
Abstract
Background
Enterocytozoon bieneusi is a zoonotic pathogen widely distributed in animals and humans. It can cause diarrhea and even death in immunocompromised hosts. Approximately 800 internal transcribed spacer (ITS) genotypes have been identified in E. bieneusi. Farmed foxes and raccoon dogs are closely associated to humans and might be the reservoir of E. bieneusi which is known to have zoonotic potential. However, there are only a few studies about E. bieneusi genotype identification and epidemiological survey in foxes and raccoon dogs in Henan and Hebei province. Thus, the present study investigated the infection rates and genotypes of E. bieneusi in farmed foxes and raccoon dogs in the Henan and Hebei provinces.
Result
A total of 704 and 884 fecal specimens were collected from foxes and raccoon dogs, respectively. Nested PCR was conducted based on ITS of ribosomal RNA (rRNA), and then multilocus sequence typing (MLST) was conducted to analyze the genotypes. The result showed that infection rates of E. bieneusi in foxes and raccoon dogs were 18.32% and 5.54%, respectively. Ten E. bieneusi genotypes with zoonotic potential (NCF2, NCF3, D, EbpC, CHN-DC1, SCF2, CHN-F1, Type IV, BEB4, and BEB6) were identified in foxes and raccoon dogs. Totally 178 ITS-positive DNA specimens were identified from foxes and raccoon dogs and these specimens were then subjected to MLST analysis. In the MLST analysis, 12, 2, 7 and 8 genotypes were identified in at the mini-/ micro-satellite loci MS1, MS3, MS4 and MS7, respectively. A total of 14 multilocus genotypes were generated using ClustalX 2.1 software. Overall, the present study evaluated the infection of E. bieneusi in foxes and raccoon dogs in the Henan and Hebei province, and investigated the zoonotic potential of the E. bieneusi in foxes and raccoon dogs.
Conclusions
These findings expand the geographic distribution information of E. bieneusi’ host in China and was helpful in preventing against the infection of E. bieneusi with zoonotic potential in foxes and raccoon dogs.
Similar content being viewed by others


Background
Microsporidia are obligate intracellular parasites with hosts ranging from protists to mammals [1]. More than 200 genera and approximately 1,500 species of microsporidia have been identified, and 17 species causes the infection of human beings. E. bieneusi is responsible for more than 90% cases of human microsporidiosis infection [1,2,3,4,5,6]. Since its discovery in an acquired immune-deficiency syndrome patient in 1985, many genotypes have been identified [7, 8]. E. bieneusi could induce diarrhea or even death of the patient, but most of the patients infected with E. bieneusi only showed slightly dysbiosis or disruption of nutrient absorption [2, 9, 10].
More than 800 genotypes of E. bieneusi have been identified using polymorphism analysis of the internal transcribed spacer (ITS) region of the rRNA gene which belongs to 13 phylogenetic groups [2, 4, 11, 12]. More than 310 genotypes are included in Group 1 which are believed to infect both human and animals. BEB4, BEB6, I, and J are the dominant genotype in Group 2 which are found in ruminants, non-ruminant animals, and humans. Genotypes in Group 3–13 infect animals and showed little effect on public health [13]. Different groups of E. bieneusi genotypes display diverse zoonotic potential and host specificity [4]. Group 1 (e.g., genotype D, Type IV, and EbpC) is the largest group of E. bieneusi genotypes and can infect different kinds of animals with high adaptability to the environment [4, 14]. Most of Group2 members of E. bieneusi, e.g., genotype I, J, BEB4, and BEB6 are the most common genotypes of E. bieneusi identified in sheep, goats, cattle, and deer [4, 6]. Most genotypes of E. bieneusi in groups 3–11 have a limited host range and thus pose a minor or unknown public health threat [4]. Nevertheless, the ITS genotyping method cannot fully reflect the genetic characteristics of E. bieneusi as it represents only a limited portion of the E. bieneusi genome (total length about 6 Mb) [15,16,17]. Multilocus sequence typing (MLST) is more discriminatory than ITS genotyping method by taking genetic polymorphisms of four mini- and microsatellites into account [8, 16, 18]. A higher genetic diversity was identified in E. bieneusi isolated from humans and animals using MLST analysis and several genetically isolating subgroups were formed within the ITS group 1 owing to their characteristics [19,20,21].
E. bieneusi can infect different animals and the zoonotic potential of E. bieneusi has been assessed in previous studies. Studies showed that the genotypes D, EbpC, and IV have considerable potential of cross-species infection due to their extremely broad host and geographic distribution [4]. Genotype D was first identified in raccoon dogs which raised the concerns regarding its potential for transmission to humans [22]. Other genotypes including CHN-DC1, WildBoar3, CHN-R1, NCF2, CHN-F1, NCR2, NCR1, Korea-WL1, Korea-WL2, Korea-WL3, Korea-D, CHG1, Peru8, Type IV, and EbpA, all belong to ITS group 1, were also found in raccoon dogs [11, 22,23,24,25,26,27]. Although the epidemiological investigation of E. bieneusi in foxes first began in 2003, it was not until 2014 when foxes were found to be infected with genotype D [28, 29]. Subsequently, genotypes CHN-F, EbpC, Type IV, Peru8, NCF1, NCF2, NCF3, NCF4, NCF5, NCF6, NCF7, CHN-DC1, SDF1, SDF2, Hum-q1, HND-1, and C, all belong to ITS group 1, were also identified in foxes [5, 22, 25, 27, 30, 31]. Previous studies showed that infection rate of E. bieneusi in raccoon dogs were 2.6–40.2% and were 7.7–30% in foxes [5, 11, 22,23,24, 26,27,28,29,30,31]. These findings suggest that E. bieneusi in raccoon dogs and foxes may be a source of E. bieneusi that causes the infection of humans.
However, there are only a few epidemiological studies on E. bieneusi in foxes and raccoon dogs in captivity worldwide. Thus, to further understand the genetic diversity of E. bieneusi in foxes and raccoon dogs, obtain geographic information, and compare the infection rates of E. bieneusi in different regions, the present study analyzed the infection rates and genotypes of E. bieneusi in farmed foxes and raccoon dogs in the Henan and Hebei provinces using MLST.
Results and discussion
As shown in Table 1, a total of 178 E. bieneusi-positive samples (11.21%, 95% CI: 9.66–12.76) were identified via nested PCR based on the ITS locus in 1588 fecal samples from foxes and raccoon dogs and the total infection rate of E. bieneusi was similar with the total infection rate of E. bieneusi in farmed blue foxes and raccoon dogs was 12.6% in the Heilongjiang and Jilin Province [27], while was higher relative to the total infection rate of farmed blue foxes and raccoon dogs in Xinjiang China (2.7%) [32]. This indicated that prevalence of E. bieneusi was associated with geographic distribution of the animals. The infection rates of E. bieneusi were 8.65% (84/971), 7.81% (21/269), and 20.98% (73/348) in samples from Xinxiang city, Hebi city, and Changli city. The infection rate in samples from Changli city was significantly higher than that from Xinxiang city and Hebi city (P < 0.01). The infection rate in foxes was 18.32% (129/704), which was significantly higher relative to that in raccoon dogs (5.54%, 49/884) (P < 0.01). The infection rate of E. bieneusi in foxes and raccoon dogs in the present study was similar with previous findings in which E. bieneusi was detected in 16.4% (18/110) farmed blue foxes and 4.1% (2/49) raccoon dogs [27]. The infection rate in pre-weaned foxes (3.51%, 95% CI: 17.84–24.70) was lower than that in young (20.00%, 95% CI: 7.04–32.96) and adult foxes (21.27%, 95% CI: 17.84–24.70). The lower infection rate in pre-weaned foxes observed in the present study might be associated with the immune status and the antibodies contained in the colostrum, but the mechanism should be elucidated further. The infection rate in male foxes was slightly lower than that in female foxes which was different from previous study in which they found no significant difference in the infection rate of E. bieneusi between male and female foxes [33]. The infection rate in male raccoon dogs (8.17%, 95% CI: 5.67–10.67) was higher than that in female raccoon dogs (2.63%, 95% CI: 1.09–4.16), and this finding is in line with the results of previous studies [24, 26]. The differences observed in the infection rate of E. bieneusi in foxes and raccoon dogs of different gender in the present study maybe associated with sample size, different animal husbandry practice and animal welfare. Study demonstrated that no effective therapeutic method was available for the treatment of E. bieneusi [34]. This might be the reason why no significant difference in the infection rate between dewormed (dewormed with Avermectin) and non-dewormed farm animals was observed in the present study.
As shown in Table 2, ten genotypes (NCF2, NCF3, D, EbpC, CHN-DC1, SCF2, CHN-F1, Type IV, BEB4, and BEB6) were identified by sequencing in the present study, among which genotype NCF2 was the dominant one, and all genotypes identified in the present study were zoonotic [34]. The genotypes NCF2, NCF3, D, CHN-DC1, and SCF2 has been identified in foxes previously [5, 22, 25, 27,28,29,30,31], but the genotypes SCF2, CHN-F1, and BEB6 were first identified in foxes in the present study. Raccoon dogs has been reported to be infected with genotypes NCF2, D, and Type IV previously [11, 22,23,24,25,26,27], but the genotypes NCF-3, EbpC, SCF-2, BEB4, and BEB6 were first identified in raccoon dogs in the present study (see Table 2). Among them, genotypes BEB4 and BEB6 belong to the ITS group 2 which have not been reported to be found in foxes and raccoon dogs. Previous studies identified genotype BEB4 in cattle, yaks, pigs, humans, and non-human primates [4], while genotype BEB6 was identified in cattle, sheep, goats, and humans [35, 36]. Thus, we hypothesized that genotypes BEB4 and BEB6 identified in foxes and raccoon dogs in the present study may be transmitted from cattle, because all the genotypes BEB4 and BEB6 identified in the present study were from the same farm which is close to a cattle farm. This transmission may be due to the contamination of the raw water by the feces of infected cattle in the farm nearby, but the prevalence of E. bieneusi in the cattle farm and raw water was not evaluated in the present study, therefore further study is still needed to clarify our hypothesis.
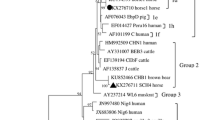
Phylogenetic analysis of the ITS loci showed that genotypes NCF2, NCF3, D, EbpC, CHN-DC1, SCF2, CHN-F1, and Type IV clustered into ITS group 1. Among them, genotypes EbpC, Type IV, and D are the most common genotypes of E. bieneusi that affect not only humans but also livestock and wild animals worldwide [4]. Although the genotypes BEB4 and BEB6 belong to ITS group 2 whose hosts are mostly ruminants, they may transmit to other hosts and lead to the infection of human beings [34]. Overall, these findings suggest that foxes and raccoon dogs may be potential sources of E. bieneusi infection in humans and other animals (Fig. 1).

Phylogenetic relationships among E. bieneusi isolates inferred with a neighbor-joining analysis based on the ITS nucleotide sequences. The reliability of cluster formation was assessed by the bootstrap analysis with 1000 replicates, and the values generated greater than 50% are shown beside the nodes. The known ITS genotypes identified in the present study are indicated by black triangles
At the MS1, MS3, MS4 and MS7 loci, 47 (43.93%), 74 (69.16%), 25 (23.36%) and 92 (85.98%) DNA specimens were amplified and sequenced successfully with 12, 2, 7, and 8 genotypes being identified, respectively. Eighteen multilocus genotypes (MLGs) were successfully amplified at all the five loci (ITS, MS1, MS3, MS4, and MS7), and 14 MLGs were formed (Table 3).
The phylogenetic analysis of microsatellite and microsatellite loci revealed that most of the E. bieneusi isolates from foxes and raccoon dogs were clustered together with the isolates from pigs and showed a close genetic match (Fig. 2). A few E. bieneusi isolates found in the present study were closest matched with the isolates from bear, Vicugna pacos, and squirrel and non from humans (Fig. 2), which is consistent with results of a previous study [18].

Phylogenetic relationships among E. bieneusi isolates inferred with a neighbor-joining analysis based on MS1, MS3, MS4 and MS7 locus, respectively. The reliability of cluster formation was assessed by the bootstrap analysis with 1000 replicates, and the values generated greater than 50% are shown beside the nodes. The types identified in the present study are indicated by black triangles
The findings of in the present study enrich the knowledge on the genetic diversity of E. bieneusi in foxes and raccoon dogs and performed the epidemiological investigation of E. bieneusi in foxes and raccoon dogs in the Henan Province and Hebei Province which has not been done in China. Currently, little information is available about the epidemiology of E. bieneusi in wild foxes and raccoon dogs; thus, wild species should be the focused in future studies.
Conclusion
In the present study, differences in the infection rates of E. bieneusi in foxes and raccoon dogs were assessed by region, breed, age, sex, and deworming condition. Ten zoonotic E. bieneusi genotypes (i.e., NCF2, NCF3, D, EbpC, CHN-DC1, SCF2, CHN-F1, Type IV, BEB4, and BEB6) were identified, and a total of 14 MLGs were formed. Findings of the present study are benefit for the control and prevention of E. bieneusi infection in foxes and raccoon dogs.
Methods
Sample collection
Fresh fecal samples were collected from the rectum of foxes and raccoon dogs using disposable chlorinated polyethylene (CPE) gloves. Then specimens were placed in an ice-cold container and transported to the laboratory immediately. Half of the fecal samples were stored at 4 °C for DNA extraction, and the remaining samples were soaked in 2.5% potassium dichromate and stored at -20℃. A total of 1588 samples were collected between June and December 2020 from eight farms in Henan and Hebei province and full name of the farms were listed in table S1 in the supplementary file. The detailed information regarding sample collection was presented in Table 1.
DNA extraction
Genomic DNA was extracted using the Stool DNA Kit (Omega Bio-Tek Inc., Norcross, GA, USA) according to the manufacturer’s instruction and the isolated DNA was stored at -20℃.
PCR amplification and MLST
Infection of E. bieneusi were evaluated by nest PCR assay based on ITS locus, and the primers used in the present study has been described in our previous study [37]. The ITS-positive samples were selected based on ITS genotype, region, breed, age, and sex, and then were subjected to MLST analysis at the MS1, MS3, MS4, and MS7 loci. The primers and annealing temperatures used in the MLST analysis of the present study were described previously [14]. The secondary PCR products were visualized by 1.5% agarose gel electrophoresis (containing 1 × 10− 5 DNA Green).
Sequencing and phylogenetic analysis
The ITS positive secondary PCR products were sent to SinoGenoMax Biotechnology Co., Ltd. (Beijing, China) for sequencing and sequences obtained were aligned with reference sequences downloaded from the GenBank (http://blast.ncbi.nlm.nih.gov) using Clustal X 2.13 (http://www.clustal.org/) to confirm different species or genotypes.
To determine the phylogenetic relationships among the detected genotypes, neighbor-joining trees were constructed using the MEGA VII program (www.megasoftware.net) based on evolutionary distances calculated with the Kimura 2-parameter model. The reliability of these trees was assessed via bootstrap analysis of 1000 replicates.
Statistical analysis
Significant differences in the prevalence of E. bieneusi among farmed foxes and raccoon dogs of different region, breed, age, sex, and deworming condition were analyzed using the chi-square test using SPSS version 26.0 (IBM Corporation, Armonk, NY, USA). Significant was defined at P < 0.05 and extremely significant defined at P < 0.01. The 95% confidence intervals (CIs) and odds ratios (ORs) were measured using SPSS version 26.0 (IBM Corporation, Armonk, NY, USA).
Data availability
The representative nucleotide sequences (ITS, MS1, MS3, MS4, and MS7) obtained in the present study are available in the [GenBank] repository, [https://submit.ncbi.nlm.nih.gov/]. The accession number of representative sequences are MW999206 - MW999220, MZ020581 - MZ020592, MZ020593 - MZ020594, MZ020595 - MZ020601, and MZ043847 - MZ043854, respectively.
Abbreviations
- ITS:
-
internal transcribed spacer
- MLST:
-
multilocus sequence typing
- MLG:
-
multilocus genotype
References
Stentiford GD, Becnel J, Weiss LM, Keeling PJ, Didier ES, Williams BP, Bjornson S, Kent ML, Freeman MA, Brown MJF, et al. Microsporidia - Emergent pathogens in the global food chain. Trends Parasitol. 2016;32(4):336–48.
Han B, Pan G, Weiss LM. Microsporidiosis in humans. Clin Microbiol Rev. 2021;34(4):e0001020.
Han Y, Gao H, Xu J, Luo J, Han B, Bao J, Pan G, Li T, Zhou Z. Innate and adaptive Immune responses against Microsporidia infection in mammals. Front Microbiol. 2020;11:1468.
Li W, Feng Y, Santin M. Host Specificity of Enterocytozoon Bieneusi and Public Health implications. Trends Parasitol. 2019;35(6):436–51.
Santín M, Calero-Bernal R, Carmena D, Mateo M, Balseiro A, Barral M, Lima Barbero JF, Habela M. Molecular characterization of Enterocytozoon Bieneusi in Wild carnivores in Spain. J Eukaryot Microbiol. 2018;65(4):468–74.
Thellier M, Breton J. Enterocytozoon Bieneusi in human and animals, focus on laboratory identification and molecular epidemiology. Parasite (Paris France). 2008;15(3):349–58.
Desportes I, Le Charpentier Y, Galian A, Bernard F, Cochand-Priollet B, Lavergne A, Ravisse P, Modigliani R. Occurrence of a new microsporidan: Enterocytozoon Bieneusi n.g., n. sp., in the enterocytes of a human patient with AIDS. J Protozoology. 1985;32(2):250–4.
Liu X, Wu Y, Yang F, Gong B, Jiang Y, Zhou K, Cao J, Zhang W, Liu A, Shen Y. Multilocus sequence typing of Enterocytozoon Bieneusi isolates from various Mammal and Bird species and Assessment of Population structure and substructure. Front Microbiol. 2020;11:1406.
Li W, Feng Y, Xiao L. Enterocytozoon Bieneusi. Trends Parasitol. 2022;38(1):95–6.
Zhong Z, Tian Y, Song Y, Deng L, Li J, Ren Z, Ma X, Gu X, He C, Geng Y, et al. Correction: molecular characterization and multi-locus genotypes of Enterocytozoon bieneusi from captive red kangaroos (Macropus Rfus) in Jiangsu province, China. PLoS ONE. 2017;12(12):e0190660.
Zhang Y, Xin L, Zhao A, Xu C, Wang T, Jing B, Qi M. Molecular detection and genotypes of Enterocytozoon Bieneusi in farmed mink (Neovison vison), blue foxes (Alopex lagopus), and raccoon dogs (Nyctereutes procyonoides) in Xinjiang, China. Int J Parasitol Parasites Wildl. 2021;14:211–5.
Koehler AV, Zhang Y, Gasser RB. A Perspective on the Molecular Identification, Classification, and Epidemiology of Enterocytozoon bieneusi of Animals. Experientia supplementum (2012) 2022, 114:389–415.
Tuo H, Zhang B, He Y, Zhao A, Zhang Z, Qi M, Yu F. Molecular characterization of Enterocytozoon bieneusi genotypes in wild Altai marmot (Marmota baibacina) in Xinjiang, China: host specificity and adaptation. Parasitol Res. 2023;123(1):7.
Li W, Feng Y, Zhang L, Xiao L. Potential impacts of host specificity on zoonotic or interspecies transmission of Enterocytozoon Bieneusi. Infect Genet Evol. 2019;75:104033.
Akiyoshi DE, Morrison HG, Lei S, Feng X, Zhang Q, Corradi N, Mayanja H, Tumwine JK, Keeling PJ, Weiss LM, et al. Genomic survey of the Non-cultivatable Opportunistic Human Pathogen, Enterocytozoon Bieneusi. PLoS Pathog. 2009;5(1):e1000261.
Feng Y, Li N, Dearen T, Lobo ML, Matos O, Cama V, Xiao L. Development of a multilocus sequence typing tool for high-resolution genotyping of Enterocytozoon Bieneusi. Appl Environ Microbiol. 2011;77(14):4822–8.
Widmer G, Akiyoshi DE. Host-specific segregation of ribosomal nucleotide sequence diversity in the microsporidian Enterocytozoon bieneusi. Infect Genet Evol. 2010;10(1):122–8.
Li W, Wan Q, Yu Q, Yang Y, Tao W, Jiang Y, Xiao L. Genetic variation of mini- and microsatellites and a clonal structure in Enterocytozoon Bieneusi population in foxes and raccoon dogs and population differentiation of the parasite between fur animals and humans. Parasitol Res. 2016;115(7):2899–904.
Karim MR, Wang R, He X, Zhang L, Li J, Rume FI, Dong H, Qi M, Jian F, Zhang S, et al. Multilocus sequence typing of Enterocytozoon Bieneusi in nonhuman primates in China. Vet Parasitol. 2014;200(1):13–23.
Li W, Cama V, Akinbo FO, Ganguly S, Kiulia NM, Zhang X, Xiao L. Multilocus sequence typing of Enterocytozoon bieneusi: lack of geographic segregation and existence of genetically isolated sub-populations. Infect Genet Evol. 2013;14:111–9.
Li W, Cama V, Feng Y, Gilman RH, Bern C, Zhang X, Xiao L. Population genetic analysis of Enterocytozoon Bieneusi in humans. Int J Parasitol. 2012;42(3):287–93.
Yang Y, Lin Y, Li Q, Zhang S, Tao W, Wan Q, Jiang Y, Li W. Widespread presence of human-pathogenic enterocytozoon bieneusi genotype D in farmed foxes (Vulpes vulpes) and raccoon dogs (Nyctereutes procyonoides) in China: first identification and zoonotic concern. Parasitol Res. 2015;114(11):4341–8.
Amer S, Kim S, Han JI, Na KJ. Prevalence and genotypes of Enterocytozoon bieneusi in wildlife in Korea: a public health concern. Parasites & Vectors. 2019;12(1):160.
Ma YY, Ma YT, Nie LB, Li TS, Peng JJ, Cong W, Zou Y, Zhu XQ. Prevalence and genotype distribution of Enterocytozoon Bieneusi in farmed raccoon dogs (Nyctereutes procyonoides) in Shandong Province, eastern China. Parasitol Res. 2020;119(6):1873–8.
Perec-Matysiak A, Leśniańska K, Buńkowska-Gawlik K, Merta D, Popiołek M, Hildebrand J. Zoonotic genotypes of Enterocytozoon Bieneusi in Wild Living Invasive and native carnivores in Poland. Pathogens (Basel Switzerland) 2021, 10(11).
Xu C, Ma X, Zhang H, Zhang XX, Zhao JP, Ba HX, Rui D, Xing XM, Wang QK, Zhao Q. Prevalence, risk factors and molecular characterization of Enterocytozoon bieneusi in raccoon dogs (Nyctereutes procyonoides) in five provinces of Northern China. Acta Trop. 2016;161:68–72.
Zhao W, Zhang W, Yang Z, Liu A, Zhang L, Yang F, Wang R, Ling H. Genotyping of Enterocytozoon Bieneusi in Farmed Blue Foxes (Alopex lagopus) and Raccoon Dogs (Nyctereutes procyonoides) in China. PLoS ONE. 2015;10(11):e0142611.
Galván-Díaz AL, Magnet A, Fenoy S, Henriques-Gil N, Haro M, Gordo FP, Millán J, Miró G, del Águila C, Izquierdo F. Microsporidia detection and genotyping study of human pathogenic E. bieneusi in animals from Spain. PLoS ONE. 2014;9(3):e92289.
Sulaiman IM, Fayer R, Lal AA, Trout JM, Schaefer FW 3rd, Xiao L. Molecular characterization of microsporidia indicates that wild mammals Harbor host-adapted Enterocytozoon spp. as well as human-pathogenic enterocytozoon bieneusi. Appl Environ Microbiol. 2003;69(8):4495–501.
Ma YY, Zou Y, Ma YT, Nie LB, Xie SC, Cong W, Xu QM, Zhu XQ. Molecular detection and genotype distribution of Enterocytozoon Bieneusi in farmed silver foxes (Vulpes vulpes) and arctic foxes (Vulpes lagopus) in Shandong Province, eastern China. Parasitol Res. 2020;119(1):321–6.
Zhang X-X, Cong W, Lou Z-L, Ma J-G, Zheng W-B, Yao Q-X, Zhao Q, Zhu X-Q. Prevalence, risk factors and multilocus genotyping of Enterocytozoon Bieneusi in farmed foxes (Vulpes lagopus), Northern China. Parasites & Vectors. 2016;9(1):72.
Zhang Y, Xin L, Zhao A, Xu C, Wang T, Jing B, Qi M. Molecular detection and genotypes of Enterocytozoon Bieneusi in farmed mink (Neovison vison), blue foxes (Alopex lagopus), and raccoon dogs (Nyctereutes procyonoides) in Xinjiang, China. Int J Parasitology: Parasites Wildl. 2021;14:211–5.
Zhang X-X, Cong W, Lou Z-L, Ma J-G, Zheng W-B, Yao Q-X, Zhao Q, Zhu X-Q. Prevalence, risk factors and multilocus genotyping of Enterocytozoon Bieneusi in farmed foxes (Vulpes lagopus), Northern China. Parasites & vectors 2016, 9.
Li W, Xiao L. Ecological and public health significance of Enterocytozoon Bieneusi. One Health (Amsterdam Netherlands). 2021;12:100209.
Karim MR, Rume FI, Li D, Li J, Zhang L. First molecular characterization of Enterocytozoon Bieneusi in children and calves in Bangladesh. Transbound Emerg Dis. 2022;69(4):1999–2007.
Taghipour A, Bahadory S, Javanmard E. The global molecular epidemiology of microsporidia infection in sheep and goats with focus on Enterocytozoon bieneusi: a systematic review and meta-analysis. Trop Med Health. 2021;49(1):66.
Li W, Diao R, Yang J, Xiao L, Lu Y, Li Y, Song M. High diversity of human-pathogenic Enterocytozoon bieneusi genotypes in swine in northeast China. Parasitol Res. 2014;113(3):1147–53.
Funding
This research was supported by the Natural Science Foundation of China (grant number 31402187), Young Backbone Teachers Project of Colleges and Universities in Henan Province (grant number 2018GGJS031), and Project of Tackling Key Problems in Science and Technology of Henan Province (grant number 92102110079). The sponsors played no role in the formulation of the study design; collection, analysis, or interpretation of the data; writing the report; or the decision to submit the article for publication.
Author information
Authors and Affiliations
Contributions
MC wrote the main manuscript. MC, HW, XL, YG, YL, LZ, GL, YS and BW collected the samples and conducted the laboratory analysis. HD, HD and LZ designed the experiment, analyzed the data and revised the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The protocol of the present study was reviewed and approved by the Research Ethics Committee of Henan Agricultural University (Zhengzhou City, China) which was performed in accordance with the Guidelines for Experimental Animals of the Ministry of Science and Technology (2006, Beijing, China). The permission has been obtained from their owners before beginning of the present study and informed consent to participate were also obtained from the animal owners. No animals were hurt during sample collection.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chen, M., Wang, H., Li, X. et al. Molecular epidemiology of Enterocytozoon bieneusi from foxes and raccoon dogs in the Henan and Hebei provinces in China. BMC Vet Res 20, 53 (2024). https://doi.org/10.1186/s12917-024-03883-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12917-024-03883-6