiality of this protein and explains why there was no counterpart found in DEG.
Even though the vast majority of proteins have homologs in DEG, this does not reduce the importance of reporting their essentiality. Considering Cp is not covered by DEG till today, the description of essentiality in this organism is novel for all 181 proteins. It is worth noting that while most essential proteins have homologs from over 20 organisms covered by DEG, three proteins have homologs in only a single organism, demonstrating either the lack of experiments which would support their essentiality, the lack of protein conservation across different species or that the essentiality of these proteins is not conserved across species [67]. These proteins are: Catalase (KatA), Endonuclease III (Nth) and Trigger factor Tig (Tig). Catalase (KatA) is homologous to KatE from Salmonella enterica. KatA is an oxidoreductase enzyme which decomposes hydrogen peroxide (H2O2). It was already studied for instance in C. glutamicum [68, 69] and C. pseudotuberculosis [70]. Endonuclease III (Nth) has a homologous counterpart in Haemophilus influenzae stored in DEG. Nth is a base excision repair enzyme [71] that participates in a pathway preventing the loss of DNA functionality e.g., by spontaneous mutagenic lesion [72] or near-UV radiations [73]. This mechanism is well studied and is conserved in the Corynebacterium species [74]. Trigger factor Tig (Tig) has a DEG homology against Pseudomonas aeruginosa and participates in the protein folding process.
Additionally, in order to propose potential biomarkers or therapeutic targets among the essential proteins, a search for homologs in the host organisms O. aries, C. hircus, B. taurus, E. caballus and H. sapiens was performed. We identified 41 non-host homologous proteins, i.e., these are essential proteins of Cp which have no homologs in one or more host organisms. Among these non-host homologous proteins, 15 are with no alignment hits against any of the five hosts, nine with no alignment hits against O. aries and C. hircus and the remaining 17 had only low identity and low coverage hits (Additional file 5: Figure S2).
The 24 non-host homologous proteins without any significant hit against at least one host are: chorismate synthase (aroC), dihydrodipicolinate reductase (dapB), DNA primase (dnaG), elongation factor P (efp), cell division protein (ftsZ), ATP phosphoribosyl transferase (hisG), dihydroxy-acid dehydratase (ilvD), aspartate kinase (lysC), UDP-N-acetylglucosamine (murA), transcription anti-termination protein (nusG), uridylate kinase (pyrH), DNA repair protein (recN), transcription termination factor (rho), 50S ribosomal protein L1 (rplA), 50S ribosomal protein L10 (rplJ), 50S ribosomal protein L31 (rpmE), DNA-directed RNA polymerase subunit alpha (rpoA), 30S ribosomal protein S3 (rpsC), 30S ribosomal protein S6 (rpsF), 30S ribosomal protein S13 (rpsM), holliday junction DNA helicase subunit (ruvA), SsrA-binding protein/SmpB superfamily (smpB), indole-3-glycerol phosphate synthase (trpC2) and anthranilate synthase (trpE). As these proteins are essential to Cp but do not occur in the host organisms, they naturally are a potential drug-targets because inhibiting these proteins is likely to be lethal for Cp whereas the host proteome remains unaffected due to the missing homologs and furthermore due to the greater potential of these proteins to participate in inter-species interactions with the host [75].
A small subset of the essential non-host homologous proteins participates in the same metabolic pathway and thus is of particular interest. These proteins are the Indole-3-glycerol phosphate synthase (trpC2), Anthranilate phosphoribosyl transferase (trpD), Anthranilate synthase (trpE) and Anthranilate synthase component II (trpG); all are involved in the metabolic pathway of tryptophan biosynthesis, which produces amino acids of biotechnological interest and are essential in human and animal nutrition [76]. This metabolic pathway involves proteins encoded by the genes of the Cp operon trpABCDGEF which was already studied and characterized in other organisms [77]. Prephenate dehydratase (pheA) is involved in the metabolic pathway of phenylalanine biosynthesis from the chorismate pathway [78]. Tryptophan, phenylalanine and tyrosine are aromatic amino acids and share the beginning of a pathway found and characterized in C. glutamicum [79] whose proteins are also partially present in the Cp biovar ovis. Additionally, the other essential proteins interacting in this network are Tryptophanyl-tRNA synthetase (trpS), Phenylalanyl-tRNA synthetase subunit alpha (pheS) and Tyrosyl-tRNA synthetase (tyrS).
Furthermore, the cluster analysis draws attention to the Cp iron acquisition system, which is well characterized and contributes to the survival and virulence of microorganisms [80, 81]. The cluster consists of proteins associated with different iron acquisition systems, a strategy to acquire iron from multiple sources in low availability [82], suggesting both, alternative metabolic pathways and alternative proteins from different operons exerting the same function. In the potential Cp networks, these multiple systems interact with each other and consist mainly of proteins from the operons fag, ciu, fec and hmu, suggesting a potential ability to import iron from the host [83, 84] (Additional file 3).
Conclusions
For the first time, we reported potential PPI networks for nine Cp ovis strains based on an in silico prediction. The employed methodology is well-established and we consider this work as the starting-point for the development novel hypothesis and the design of upcoming wet-lab studies. Nevertheless, it is important to notice that the in silico predictions only represent a candidate list of potential interactions and may contain false-positives, in particular when considering that the original interactions utilized for the prediction also contain false-positives themselves.
The main contribution and analysis of this work is the identification of potentially essential genes which have a very high node-degree in the network reducing the impact of sporadic false-positives. In total, we identified 181 essential proteins, 41 of them being non-host homologous, hence becoming good candidates for drug development or CLA diagnosis. Since the essential proteins interact with many others, it is natural to assume they are associated with various biological processes, in their own species as well as in the host, and hence are attractive targets for therapeutic and diagnostic proposes [85]. Especially each predicted interaction of an essential protein is a potential candidate for the identification of inhibitors [86, 87] and thus opening several drug development opportunities targeting C. pseudotuberculosis. Especially the non-host homologous essential proteins might serve as potential targets for inhibiting interaction class drugs [40, 86, 88]. Generally, all reported potential interactions might allow for searching small molecule inhibitors of binding interactions [45, 86, 89], making modern drug discovery research possible [90]. By knowing the interaction partners of a protein, it is hence possible to provide a systemic view of the organism [91]. To sum up, the PPI networks reported here are valuable tools for researchers to identify proteins or interactions as potential drug targets.
Methods
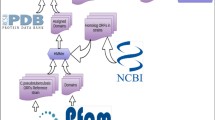
We have employed the workflow depicted in Fig. 1 for deriving the candidate list of potential protein-protein interactions. We will give a brief summary of the method before describing the details in the subsequent chapters: We have extracted known regulations of publicly available databases and used those as the basis for our predictions (see subchapter data sources). For each interaction we have searched for conserved counterparts in the Cp strains. In case both interaction partners were sufficiently conserved (refer to the subchapter interolog mapping) we assumed the interaction to be a candidate for a potential interaction in the corresponding Cp strain. The networks derived with this method then were briefly checked for sanity in silico. We continued the analysis by using only potential interactions predicted for all nine Cp strains, i.e., the core-interactome. Here, we extracted the 15 % top ranking nodes with respect to the node degree in the networks which represent potentially essential proteins. In fact, we found for all extracted proteins but one an entry of a homologous protein in the DEG database. Further, we compared the potentially essential proteins against the proteome of the host organisms in order to discover essential proteins exclusive to Cp which comprise potential drug targets.

Overview of the workflow utilized in this manuscript. a We extracted known interactions from the STRING, DIP, and IntAct databases. b For each interaction we searched for conserved counterparts in the nine Cp strains and mapped the interaction in case an ICS score of 0.5625 or larger was achieved (c). The mapped interactions form the candidate networks for the Cp strains (d). For the further investigation we only used those interactions which are present in all nine strains (e) and extracted the top 15 % proteins with the highest interaction degree (f) as they are most likely to be essential proteins. Of the selected 181 proteins were 180 indeed present in the DEG database and furthermore, 41 of those have no homologous counterpart in the host organisms and thus are promising potential drug targets.
Data sources
The prediction of the PPI networks is based on the protein sequence similarity and the information of already known PPIs. The protein sequences were downloaded from NCBI and the known PPIs were retrieved from three publicly available databases (Table 2). The STRING database [54] is composed of known and predicted PPIs, including direct (physical) and indirect (functional) associations derived mainly from genomic context, high-throughput experiments, co-expression and computational prediction methods. The DIP database [53] contains experimentally determined PPIs that are automatically or manually curated. The IntAct [55] database consists of molecular interaction data derived from literature or direct submissions. The DIP and IntAct databases are curated by the IMEx (International Molecular Exchange) consortium [92]. It is important to note that all databases may contain false-positives, i.e., report an interaction when in fact there is none. This might in particular be true for the largest of the databases, the STRING database. There have been several attempts to filter out false-positives from such databases, e.g., by means of integrating several scores with Bayesian methods [93] or by incorporating inter-species confirmation (i.e., regulations which have been experimentally confirmed in different species) [94]. In this work, the main focus is on the identification of essential proteins (i.e., proteins with a high node degree in the network) thus impact of a limited number of false-positives is reduced. We only employ interactions from the STRING database with a score of above 0.700 (i.e., high-confidence interactions) [95]. Only approximately 10 % of the interactions (around 29 million) are classified in the high or highest confidence categories.
The interolog mapping
In order to transfer the known interactions to the Cp strains, we employed the so-called interolog mapping which was already successfully utilized in several other studies [48–50] and essentially corresponds to the “generalized interolog mapping” method as described in Yu et al. [57].
The main assumption of the method is that if two interacting proteins (A and B) have respective orthologous proteins (A’ and B’) in another organism, the orthologous pair also interacts [96]. For the homology detection, we utilized NCBI BLAST [97]. As we are aiming to base our predictions on a wide basis, we employed an as generous E-value cut-off as computationally feasible. We set the E-value parameter to 1e−5 for proteins from DIP and IntAct databases and to 1e−9 for proteins from the STRING database due to its sheer size. We performed a reciprocal search, meaning each protein was used as subject in one run and as query in the other run (i.e., we search in both directions). For the remainder, we only consider protein pairs that yield a hit in both directions (reciprocal hits). For each reciprocal hit, we compute the prediction score (PS):
where A ' represents a protein of Cp and A the homologous protein of the interaction database, id(A ' → A) the percentage of matching letters in the pair-wise alignment of the sequences and cov(A ' → A) the length of the alignment compared to the protein length. Finally, we assigned an interaction conservation score (ICS) to each known interaction having homologous proteins in Cp:
We considered interactions with an ICS(AB) greater than 0.5625 (corresponds to at least 75 % identity and 75 % coverage on average) as conserved. This threshold was derived in a previous study [52] as described in the Background section. When redundant interactions were found (e.g., through a homologous interaction pair of a different organism), the one with highest ICS(AB) was used to compose the PPI network.
In silico PPI network validation
As a first sanity check of our potential PPI network, we aimed to show that the predicted networks show realistic and typical network properties. Therefore, we computed several network statistics and compared them to those of known biological networks. We utilized the Cytoscape [98] plugin NetworkAnalyzer [99] and calculated the shortest path [33, 62, 100], the degree distribution [28], the network topology and the Shapiro-Wilk normality test [59].
Furthermore, we investigated the inherent network structure by performing a cluster analysis. We employed Markov Clustering (MCL) [101] implemented in the Cytoscape plug-in ClusterMaker [102]. We used an inflation parameter of 3.0 for the clustering. To reinforce that these interactions do occur in Cp, a literature search was performed to verify the existence of these clusters in phylogenetically close organisms (Additional file 3).
Essential proteins
In Saccharomyces cerevisiae it was shown that the node degree of a protein (i.e., the number of interactions of that protein) is correlated with the lethality of removing that protein from the network [62, 63]. Nodes with a high node degree are called hubs, but a clear definition of what node degree should be regarded as “high” is missing [103]. Nevertheless, identifying nodes with a larger degree is a means for identifying essential proteins [104–106], since the knockout of hub proteins most likely cause a substantial disruption in the interaction network [85]. We decided to classify proteins as essential when they are among the top 15 % proteins with respect to the node degree, a threshold commonly used [103]. Next, to validate essential hub proteins, we searched for homologous sequences stored in DEG [64, 65] (v11.2, updated on July 3, 2015), a database of bacterial essential genes. For the homology detection we again employed BLAST with the following parameters: e-value = 1e−5, low complexity filter = false and matrix = BLOSUM62. We also aligned the essential proteins of Cp against the all proteins of the five host organisms Ovis aries (taxid: 9940), Capra hircus (taxid: 9925), Bos Taurus (taxid: 9913), Equus caballus (taixd: 9796), and Homo sapiens (taxid: 9606).
Abbreviations
- AUC:
-
Area under curve
- CLA:
-
Caseous lymphadenitis
- CMNR:
-
Corynebacterium Mycobacterium, Nocardia, Rhodococcus
- Cp:
-
Corynebacterium pseudotuberculosis
- ICS:
-
Interaction conservation score
- MCL:
-
Markov clustering
- PPI:
-
Protein-protein interactions
- PS:
-
Prediction score
References
Butler W, Ahearn D, Kilburn J. High-performance liquid chromatography of mycolic acids as a tool in the identification of Corynebacterium, Nocardia, Rhodococcus, and Mycobacterium species. J Clin Microbiol. 1986;23(1):182–5.
Selim S. Oedematous skin disease of buffalo in Egypt. J Vet Med B. 2001;48(4):241–58.
Songer JG, Beckenbach K, Marshall MM, Olson GB, Kelley L. Biochemical and genetic characterization of Corynebacterium pseudotuberculosis. Am J Vet Res. 1988;49(2):223–6.
Ivanović S, Žutić M, Pavlović I, Žujović M. Caseous lymphadenitis in goats. Biotechnol Animal Husbandry. 2009;25(5-6-2):999–1007.
Hémond V, Rosenstingl S, Auriault M, Galanti M, Gatfosse M. Lymphadénite axillaire à Corynebacterium pseudotuberculosis chez une patiente de 63 ans. Med Mal Infect. 2009;39(2):136–9.
Oreiby A, Hegazy Y, Osman S, Ghanem Y, Al-Gaabary M. Caseous lymphadenitis in small ruminants in Egypt. Tierärztliche Praxis Großtiere. 2014;42(5):271–7.
Windsor PA. Control of caseous lymphadenitis. Vet Clin N Am Food Anim Pract. 2011;27(1):193–202.
Voigt K, Baird GJ, Munro F, Murray F, Brülisauer F. Eradication of caseous lymphadenitis under extensive management conditions on a Scottish hill farm. Small Rumin Res. 2012.
Hassan SS, Guimarães LC, de Pádua Pereira U, Islam A, Ali A, Bakhtiar SM, Ribeiro D, Dos Santos AR, de Castro Soares S, Dorella F. Complete genome sequence of Corynebacterium pseudotuberculosis biovar ovis strain P54B96 isolated from antelope in South Africa obtained by Rapid Next Generation Sequencing Technology. Stand Genomic Sci. 2012;7(2):189.
Ruiz JC, D'Afonseca V, Silva A, Ali A, Pinto AC, Santos AR, Rocha AAMC, Lopes DO, Dorella FA, Pacheco LGC. Evidence for reductive genome evolution and lateral acquisition of virulence functions in two Corynebacterium pseudotuberculosis strains. PLoS One. 2011;6(4), e18551.
Lopes T, Silva A, Thiago R, Carneiro A, Dorella FA, Rocha FS, dos Santos AR, Lima ARJ, Guimarães LC, Barbosa EG. Complete genome sequence of Corynebacterium pseudotuberculosis strain Cp267, isolated from a llama. J Bacteriol. 2012;194(13):3567–8.
Pethick FE, Lainson AF, Yaga R, Flockhart A, Smith DG, Donachie W, Cerdeira LT, Silva A, Bol E, Lopes TS. Complete genome sequences of Corynebacterium pseudotuberculosis strains 3/99-5 and 42/02-A, isolated from sheep in scotland and Australia, respectively. J Bacteriol. 2012;194(17):4736–7.
Trost E, Ott L, Schneider J, Schröder J, Jaenicke S, Goesmann A, Husemann P, Stoye J, Dorella FA, Rocha FS. The complete genome sequence of Corynebacterium pseudotuberculosis FRC41 isolated from a 12-year-old girl with necrotizing lymphadenitis reveals insights into gene-regulatory networks contributing to virulence. BMC Genomics. 2010;11(1):728.
Silva A, Schneider MPC, Cerdeira L, Barbosa MS, Ramos RTJ, Carneiro AR, Santos R, Lima M, D'Afonseca V, Almeida SS. Complete genome sequence of Corynebacterium pseudotuberculosis I19, a strain isolated from a cow in Israel with bovine mastitis. J Bacteriol. 2011;193(1):323–4.
Cerdeira LT, Pinto AC, Schneider MPC, de Almeida SS, Dos Santos AR, Barbosa EGV, Ali A, Barbosa MS, Carneiro AR, Ramos RTJ. Whole-genome sequence of Corynebacterium pseudotuberculosis PAT10 strain isolated from sheep in Patagonia, Argentina. J Bacteriol. 2011;193(22):6420–1.
Colom-Cadena A, Velarde R, Salinas J, Borge C, García-Bocanegra I, Serrano E, Gassó D, Bach E, Casas-Díaz E, López-Olvera JR. Management of a caseous lymphadenitis outbreak in a new Iberian ibex (Capra pyrenaica) stock reservoir. Acta Vet Scand. 2014;56(1):83.
Mira C, Fatima BK, Fadhela S, Kada K, Yacine T. Epidemiological and histopathological studies on Caseous Lymphadenitis in slaughtered goats in Algeria. Lung. 2014;6:26.25.
Jung BY, Lee S-H, Kim H-Y, Byun J-W, Shin D-H, Kim D, Kwak D. Serology and clinical relevance of Corynebacterium pseudotuberculosis in native Korean goats (Capra hircus coreanae). Tropl Anim Health Prod. 2015;47(4):657–61.
Osman AY, Abdullah FFJ, Chung ELT, Abba Y, Sadiq MA, Mohammed K, Lila MAM, Haron AW, Saharee AA. Caseous lymphadenitis in a goat: a case report. International Journal of Livestock Research. 2015;5(3):128–32.
Seyffert N, Guimarães A, Pacheco L, Portela R, Bastos B, Dorella F, Heinemann M, Lage A, Gouveia A, Meyer R. High seroprevalence of caseous lymphadenitis in Brazilian goat herds revealed by Corynebacterium pseudotuberculosis secreted proteins-based ELISA. Res Vet Sci. 2010;88(1):50–5.
Hariharan H, Tiwari K, Kumthekar S, Thomas D, Hegamin-Younger C, Edwards B, Sharma R. Serological detection of caseous lymphadenitis in sheep and goats using a commercial ELISA in Grenada, West Indies. 2014.
Baird GJ, Fontaine MC. Corynebacterium pseudotuberculosis and its role in Ovine Caseous Lymphadenitis. J Comp Pathol. 2007;137(4):179–210.
Dorella FA, Pacheco LGC, Oliveira SC, Miyoshi A, Azevedo V. Corynebacterium pseudotuberculosis: microbiology, biochemical properties, pathogenesis and molecular studies of virulence. Vet Res. 2006;37(2):201–18.
Santarosa BP, Dantas GN, Amorim RL, Chiacchio SB, Oliveira-Filho JP, Amorim RM, Ribeiro MG, Gonçalves RC. Meningoencefalite supurativa por Corynebacterium pseudotuberculosis em cabra com linfadenite caseosa: Relato de caso. Veterinária e Zootecnia. 2015;21(4):537–42.
Garma L, Mukherjee S, Mitra P, Zhang Y. How many protein-protein interactions types exist in nature? PLoS One. 2012;7(6), e38913.
Flórez A, Park D, Bhak J, Kim BC, Kuchinsky A, Morris J, Espinosa J, Muskus C. Protein network prediction and topological analysis in Leishmania major as a tool for drug target selection. BMC bioinformatics. 2010;11(1):484.
Sharan R, Suthram S, Kelley RM, Kuhn T, McCuine S, Uetz P, Sittler T, Karp RM, Ideker T. Conserved patterns of protein interaction in multiple species. Proc Natl Acad Sci U S A. 2005;102(6):1974–9.
Barabási AL, Oltvai ZN. Network biology: understanding the cell's functional organization. Nat Rev Genet. 2004;5(2):101–13.
Gonzalez MW, Kann MG. Protein interactions and disease. PLoS Comput Biol. 2012;8(12), e1002819.
Wetie N, Armand G, Sokolowska I, Woods AG, Roy U, Loo JA, Darie CC. Investigation of stable and transient protein–protein interactions: Past, present, and future. Proteomics. 2013.
Peng W, Wang J, Cai J, Chen L, Li M, Wu F-X. Improving protein function prediction using domain and protein complexes in PPI networks. BMC Syst Biol. 2014;8(1):35.
De Las Rivas J, Fontanillo C. Protein-protein interaction networks: unraveling the wiring of molecular machines within the cell. Brief Funct Genomics. 2012.
Wang J, Li M, Deng Y, Pan Y. Recent advances in clustering methods for protein interaction networks. BMC Genomics. 2010;11 Suppl 3:S10.
Braun P, Gingras AC. History of protein-protein interactions: from egg-white to complex networks. Proteomics. 2012;12(10):1478–98.
Zhang X, Xu J, Xiao W-x. A new method for the discovery of essential proteins. PLoS One. 2013;8(3), e58763.
Lu Y, Lu Y, Deng J, Peng H, Lu H, Lu LJ. A novel essential domain perspective for exploring gene essentiality. Bioinformatics. 2015;31(18):2921–9.
Cui T, He Z-G. Improved understanding of pathogenesis from protein interactions in Mycobacterium tuberculosis. Expert Rev Proteomics. 2014;11(6):745–55.
Wetie AGN, Sokolowska I, Woods AG, Roy U, Deinhardt K, Darie CC. Protein-protein interactions: switch from classical methods to proteomics and bioinformatics-based approaches. Cell Mol Life Sci. 2014;71(2):205–28.
Mulder NJ, Akinola RO, Mazandu GK, Rapanoel H. Using biological networks to improve our understanding of infectious diseases. Comput Struct Biotechnol J. 2014.
Li H, Kasam V, Tautermann CS, Seeliger D, Vaidehi N. A computational method to identify druggable binding sites that target protein-protein interactions. J Chem Inf Model. 2014;54(5):1391–400.
Li M, Zhang H, Wang J-x, Pan Y. A new essential protein discovery method based on the integration of protein-protein interaction and gene expression data. BMC Syst Biol. 2012;6(1):15.
Häuser R, Ceol A, Rajagopala SV, Mosca R, Siszler G, Wermke N, Sikorski P, Schwarz F, Schick M, Wuchty S. A second-generation protein–protein interaction network of helicobacter pylori. Mol Cell Proteomics. 2014;13(5):1318–29.
Lage K. Protein-protein interactions and genetic diseases: the interactome. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease. 2014;1842(10).
Mosca R, Pons T, Céol A, Valencia A, Aloy P. Towards a detailed atlas of protein–protein interactions. Curr Opin Struct Biol. 2013;23(6):929–40.
Zoraghi R, Reiner NE. Protein interaction networks as starting points to identify novel antimicrobial drug targets. Curr Opin Microbiol. 2013;16(5):566–72.
Baumbach J. On the power and limits of evolutionary conservation--unraveling bacterial gene regulatory networks. Nucleic Acids Res. 2010;38(22):7877–84.
Baumbach J, Apeltsin L. Linking Cytoscape and the corynebacterial reference database CoryneRegNet. BMC Genomics. 2008;9:184.
Liu Z-P, Wang J, Qiu Y-Q, Leung RK, Zhang X-S, Tsui SK, Chen L. Inferring a protein interaction map of Mycobacterium tuberculosis based on sequences and interologs. BMC bioinformatics. 2012;13 Suppl 7:S6.
Rezende AM, Folador EL, Resende DM, Ruiz JC. Computational prediction of protein-protein interactions in Leishmania predicted proteomes. PLoS One. 2012;7(12), e51304.
Lo Y, Huang S, Luo Y, Lin C, Yang J. Reconstructing genome-wide protein-protein interaction networks using multiple strategies with homologous mapping. PLoS One. 2015;10(1), e0116347.
Han YC, Song JM, Wang L, Shu CC, Guo J, Chen LL. Prediction and characterization of protein-protein interaction network in Bacillus licheniformis WX-02. Sci Rep. 2016;6:19486.
Folador EL, Hassan SS, Lemke N, Barh D, Silva A, Ferreira RS, Azevedo V. An improved interolog mapping-based computational prediction of protein-protein interactions with increased network coverage. Integr Biol. 2014;6(11):1080–7.
Xenarios I, Rice DW, Salwinski L, Baron MK, Marcotte EM, Eisenberg D. DIP: the database of interacting proteins. Nucleic Acids Res. 2000;28(1):289–91.
Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C. STRING v9. 1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41(D1):D808–15.
Hermjakob H, Montecchi-Palazzi L, Lewington C, Mudali S, Kerrien S, Orchard S, Vingron M, Roechert B, Roepstorff P, Valencia A. IntAct: an open source molecular interaction database. Nucleic Acids Res. 2004;32 suppl 1:D452–5.
Gong S, Yoon G, Jang I, Bolser D, Dafas P, Schroeder M, Choi H, Cho Y, Han K, Lee S, et al. PSIbase: a database of Protein Structural Interactome map (PSIMAP). Bioinformatics. 2005;21(10):2541–3.
Yu H, Luscombe NM, Lu HX, Zhu X, Xia Y, Han JD, Bertin N, Chung S, Vidal M, Gerstein M. Annotation transfer between genomes: protein-protein interologs and protein-DNA regulogs. Genome Res. 2004;14(6):1107–18.
Michel A, Koch-Koerfges A, Krumbach K, Brocker M, Bott M. Anaerobic growth of Corynebacterium glutamicum via mixed-acid fermentation. Appl Environ Microbiol. 2015;81(21):7496–508.
Shapiro SS, Wilk MB. An analysis of variance test for normality (complete samples). Biometrika. 1965;52(3/4):591–611.
Galeota E, Gravila C, Castiglione F, Bernaschi M, Cesareni G. The hierarchical organization of natural protein interaction networks confers self-organization properties on pseudocells. BMC Syst Biol. 2015;9 Suppl 3:S3.
Soares SC, Silva A, Trost E, Blom J, Ramos R, Carneiro A, Ali A, Santos AR, Pinto AC, Diniz C. The pan-genome of the animal pathogen Corynebacterium pseudotuberculosis reveals differences in genome plasticity between the biovar ovis and equi strains. PLoS One. 2013;8(1), e53818.
Hawoong J, Mason SP, Barabási A-L, Oltvai ZN. Lethality and centrality in protein networks. Nature. 2001;411(6833):41-2.
Estrada E. Virtual identification of essential proteins within the protein interaction network of yeast. Proteomics. 2006;6(1):35–40.
Luo H, Lin Y, Gao F, Zhang C-T, Zhang R. DEG 10, an update of the database of essential genes that includes both protein-coding genes and noncoding genomic elements. Nucleic Acids Res. 2014;42(D1):D574–80.
Zhang R, Ou HY, Zhang CT. DEG: a database of essential genes. Nucleic Acids Res. 2004;32 suppl 1:D271–2.
Eisen JA, Hanawalt PC. A phylogenomic study of DNA repair genes, proteins, and processes. Mutat Res/DNA Repair. 1999;435(3):171–213.
Caufield JH, Abreu M, Wimble C, Uetz P. Protein complexes in bacteria. PLoS Comput Biol. 2015;11(2):1-23. doi:10.1371/journal.pcbi.1004107.
Milse J, Petri K, Rückert C, Kalinowski J. Transcriptional response of Corynebacterium glutamicum ATCC 13032 to hydrogen peroxide stress and characterization of the OxyR regulon. J Biotechnol. 2014;190:40–54.
Park H-S, Um Y, Sim SJ, Lee SY, Woo HM. Transcriptomic analysis of Corynebacterium glutamicum in the response to the toxicity of furfural present in lignocellulosic hydrolysates. Process Biochem. 2014;50(3):347–56.
Pinto AC, de Sá PHCG, Ramos RT, Barbosa S, Barbosa HPM, Ribeiro AC, Silva WM, Rocha FS, Santana MP, de Paula Castro TL. Differential transcriptional profile of Corynebacterium pseudotuberculosis in response to abiotic stresses. BMC Genomics. 2014;15(1):14.
Sahbani SK, Girouard S, Cloutier P, Sanche L, Hunting DJ. The relative contributions of DNA strand breaks, base damage and clustered lesions to the loss of DNA functionality induced by ionizing radiation. Radiat Res. 2014;181(1):99–110.
Saito Y, Uraki F, Nakajima S, Asaeda A, Ono K, Kubo K, Yamamoto K. Characterization of endonuclease III (nth) and endonuclease VIII (nei) mutants of Escherichia coli K-12. J Bacteriol. 1997;179(11):3783–5.
Serafini DM, Schellhorn HE. Endonuclease III and endonuclease IV protect Escherichia coli from the lethal and mutagenic effects of near-UV irradiation. Can J Microbiol. 1999;45(7):632–7.
Resende B, Rebelato A, D'Afonseca V, Santos A, Stutzman T, Azevedo V, Santos L, Miyoshi A, Lopes D. DNA repair in Corynebacterium model. Gene. 2011;482(1):1–7.
Zhou H, Gao S, Nguyen NN, Fan M, Jin J, Liu B, Zhao L, Xiong G, Tan M, Li S, Wong L. Stringent homology-based prediction of H. sapiens-M. tuberculosis H37Rv protein-protein interactions. Biol Direct. 2014;9(1):5. http://dx.doi.org/10.1186/1745-6150-9-5.
Mitsuhashi S. Current topics in the biotechnological production of essential amino acids, functional amino acids, and dipeptides. Curr Opin Biotechnol. 2014;26:38–44.
Merino E, Jensen RA, Yanofsky C. Evolution of bacterial trp operons and their regulation. Curr Opin Microbiol. 2008;11(2):78–86.
Hsu S-K, Lin L-L, Lo H-H, Hsu W-H. Mutational analysis of feedback inhibition and catalytic sites of prephenate dehydratase from Corynebacterium glutamicum. Arch Microbiol. 2004;181(3):237–44.
Ikeda M. Towards bacterial strains overproducing L-tryptophan and other aromatics by metabolic engineering. Appl Microbiol Biotechnol. 2006;69(6):615–26.
Köster W. ABC transporter-mediated uptake of iron, siderophores, heme and vitamin B 12. Res Microbiol. 2001;152(3):291–301.
Kunkle CA, Schmitt MP. Analysis of a DtxR-regulated iron transport and siderophore biosynthesis gene cluster in Corynebacterium diphtheriae. J Bacteriol. 2005;187(2):422–33.
Wandersman C, Delepelaire P. Bacterial iron sources: from siderophores to hemophores. Annu Rev Microbiol. 2004;58:611–47.
Allen CE, Schmitt MP. HtaA is an iron-regulated hemin binding protein involved in the utilization of heme iron in Corynebacterium diphtheriae. J Bacteriol. 2009;191(8):2638–48.
Allen CE, Schmitt MP. Novel hemin binding domains in the Corynebacterium diphtheriae HtaA protein interact with hemoglobin and are critical for heme iron utilization by HtaA. J Bacteriol. 2011;193(19):5374–85.
Han J-DJ, Bertin N, Hao T, Goldberg DS, Berriz GF, Zhang LV, Dupuy D, Walhout AJ, Cusick ME, Roth FP. Evidence for dynamically organized modularity in the yeast protein–protein interaction network. Nature. 2004;430(6995):88–93.
Villoutreix BO, Kuenemann MA, Poyet JL, Bruzzoni-Giovanelli H, Labbe C, Lagorce D, Sperandio O, Miteva MA. Drug-like protein-protein interaction modulators: challenges and opportunities for drug discovery and chemical biology. Mol Inf. 2014;33(6–7):414–37.
Gowthaman R, Lyskov S, Karanicolas J. DARC 2.0: improved docking and virtual screening at protein interaction sites. PLoS One. 2015;10(7):e0131612.
Bier D, Thiel P, Briels J, Ottmann C. Stabilization of protein-protein interactions in chemical biology and drug discovery. Prog Biophys Mol Biol. 2015;119(1):10–9.
Mora A, Donaldson IM. Effects of protein interaction data integration, representation and reliability on the use of network properties for drug target prediction. BMC bioinformatics. 2012;13(1):294.
Sheng C, Dong G, Miao Z, Zhang W, Wang W. State-of-the-art strategies for targeting protein–protein interactions by small-molecule inhibitors. Chem Soc Rev. 2015;44(22):8238–59.
Anh NH, Long VC, Phuong TM, Lam BT. Discovery of pathways in protein-protein interaction networks using a genetic algorithm. Data Knowl Eng. 2015;96:19–31.
Orchard S, Kerrien S, Abbani S, Aranda B, Bhate J, Bidwell S, Bridge A, Briganti L, Brinkman FS, Cesareni G. Protein interaction data curation: the International Molecular Exchange (IMEx) consortium. Nat Methods. 2012;9(4):345–50.
Lee I, Date SV, Adai AT, Marcotte EM. A probabilistic functional network of yeast genes. Science. 2004;306(5701):1555–8.
Rolland T, Tasan M, Charloteaux B, Pevzner SJ, Zhong Q, Sahni N, Yi S, Lemmens I, Fontanillo C, Mosca R, et al. A proteome-scale map of the human interactome network. Cell. 2014;159(5):1212–26.
von Mering C, Jensen LJ, Snel B, Hooper SD, Krupp M, Foglierini M, Jouffre N, Huynen MA, Bork P. STRING: known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. 2005;33(Database issue):D433–7.
Sahu SS, Weirick T, Kaundal R. Predicting genome-scale Arabidopsis-Pseudomonas syringae interactome using domain and interolog-based approaches. BMC bioinformatics. 2014;15 Suppl 11:S13.
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden T. BLAST+: architecture and applications. BMC bioinformatics. 2009;10(1):421.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
Assenov Y, Ramírez F, Schelhorn S-E, Lengauer T, Albrecht M. Computing topological parameters of biological networks. Bioinformatics. 2008;24(2):282–4.
Taylor IW, Wrana JL. Protein interaction networks in medicine and disease. Proteomics. 2012;12(10):1706–16.
Van Dongen S. A cluster algorithm for graphs. Report-Information systems. 2000;10:1–40.
Morris JH, Apeltsin L, Newman AM, Baumbach J, Wittkop T, Su G, Bader GD, Ferrin TE. clusterMaker: a multi-algorithm clustering plugin for Cytoscape. BMC Bioinf. 2011;12(1):436.
Delprato A. Topological and functional properties of the small GTPases protein interaction network. Plos one. 2012;7(9), e44882.
Betul K, Eric A. Experimental evolution of protein-protein interaction networks. Biochem J. 2013;453(3):311–9.
Tang Y, Li M, Wang J, Pan Y, Wu F-X. CytoNCA: a cytoscape plugin for centrality analysis and evaluation of protein interaction networks. Biosystems. 2014.
Khuri S, Wuchty S. Essentiality and centrality in protein interaction networks revisited. BMC Bioinf. 2015;16(1):109.
Acknowledgments
We acknowledge the support of the National Center for High Performance Computing (CENAPAD-MG).
Funding
This work was supported by Coordenação de Aperfeiçoamento de Pessoal de Ensino Superior (CAPES) and Conselho Nacional de Pesquisa (CNPq): The authors AS, RSF, WMS, PVSDC, and ELF receive support from CAPES; the authors VA, RSF, and ELF freceive support from the CNPq program.
Availability of data and materials
All necessary information is contained in the manuscript and the Supporting Material.
Authors’ contributions
Conceived and designed the experiments: ELF. Designed and modeled the database in Postgres DBMS: ELF. Developed routines in PL/PgSQL: ELF. Performed the experiments: ELF. Analyzed the data: ELF. Structured the paper: ELF, MG, RR, DB. Wrote the paper: ELF. Performed the clusters description: PVSDC, WMS. Performed the essential protein description ELF. Participated in revising the draft: ALL. Contributed materials/analysis tools/structure: JB, MG, RR, RSF, AS, DB and VA. All authors read and approved the final manuscript.
Authors’ information
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Figure S1.
Source organisms of the mapped interactions. (JPG 374 kb)
Additional file 2:
Shortest path and degree distribution analysis. Shortest Path analysis of the nine Corynebacterium pseudotuberculosis serovar ovis strains and Degree distribution analysis of the nine C. pseudotuberculosis serovar ovis strains. (PDF 1004 kb)
Additional file 3:
Protein complex analysis. Literature-based description of protein complexes found in the interaction network. (PDF 1723 kb)
Additional file 4:
List of 181 essential proteins. The amino acid sequence of hubs proteins was compared against bacterial proteins sequence from Database of Essential Genes (DEG). (TXT 62 kb)
Additional file 5: Figure S2.
Homology distribution of Cp essential proteins aligned against hosts. Dark green: proteins homologous to host; Yellow: Proteins with low identity against hosts (identity < 30 %). Dark red: non-host homologous proteins, proteins with low identity and low coverage alignment against hosts (identity x coverage < = 10 %). Dark blue: non-host homologous proteins, proteins with no alignment hits against O. aires and C. hircus. Light blue: non-host homologous proteins, proteins with no alignment hits against the five hosts. The alignment summary is depicted in Additional file 6. (JPG 318 kb)
Additional file 6:
Essential protein alignment against host. Blast alignment summary of 181 essential proteins of Corynebacteria pseudotuberculosis against the host. (TXT 65 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
{kind=link}
Cite this article
Folador, E.L., de Carvalho, P.V.S.D., Silva, W.M. et al. In silico identification of essential proteins in Corynebacterium pseudotuberculosis based on protein-protein interaction networks. BMC Syst Biol 10, 103 (2016). https://doi.org/10.1186/s12918-016-0346-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12918-016-0346-4