
Abstract
Background
Varicellovirus equidalpha1 (formerly Equid alphaherpesvirus 1, EqAHV-1) is among the most important viruses responsible for respiratory disease outbreaks among horses throughout the world. No reports to date have detailed the association between EqAHV-1 and respiratory disease among horses in China. This study described one such outbreak among a population of horses in north Xinjiang that occurred from April 2021 - May 2023.
Results
qPCR revealed that EqAHV-1 was detectable in all samples and this virus was identified as a possible source of respiratory disease, although a limited subset of these samples were also positive for EqAHV-2, EqAHV-4, and EqAHV-5. In total, three EqAHV-1 strains responsible for causing respiratory illness in horses were isolated successfully, and full-length ORF33 sequence comparisonsand phylogenetic analyses indicated that these isolates may have originated from EqAHV-1 strains detected in Yili horse abortions. ORF30 sequence data additionally suggested that these strains were neuropathic, as evidenced by the presence of a guanine residue at nucleotide position 2254 corresponding to the aspartic acid present at position 752 in the DNA polymerase encoded by this virus.
Conclusion
This study is the first report of an outbreak of respiratory disease among horses in China caused by EqAHV-1. ORF30 sequence characterization revealed that these EqAHV-1 strains harbored a neuropathogenic genotype. Given the detection of this virus in horses suffering from respiratory disease, concern is warranted with respect to this neuropathogenic EqAHV-1 outbreak.
Similar content being viewed by others

Background
Varicellovirus equidalpha1 formerly know as Equid alphaherpesvirus 1 is an Alphaherpesvirinae subfamily member of the Varicellovirus genus that represents an important cause of equine disease, frequently causing abortion, respiratory illness, neurological disease, and ocular disease [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20]. Given the potential severity of these symptoms, EqAHV-1 thus represents an important threat to the health of individual horses and to the global equine industry as a whole [2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20]. The EqAHV-1 genome is 150 kbp in length, encoding as many as 80 open reading frames (ORFs) [20,21,22]. ORF30 codes for the viral DNA polymerase, and the nucleotide present at position 2254 (A/G/C) is strongly, albeit not exclusively, associated with whether or not the EqAHV-1 strain is neuropathogenic [6,7,8,9,10,11,12,13,14,15,16,17,18,19]. ORF33 encodes the highly conserved envelope glycoprotein B (gB), which is often targeted when detecting EqAHV-1 [6, 16, 20,21,22].
An outbreak of acute respiratory disease with an incubation period of under 24 h was detected among Yili mares, thoroughbred stallions, and thoroughbred racehorses at the Hengxing Equestrian Club in Changji, northern Xinjiang, China in April 2021. This respiratory disease has since spread rapidly through breeding mares on other farms in cities throughout northern Xinjiang including Changji, Kelamayi, Shihezi, and Urumqi. An outbreak of acute respiratory illness additionally occurred during horse racing in Zhaosu County, Yining in northern Xinjiang in August 2022. In May of 2023, this same respiratory disease again spread among horses in Zhaosu County during breeding season. The morbidity rate of the respiratory diaease ranges from 50 to 70%. Symptoms of this disease reportedly include pyrexia (> 40 °C), cough, purulent nasal discharge, reduced appetite, and weight loss, but lack neurological signs. Following veterinary isolation in new premises, sick horses are treated with both intramuscular (Sulfamethoxydiazine Sodium and Astragalus polysaccharides) and intravenous (Sodium salicylate, Urotropine, and Calcium gluconate) injections, all horses recovered from this illness. At present, horses in Xinjiang do not undergo any routinely scheduled vaccination.
In the present study, there was no evidence that these horses were infected with the H3N8 equine influenza virus, which has been reported as a cause of respiratory disease in horses in China [23]. To identify viruses potentially responsible for these outbreaks of respiratory disease, nasal swabs from the majority of sick horses were collected and analyzed via PCR or qPCR to detect EqAHV-1, -4, -2, and − 5 in these samples. The ORF30 and ORF33 sequences from identified EqAHV-1 isolates were then subject to further characterization. This study is the first to have been conducted with a focus on EqAHV-1 isolates responsible for equine respiratory disease in China, will strongly promote EqAHV-1 vaccine development.
Materials and methods
Sample collection
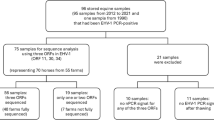
In April 2021, August 2022, and May 2023, 162 total nasal swabs were collected from symptomatic horses during respiratory disease outbreaks in Changji (n = 67), Shihezi (n = 12), Kelamayi (n = 11), Urumqi (n = 48), and Yining (n = 24) in north Xinjiang, China. An additional 416 nasal swabs were collected from clinically healthy control horses. Each of these sample was collected, and placed in a tube containing 1.5 mL of phosphate buffer and stored at -80 °C by the farm’s veterinarian according to the approved procedures.
EqAHV detection
Nasal swab samples were vortexed, centrifuged, and a 200 µL supernatant sample was used to extract viral nucleic acids using a kit (Geneaid Biotech Co.) based on provided instructions. The presence of EqAHV-1 DNA in these samples was further confirmed through a TaqMan-MGB qPCR assay targeting a portion of ORF68 using the primers and probes listed in Table 1. Target genes were amplified in qPCR assays using SuperReal PreMix (Tiangen Biotech Co.) with the following thermocycler settings: 95 °C for 15 min; 45 cycles of 94 °C 30 s and 60 °C 30 s. EqAHV-2 (716 nt), EqAHV-4 (587 nt), and EqAHV-5 (881 nt) DNA was additionally detected in these samples via PCR using primers targeting portions of ORF33 (EqAHV-4) or ORF8 (EqAHV-5 or EqAHV-2) encoding the gB in Table 1.
Isolation, screening, and electron microscopy of EqAHV-1
MDBK cells were used for the in vitro isolation of EqAHV-1 as reported previously [16]. Briefly, PCR-positive samples were centrifuged for 3 min at 12,000 x g, after which supernatants were passed through a 0.22 μm filter, used to inoculate MDBK cells for 2 h at 37 °C in a 5% CO2 incubator, and the inoculum was subsequently discarded and replaced with DMEM supplemented with 2% fetal bovine serum (FBS). Cells were then maintained for 72 h, followed by three rounds of freezing and thawing to harvest viruses, followed by repeated inoculation for six cell passages. Cytopathic effect (CPE) testing was then performed daily following inoculation, and the full-length OFR33 and partial ORF30 sequences of EqAHV-1 were amplified using the primers provided in Table 1. Positive PCR amplicons were then inserted into the pESI-T vector (Yeasen Biotech) and used for the transformation of E. coli DH5α Chemically Competent Cell (Weidi Biotech), after which three clones per amplicon were selected for Sanger sequencing (Sangon Biotech). For electron microscopy, EqAHV-1 particles purified by sucrose density gradient centrifugation as previously described [16] were negatively stained with 2% phosphotungstic acid.
Multiple sequence alignment and phylogenetic analyses
Additional information, including the GenBank accession numbers, for the sequences in this study is provided in Fig. 1. All full-length ORF33 and partial ORF30 EqAHV-1 nucleotide sequences isolated in this study were submitted to GenBank, with the following accession numbers: OQ886071-OQ886076. The MegAlign software was used to analyze these sequences in Lasergene v7.1, and a maximum-likelihood phylogenetic network of all target sequences was constructed in MEGA7 with the Tamura-Nei model. Tree topological accuracy was assessed with 1,000 bootstrap replicates [25].

A maximum-likelihood phylogenic network developed based on EqAHV-1 ORF33 gene sequences and the Tamura–Nei model (MEGA7). EqAHV-1 strains represented in the present study are represented with black circles
Results
EqAHV detection
Initial PCR testing confirmed that EqAHV-1, -2, -4, and − 5 were detectable in nasal swabs from horses suffering from respiratory disease, with respective positivity rates of 100% (162/162), 13.6% (22/162), 9.3% (15/162), and 9.3% (15/162). In contrast, the positivity rates in nasal swabs from healthy horses were 0% (0/416), 11.1% (46/416), 5.1% (21/416), and 16.6% (69/416), respectively. Seven sick horses were co-infected with all four of these EqAHV varieties, while three healthy horses were positive for co-infection with EqAHV-2, EqAHV-4, and EqAHV-5. These results thus suggested that EqAHV-1 was the potential causative pathogen responsible for these outbreaks of respiratory disease among horses in northern Xinjiang, China.
EqAHV-1 isolation
Next, viral isolation was performed using three nasal swabs collected from symptomatic horses that were only positive for EqAHV-1. Using MDBK cells as recipients, all three of these viral isolates exhibited clear CPEs by passage six, and PCR subsequently confirmed the successful isolation of three EqAHV-1 isolates (Zhaosu/2022, Changji/2021-4, and Changji/2021-7) after eight passages. Relative to control cells that underwent mock infection, MDBK cells that were infected with EqAHV-1 exhibited rounding, clustering, fusion, shedding, and void formation at 24 h post-infection. Transmission electron microscopy revealed that the viral particles in the EqAHV-1 Zhaosu/2022 isolate exhibited the typical morphological characteristics of EqAHV-1, with virions ~200 nm in diameter.
ORF33 and ORF30 sequences analyses
The full-length (2793 nt) ORF33 sequences from these three EqAHV-1 isolates were next compared, revealing respective nucleotide and amino acid similarity levels of 99.9–100% and 99.8–100%, (accession numbers: OQ886071-OQ886073), confirming the high degree of evolutionary conservation of this gene. These sequences were additionally compared to EqAHV-1 reference strains from China (accession numbers: MT063054, and MZ561483-MZ561525), the United Kingdom (V592: accession number. AY464052, Ab4: accession number. AY665713, and Suffolk/123/2005: accession number. KU206480), Japan (NY03: accession number. KF644569), the USA (T953_P210/2015: accession number. KR047045), India (Hisar-14/2014: accession number. MN912433), and Belgium (BE/21P43_BD5: accession number. MW855960). These results revealed DNA and amino acid sequence similarity levels ranging from 99.5 to 100% and 99–100% (Table 2), respectively. A phylogenetic network was then constructed based on these sequences, revealing the classification of the EqAHV-1 Zhaosu/2022 strain in one of six known clades [7], whereas EqAHV-1 isolates Changji/2021-4 and Changji/2021-7 formed a novel clade that was designated as cluster seven (Fig. 1).
To further explore the potential neuropathogenicity of these three EqAHV-1 isolates from horses with respiratory disease in China, a 559 nucleotide fragment of the EqAHV-1 ORF30 was next amplified using previously reported primers [7, 16]. These three partial ORF30 sequences exhibited 100% nucleotide and amino acid sequence identity (accession numbers: OQ886074-OQ886076), consistent with the high degree of genetic conservation. Consistent with the ZS01, ZS02, ZS05, ZS08, ZS10, ZS11, ZS16, ZS17, Ab4, and T953_P210 neuropathogenic EqAHV-1 reference strains, all three of these EqAHV-1 isolates harbored a G at position 2254 (D in position 752 of the viral DNA polymerase) (Fig. 2), suggesting that these strains represent neuropathogenic EqAHV-1 isolates.

ORF30 gene sequence analyses. (A) The G2254 polymorphism detected in all EqAHV-1 strains from this study. (B) The D752 polymorphism detected in all EqAHV-1 strains from this study. Dots represent the sequence identity when aligned with the EqAHV-1 Suffolk/123 isolate (CLC Sequence Viewer 8)
Discussion
Outbreaks of respiratory disease have a substantial negative impact on the global equine industry, resulting in the disruption of stallion mating, impaired training, the need for horses to bow out of competitions, and the high costs associated with quarantine/isolation and the treatment of infected horses [8,9,10,11,12,13]. Symptoms of the sick horses analyzed in this study included pyrexia, cough, purulent nasal discharge, reduced appetite, and weight loss, disrupting horse breeding, trading, training, and racing, thereby resulting in substantial economic losses for the equine industry in Xinjiang.
Multiple prior reports have identified equine influenza virus (EIV), EqAHV-1, EqAHV-4, equine rhinitis viruses A (ERAV) and B (ERBV), and equine arteritis virus (EAV) as the most common causes of respiratory illness among horses [11,12,13]. H3N8 EIV has previously been identified as being associated with respiratory illness among Chinese horses [23], but this virus was undetectable in nasal swabs from horses in north Xinjiang during the 2021–2023 outbreak discussed herein. Another recent publication focused on Xinjiang detected EqAHV-4, -2, and − 5 in nasal swabs from thoroughbred foals exhibiting respiratory symptoms [24]. In the present study, EqAHV-4, -2, and − 5 were respectively detected in 9.4%, 13.8%, and 9.4% of nasal swabs from horses with respiratory symptoms, but they were also detected in 5.1%, 11.1%, and 16.6% of nasal swabs from healthy horses, thus suggesting that these three viruses were not responsible for this outbreak of equine respiratory disease.
EqAHV-1 is a viral pathogen that has previously been demonstrated to cause respiratory illness, abortion, and stillbirth among horses [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20]. However, there have only been four publications from 2016 to 2022 indicating the presence of EqAHV-1 in aborted fetal lung tissue samples from mares in China. Three of these studies determined that EqAHV-1 was responsible for Yili horse abortions at the Chinese State Studs in Zhaosu County in 2015 [5] and 2021 [7, 16], while one detected this virus in aborted tissue samples from a Przewalski’s horse conservation center in Jimusaer County in 2019 [17]. No studies to date have documented a role for EqAHV-1 as a cause of respiratory disease among horses in China. The present qPCR data, however, confirmed that 100% of nasal swabs from symptomatic horses suffering from respiratory disease in the analyzed outbreaks were positive for EqAHV-1, whereas 0% of samples from healthy horses were positive for this virus.
These results suggested that EqAHV-1 is a strong candidate as the pathogen responsible for the high rates of respiratory disease among the horse population in north Xinjiang from 2021 to 2023. This study was the first report of the detection of EqAHV-1 in nasal swabs from horses in China, supporting the tentative identification of this virus as the cause of equine respiratory illness. Future molecular and epidemiological studies will be conducted with the goal of more broadly evaluating the relationship between EqAHV-1 and respiratory disease among the Chinese horse population. The EqAHV-1 isolates detected in the present study and a previous report [16] will additionally be leveraged by our team in an effort to design an attenuated EqAHV-1 vaccine that will be tested for its ability to prevent respiratory disease and abortion incidence among horses in China.
An outbreak of abortions caused by EqAHV-1 was detected among Yili mares at the Chinese State Studs in Zhaosu County in January 2021. The first clusters of respiratory disease reported in the present study occurred in April 2021 among Yili mares, thoroughbred stallions, and thoroughbred racehorses, followed by a second event in August 2022 during Yili horse racing, and a third outbreak of acute respiratory disease among Yili mares in May 2023. Subsequent epidemiological analyses suggested that the transportation of Yili horses across regions for the purposes of racing and breeding contributed to the spread of disease among farms in northern Xinjiang from 2021 to 2023.
Further comparisons of the full-length ORF33 sequences from the three EqAHV-1 isolates in the present study and reference EqAHV-1 strains revealed a high degree of similarity (99.5–100% DNA sequence similarity, 99–100% amino acid sequence similarity) among strains associated with disease in Yili horses (Table 2). The EqAHV-1 Zhaosu/2022 strain isolated in this study was clustered into one of six different clades constructed based on all EqAHV-1 sequences detected in Yili mare storm. Overall, these results suggested that the EqAHV-1 isolates responsible for the more recent outbreaks of respiratory disease may have originated from EqAHV-1 strains responsible for the incidence of abortion among Yili horses.
In prior studies, the nucleotide at position 2254 in ORF30 (A/G/C) has been closely tied to the incidence of neurological disease in animals infected with EqAHV-1 [6,7,8,9,10,11,12,13,14,15,16,17,18,19]. Most viral samples isolated from aborted fetuses, for example, are non-neuropathogenic strains harboring an A at this position which translates to an asparagine (N) at position 752 in the final protein [13]. Prior to 2019, as reported by Yang et al. [5] who detected the EqAHV-1-XJ2015 strain in aborted fetal lung tissue samples isolated from Yili horses and Hu et al. [17] who identified the YM2019 strain in aborted fetal lung tissue samples from Przewalski’s horses, these A2254 strains are then only EqAHV-1 isolates reported in aborted horses in China [15]. However, a report published by Tong et al. [6] identified G2254 strains that were associated with high rates of abortion among Yili horses in north Xinjiang in 2021. In line with their report, the EqAHV-1 isolates associated with equine respiratory disease in north Xinjiang in the present study harbored a G at position 2254 (D in position 752), and these isolates were thus classified as a neuropathogenic EqAHV-1 strain. These results thus strongly suggest that neuropathogenic EqAHV-1 can cause both abortions and respiratory disease among horses in China.
Many reports have previously noted that relative to neuropathogenic EqAHV-1 strains (G2254), non-neuropathogenic strains (A2254) are more closely associated with respiratory disease in horses [14, 18]. Four recent reports published in the USA and Europe identified a novel ORF30 EqAHV-1 DNA polymerase genotype (C2254/H752) closely associated with respiratory illness [9, 12, 13, 15]. In contrast, a neuropathogenic strain was isolated from horses with respiratory ailments in this study. Future reports will seek to explore the incidence of infections caused by the A2254 and C2254 genotypes of EqAHV-1 among horses in China with respiratory disease.
Conclusion
In conclusion, EqAHV-1 was herein detected in samples collected from horses suffering from respiratory symptoms such that it was identified as the likely cause of an outbreak of equine respiratory disease that occurred in north Xinjiang from 2021 to 2023. Subsequent characterization of these EqAHV-1 isolates suggested that the neuropathogenic EqAHV-1 isolates initially identified as drivers of respiratory disease may have also contributed to an increase in abortions affecting Yili horses. These results are expected to raise awareness regarding the potential for neuropathogenic EqAHV-1 to contribute to respiratory illness among horses in China, spurring efforts to design a vaccine that can reliably combat the spread and severity of the associated disease.
Data availability
All data generated or analyzed during this study are included in this published article and its additional files. Sequences of EqAHV-1 isolates in this study have been submitted to GenBank under accession numbers: OQ886071-OQ886076.
Abbreviations
- EqAHV:
-
Varicellovirus equidalpha1
- ORF:
-
Open reading frame
- G:
-
Guanidine
- A:
-
Adenine
- N:
-
Asparagine
- D:
-
Aspartic acid
- C:
-
Cytosine
- H:
-
Histidine
References
Gatherer D, Depledge DP, Hartley CA, Szpara ML, Vaz PK, Benkő M, et al. ICTV Virus Taxonomy Profile: Herpesviridae 2021. J Gen Virol. 2021;102(10):001673.
Smith KC, Blunden AS, Whitwell KE, Dunn KA, Wales AD. A survey of equine abortion, stillbirth and neonatal death in the UK from 1988 to 1997. Equine Vet J. 2003;35(5):496–501.
Laval K, Poelaert KCK, Van Cleemput J, Zhao J, Vandekerckhove AP, Gryspeerdt AC, et al. The pathogenesis and immune evasive mechanisms of equine herpesvirus type 1. Front Microbiol. 2021;12:662686.
Khusro A, Aarti C, Rivas-Caceres RR, Barbabosa-Pliego A. Equine herpesvirus-I infection in horses: recent updates on its pathogenicity, vaccination, and preventive management strategies. J Equine Vet Sci. 2020;87:102923.
Yang YL, Liu JH, Song HT, Li J, Lu YB, Hu Y, et al. Isolation and identification of equine herpesvirus 1 in Xinjiang. Chin J Prevent Vet Med. 2016;38(7):550–3. (In Chinese).
Fritsche AK, Borchers K. Detection of neuropathogenic strains of equid Herpesvirus 1 (EHV-1) associated with abortions in Germany. Vet Microbiol. 2011;147(1–2):176–80.
Tong PP, Duan RL, Palidan NRL, Deng HF, Duan LY, Ren ML, et al. Outbreak of neuropathogenic equid herpesvirus 1 causing abortions in Yili horses of Zhaosu, North Xinjiang, China. BMC Vet Res. 2022;18(1):83.
Garvey M, Lyons R, Hector RD, Walsh C, Arkins S, Cullinane A. Molecular characterisation of equine herpesvirus 1 isolates from cases of abortion, respiratory and neurological disease in Ireland between 1990 and 2017. Pathogens. 2019;8(1):7.
Sutton G, Thieulent C, Fortier C, Hue ES, Marcillaud-Pitel C, Pléau A, et al. Identification of a new equid herpesvirus 1 DNA polymerase (ORF30) genotype with the isolation of a C2254/H752 strain in French horses showing no major impact on the strain behaviour. Viruses. 2020;12(10):1160.
Wilcox A, Barnum S, Wademan C, Corbin R, Escobar E, Hodzic E, et al. Frequency of detection of respiratory pathogens in clinically healthy show horses following a multi-county outbreak of equine herpesvirus-1 myeloencephalopathy in California. Pathogens. 2022;11(10):1161.
Laing G, Christley R, Stringer A, Aklilu N, Ashine T, Newton R, et al. Respiratory disease and sero-epidemiology of respiratory pathogens in the working horses of Ethiopia. Equine Vet J. 2018;50(6):793–9.
Pusterla N, James K, Barnum S, Bain F, Barnett DC, Chappell D, et al. Frequency of detection and prevalence factors associated with common respiratory pathogens in equids with acute onset of fever and/or respiratory signs (2008–2021). Pathogens. 2022;11(7):759.
Pusterla N, Barnum S, Lawton K, Wademan C, Corbin R, Hodzic E. Investigation of the EHV-1 genotype (N752, D752, and H752) in swabs collected from equids with respiratory and neurological disease and abortion from the United States (2019–2022). J Equine Vet Sci. 2023;123:104244.
Allen GP, Bolin DC, Bryant U, Carter CN, Giles RC, Harrison LR, et al. Prevalence of latent, neuropathogenic equine herpesvirus-1 in the Thoroughbred broodmare population of central Kentucky. Equine Vet J. 2008;40:105–10.
Pusterla N, Barnum S, Miller J, Varnell S, Dallap-Schaer B, Aceto H, et al. Investigation of an EHV-1 outbreak in the United States caused by a new H752 genotype. Pathogens. 2021;10(6):747.
Duan RL, Tong PP, Deng HF, Palidan NEL, Jia CY, Yang EH, et al. Isolation and identification of neuropathogenic equid herpesvirus 1 within group VIII. Chin J Prevent Vet Med. 2022;44(9):940–5. (In Chinese).
Hu Y, Jia Q, Liu J, Sun W, Bao Z, Che C, et al. Molecular characteristics and pathogenicity of an equid alphaherpesvirus 1 strain isolated in China. Virus Genes. 2022;58(4):284–93.
Preziuso S, Sgorbini M, Marmorini P, Cuteri V. Equid Alphaherpesvirus 1 from Italian horses: evaluation of the variability of the ORF30, ORF33, ORF34 and ORF68 genes. Viruses. 2019;11(9):851.
Bueno I, Pearce P, Dunowska M. Frequency of latent equine herpesvirus type-1 infection among a sample of horses in the central north island of New Zealand. N Z Vet J. 2020;68(1):23–30.
Patel JR, Heldens J. Equine herpesviruses 1 (EHV-1) and 4 (EHV-4)--epidemiology, disease and immunoprophylaxis: a brief review. Vet J. 2005;170(1):14–23.
Burton EA, Wechuck JB, Wendell SK, Goins WF, Fink DJ, Glorioso JC. Multiple applications for replication-defective herpes simplex virus vectors. Stem Cells. 2001;19(5):358–77.
Telford EA, Watson MS, McBride K, Davison AJ. The DNA sequence of equine herpesvirus-1. Virology. 1992;189(1):304–16.
Zhu C, Li Q, Guo W, Lu G, Yin X, Qi T, et al. Complete genomic sequences of an H3N8 equine influenza virus strain isolated in China. Genome Announc. 2013;1(4):e00654–13.
Xie JX, Tong PP, Zhang L, Ren ML, Song XZ, Jia CY, et al. First detection and genetic characterization of equid herpesvirus 4, 2 and 5 in China. Arch Virol. 2021;166(5):1421–6.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33(7):1870–4.
Acknowledgements
Not applicable.
Funding
This study was supported by a National Natural Science Foundation of China (32060808), The Natural Science Foundation of Xinjiang Uyghur Autonomous Region (2019D01A47, 2022D01A167), China Postdoctoral Science Foundation (2019M653901XB), Post-doctoral Science Foundation of Xinjiang Agricultural University (XJAU20180723) and Xinjiang Uygur Autonomous Region High-Level Talent Introduction grants (XJ20171123).
Author information
Authors and Affiliations
Contributions
P.T. and J.X. performed the research, analyzed the data, and drafted the manuscript. B.L., Y.S., N.P. contributed to the collection of samples. E.Y., S.T., J.P., Y.D., C.J., L.K. contributed to the detection of PCR. P.T. and J.X. revised the manuscript. J.X. conceived the study, carried out additional analyses and finalized the manuscript. All authors contributed to the revising of the manuscript. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All experimental procedures involving animals were approved by the Animal Care and Use Committee of Xinjiang Agricultural University, Urumqi, Xinjiang, China under animal protocol number: 2020012 and performed according to the Animal Ethics Procedures and Guide-lines of the Ministry of Agriculture of China. The owners gave informed written consent for the nasal swab samples to be included in the study. The study was carried out in compliance with the ARRIVE guidelines.
Consent for publication
No applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Tong, P., Yang, E., Liu, B. et al. Identification of neuropathogenic Varicellovirus equidalpha1 as a potential cause of respiratory disease outbreaks among horses in North Xinjiang, China, from 2021-2023. BMC Vet Res 20, 77 (2024). https://doi.org/10.1186/s12917-024-03925-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12917-024-03925-z