
Abstract
Background
Q fever and toxoplasmosis are economically important zoonoses as they cause considerable losses in livestock (cattle, sheep and goats) and wildlife (antelopes, giraffes, lions, and cheetahs) through reproductive disorders such as abortions and stillbirths. Q fever and toxoplasmosis testing in South Africa is conducted by the Agricultural Research Council-Onderstepoort Veterinary Research (ARC-OVR). However, both zoonoses are understudied and not monitored in South Africa as they are not considered controlled or notifiable diseases in the Animal Disease Act 35 of 1984. A retrospective study was conducted on Q fever (2007–2009) and toxoplasmosis (2007–2017) using diagnostic laboratory data at the ARC-OVR. Also, we report on sporadic abortion and stillbirth cases in livestock from diagnostic tissue samples submitted for Coxiella burnetii polymerase chain reaction (PCR) detection at the ARC-OVR.
Results
During 2007 to 2009, 766 animal samples were tested for C. burnetii antibodies and seropositivity was 0.9% (95%CI: 0.3–1.7) with sheep (1.9%; 95%CI: 0.6–4.4) having the highest seropositivity followed by cattle (0.7%; 95%CI: 0.09–2.6), while all goats (0.0%; 95%CI: 0.0–4.2) and wildlife (0.0%; 95%CI: 0.0–2.5) tested were negative. From 2007 to 2017, 567 sera were tested for T. gondii antibodies; overall seropositivity was 12.2% (95%CI: 9.6–15). Wildlife had highest seropositivity to T. gondii antibodies (13.9%; 95%CI: 9.0–19.7) followed by goats (12.9%; 95%CI: 9.2–17.4) and sheep (12.3%; 95%CI: 5.1–23.8) while seropositivity in cattle was 2.4% (95%CI: 0.06–12.9). Of 11 animals tested by C. burnetii PCR detection (2021–2022), 10 (91.0%) were positive. The amplicon sequences showed similarity to Coxiella burnetii strain 54T1 transposase gene partial coding sequence.
Conclusions
We have confirmed the occurrence of the causative agents of Q fever and toxoplasmosis in livestock and wildlife in South Africa, with data limitations. These zoonoses remain of importance with limited information about them in South Africa. This study provides baseline information for future studies on Q fever and toxoplasmosis in South African livestock and wildlife, as well other African countries. Due to limited data collection experienced in this study, it is recommended that improvements in data collection samples tested should include associated factors such as sex, age, and breed of the animals.
Similar content being viewed by others


Background
Q fever is distributed worldwide except in New Zealand and is caused by Coxiella burnetii [1]. Q fever causes congenital effects such as late abortions, stillbirths, and endometritis in infected animals, resulting in substantial economic losses [2]. For instance, Q fever outbreaks in the Netherlands caused agricultural losses of approximately 35,000 Euro per disability-adjusted life year (DALY), indicating the economic significance of the zoonosis [3]. The most common reservoirs of C. burnetii are cattle, sheep, and goats, while the bacterium can also infect rodents, cats, dogs, and arthropods [4]. The most common methods for Q fever serological testing are complement fixation test (CFT), enzyme-linked immunosorbent assay (ELISA) [5], indirect haemolysis test, and immunofluorescence assay (IFA) [6]. Previously, CFT was the gold standard for Q fever diagnosis. However, lately, ELISA and IFA have replaced CFT as preferred methods for Q fever serological testing in animals due to increased sensitivity and specificity [6].
Like Q fever, prevalence of toxoplasmosis in livestock and wildlife is important because the disease is considered a public health risk in humans from consumption of raw milk and improperly cooked meat from infected animals, also causing significant economic losses [7, 8]. In Britain and Uruguay, T. gondii infections caused US $ 5–15 million losses annually [9]. Toxoplasma gondii, the causative agent of toxoplasmosis, infects a wide range of warm-blooded animals, including livestock and wildlife. Infections by the protozoan cause congenital abnormalities, late abortions and fetal death in livestock after several replication cycles of the tachyzoites [10].
Previously, toxoplasmosis diagnosis was mainly based on bioassays and microscopy, but these methods lacked sensitivity and were considered laborious and time-consuming [11]. The Sabin-Feldman test proved to be more efficient and specific. However, this test required live tachyzoites, which posed occupational hazards to laboratory workers [11]. This method was followed by the development of ELISA for serological diagnosis of toxoplasmosis. However, ELISA required species-specific antigens which might be difficult to obtain [7]. The development of direct agglutination tests, such as the latex agglutination test (LAT) and modified agglutination test [12] that used formalin-killed tachyzoites instead of live ones was the breakthrough in the veterinary diagnosis of toxoplasmosis [13]. However, lately, these techniques have become less commercially available. This led to the production of recombinant toxoplasmosis antigens in several serological assays such as LAT and ELISA, which greatly improved the diagnosis of toxoplasmosis [7, 14].
In South Africa, various laboratories, including the Agricultural Research Council-Onderstepoort Veterinary Research (ARC-OVR), are designated facilities for serological testing of both Q fever and toxoplasmosis and consequently keep records. However, currently, both Q fever and toxoplasmosis are not regularly monitored. This is because both Q fever and toxoplasmosis are not recognized as controlled or notifiable diseases in the Animal Diseases Act of 1984 (ACT 35 1984) and the Animal Diseases Regulations; R.2026 of 1986 Government in Gazette No. 10469 of 26 September 1986 [15]This is despite scientific evidence that the two zoonoses may cause huge losses through late abortions and stillbirths in livestock and wildlife [2, 7]. This means that Q fever and toxoplasmosis infections in livestock and wildlife might occur unnoticed since no routine surveillance is conducted. Furthermore, studies on Q fever and toxoplasmosis are still limited, and there are no records of retrospective studies on Q fever and toxoplasmosis in livestock and wildlife in South Africa. Thus, the study aimed to determine the occurrence of C. burnetii (2007–2009) and T. gondii antibodies (2007–2017) in various provinces of South Africa by analyzing and reporting on Q fever and toxoplasmosis serological data in the ARC-OVR database. The study also reported on factors associated with seropositivity, such as the origin of samples, species and year of testing. We also analyzed diagnostic tissue samples submitted for C. burnetii PCR detection for possible sporadic abortion and stillbirth cases in livestock caused by C. burnetii infections.
Results
Demographic distribution of samples
For seropositivity to C. burnetii antibodies, between 2007 and 2009, 766 sera were tested using CFT (Fig. 1). Most of the sera submitted for Q fever testing were from cattle (35.5%), closely followed by sheep (34.3%), while the fewest samples were from goats (11.2%) as shown in Table 2. A large proportion of sera (42.7%) were tested for seropositivity to C. burnetii antibodies in 2008, with the least tested in 2009 (22.7%) and 2007 (21.6%), as demonstrated in Table 2. For toxoplasmosis, a large proportion of the sera tested was from goats (49.4%,) followed by wildlife (30.9%) while 2.4% of the sera were from other species (Table 3 and Fig. 1).

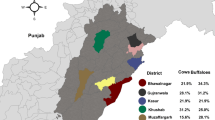
Map of South African provinces showing origins of sera tested for Q fever (2007–2009) and toxoplasmosis (2007- 2017) serology testing
The most frequent origin of diagnostic tissue samples (n=11 animals) for testing by C. burnetii PCR was the Eastern Cape (4/11), followed by Gauteng and North West Provinces (2/11) as shown in Table 1. Small ruminants (sheep and goats) accounted for most of animals tested for C. burnetii positivity by PCR (8/11) while cattle accounted for the rest. Most of the animals tested were due to abortions (9/11) while 2/11 were because of stillbirths (Table 1).
Seropositivity to C. burnetii and T. gondii antibodies and risk factor analysis
There were no significant differences in seropositivity to C. burnetii antibodies among species (p = 0.22) and provinces (p = 0.39), while the differences were substantial between years of testing (p = 0.001), being highest in 2007 (Table 2). There was no association between seropositivity to T. gondii antibodies and years of testing (p = 0.13), while there were significant differences between provinces (p < 0.001), with the highest odds of seropositivity in Limpopo, Western Cape and Free State (Table 3). Compared to cattle, which showed the lowest seroprevalence the likelihood of seropositivity to T. gondii antibodies was significantly higher in goats (p = 0.01) and sheep (p = 0.04).
PCR detection of C. burnetii
Detection of C. burnetii by IS1111 PCR showed that samples from 10/11 animals were positive (Fig. 2). Sequence analysis of the IS1111 PCR products revealed sequence similarity with C. burnetii strain 54T1 transposase gene partial coding sequence (MT268532.1) (Table 4).

Detection of C. burnetii in diagnostic tissue sample by IS1111 PCR. The first lane is Quick-Load® 100 bp DNA Ladder (New England Biolabs, Massachusetts, USA). The Coxiella gene fragment (gblock) from Integrated DNA Technologies (IDT, Iowa, USA) was used as template positive control while distilled water used as template negative control in the reaction. The blank is an empty unloaded lane. Samples 2271(a), 2271(b), 5269, 5030, 12,072(a), 12,072(b), 4234, 4322(a), 4322(b), 4322(c), 4322(d), 4460(a), 4460(b), 6450, 6451(a) and 6451(b)are diagnostic tissue samples submitted for C. burnetii PCR detection at Bacterial PCR laboratory, ARC-OVR
Discussion and conclusions
In this study, we obtained and analyzed data from ARC-OVR laboratories to establish the occurrence of the causal agents of both Q fever and toxoplasmosis in South Africa. Currently, in South Africa, both zoonoses are not regarded as notifiable or controlled diseases; therefore, there is no continuous surveillance for these zoonoses [15]. However, in other countries such as the USA, Q fever testing is a requirement for the export and import of livestock such as cattle and sheep [16], while it is not the case in South African livestock and wildlife. This is the first retrospective report on Q fever in South Africa. Seroprevalence of Q fever was reported by [17] to be 7.8% in cattle in the then Transvaal, now Gauteng province, while [18] and [17] reported Q fever seroprevalences in Mpumalanga and Gauteng provinces respectively, providing evidence of C. burnetii infections in South Africa. Despite this evidence, information on Q fever remains limited, considering that only ARC-OVR in South Africa was conducting serological testing which was stopped in 2009. Moreover, the few samples submitted for Q fever serology in this study between 2007 and 2009 indicate that the disease is not considered significant in South Africa. Also, the limited or lack of information about the reasons for submitting samples for Q fever testing further shows that there is limited knowledge on Q fever in South African livestock. Furthermore, there is scientific evidence that Q fever causes significant losses in livestock and wildlife resulting in substantial economic losses [18, 19]. Q fever was first reported in humans in South Africa in 1950 [20]. Lately, [21, 22] it is recommended that the occurrence of Q fever be continuously monitored and the relevant data accurately recorded in the testing laboratories to better understand the status of the disease in the country. The provision of an accurate database on Q fever will facilitate the decision-making process on the potential continuous surveillance of the zoonosis in South Africa.
Like Q fever, toxoplasmosis is not continuously monitored in South Africa despite evidence that the disease is present in the country. There are few reports on toxoplasmosis in the country, dating back to 2007 when [23] reported a seroprevalence of 5.6% in sheep and 37.0% in cats in 2015 [24]. Recently, [25] reported a seroprevalence of 32.6% in cattle sampled in Mpumalanga province. Moreover, between 2007 and 2017, only 567 animal samples were submitted to ARC-OVR for testing, further showing that the zoonosis is not considered significant in South Africa as only serological testing is conducted. However because toxoplasmosis is not listed as a notifiable or controlled disease in South Africa, there is no continuous surveillance. Also limited knowledge about the disease to due limited studies on toxoplasmosis might be the cause of the low flow of samples. Thus this study seeks to create awareness about the existence and toxoplasmosis in South Africa and consequences of infections. There is also evidence that toxoplasmosis infections may cause congenital disorders such as abortions in infected animals, resulting in significant economic losses in livestock and wildlife sector [26]. On the basis of the current results only, we cannot recommend that toxoplasmosis be included in the notifiable and controlled disease register in South Africa. More studies are required. However, continuous surveillance and record-keeping is required across different laboratories in South Africa so that disease is routinely monitored.
In the study, we detected C. burnetii in 10/11 animals tested by IS1111 PCR. The high detection frequency in the present study may be because samples submitted are from animals that displayed possible clinical symptoms of C. burnetii infection, such as abortions and stillbirths [2] and may not reflect the true PCR prevalence of the disease. However, the PCR data confirms the presence of C. burnetii in various parts of South Africa, particularly in North West, Kwazulu-Natal, and Eastern Cape provinces where this is the first report on the zoonosis in South African livestock and wildlife. Another study by [27] also reported PCR C. burnetii frequency of detection from aborted materials from livestock in Iran, suggesting that C. burnetii PCR detection may be multifaceted and affected by risk factors such as location or origin of specimen. Other studies have reported similar findings elsewhere. For instance, [28] reported C. burnetii PCR detection in aborted goat material and cattle. Moreover, [27] also reported a C. burnetii detection (100.0%) in goat abortion material, cattle and sheep (21.3%), in Iran, which is consistent with observations in the current study of the samples that tested positive by Coxiella IS1111 PCR. Most tissues tested in the study were from goats and sheep as compared to cattle and mainly due to abortion cases as compared to cattle which were stillbirth cases. This finding may suggest that C. burnetii infections may be responsible for abortion cases in small ruminants such as goats and sheep as compared to cattle as previously observed by [17] in the Free State province. Moreover, South Africa still has many undiagnosed abortion and stillbirth cases, caused possibly by C. burnetii infections. This is because state authorities usually focus on controlled diseases like brucellosis and chlamydiosis in cases of abortions, until recently where samples from some abortion cases are also submitted for C. burnetii PCR detection with positive results. This may reflect that although Q fever is not yet considered a notifiable or controlled disease in South Africa, there is progress in the knowledge of the disease. However, more studies need to be conducted. Although this was a national study from all nine provinces of South Africa, it is based on unrelated past and current data and the total number of animals is limited, which is one of the limitations of the study.
The study demonstrated the presence of the causative agents of both Q fever and toxoplasmosis in South Africa, laying a foundation for more studies on these zoonoses. Q fever and toxoplasmosis are important and should be regularly monitored. Proper record-keeping in various laboratories should be practised, and the records should be readily accessible. Diagnostic tissue samples were submitted for C. burnetii PCR detection because animals were showing clinical symptoms such as abortions or stillbirths. Thus, C. burnetii PCR detection frequencies in the current study do not reflect the true prevalence of the disease in the country; however, it confirms the existence of C. burnetii infections specifically in areas where this is the first report on the bacterium. This will pave the way for future in-depth epidemiological studies.
Materials and methods
Study design
The study design was to collect and analyze diagnostic laboratory data (DLD) from the ARC-OVR database of samples tested for seropositivity to C. burnetii and T. gondii antibodies and also to investigate sporadic abortion and stillbirth cases in livestock caused by C. burnetii using tissue samples submitted for C. burnetii PCR detection. Q fever and toxoplasmosis DLD were obtained from the ARC-OVR Bacterial Serology and Epidemiology, Parasites, and Vectors (EPV) laboratories, respectively. Animal samples tested included cattle, sheep, goats, and dogs. Antelope, giraffe, lion, and cheetah serum samples were grouped and referred to as wildlife. Due to low numbers, pigs (10), dogs (2), cats (1), and horses (1) were grouped together and collectively referred to as other species. Diagnostic tissue samples submitted for C. burnetii PCR detection were obtained from the Bacterial PCR laboratory at the ARC-OVR and analyzed for their places of origin, species, and tissues submitted as well as the reason(s) for testing (Table 1).
Study area
South Africa is situated on the southern tip of Africa and has an area of 1,221,037 km2. The country has nine provinces with approximately 59 million human population (Fig. 1). The ARC-OVR Bacterial Serology and EPV diagnostic laboratories are situated in Gauteng, the smallest province in South Africa.
Sampling
The samples were from farms, veterinary clinics, and provincial veterinary laboratories. The samples were submitted for testing for various reasons, including diagnostic, breeding, and screening, and to meet mandatory export requirements. The retrieved data consisted of tests conducted on sera, results of tests, origin of samples, year of sampling, and species. Other risk factors such as the age of animals, sex, and reasons for testing could not be obtained as the information was not included in the databases and the sample submission forms. Information on the exact origins of the samples in the form of postal codes or geographic coordinates was not available.
For PCR detection of C. burnetii, 20 diagnostic tissue samples from 11 animals submitted for C. burnetii PCR detection were obtained from the Bacterial PCR laboratory, ARC-OVR (2021–2022). These samples were composed of tissues from different species and provinces of South Africa and submitted for various reasons such as stillbirth and abortion cases, as described in Table 1.
Laboratory serology test data
Complement fixation test (CFT), which has relative sensitivity (Se) of 99.96% in cattle and specificity (Sp) of 99.94% relative to ELISA in ruminants, was used to test for C. burnetii antibodies. This method also has relative Se of 26.56% and Sp of 99.97% relative to ELISA in cattle [29]. Card agglutination test (CAT) which has relative sensitivity (Se) of 100% and specificity (Sp) of 94.3% was used to test for T. gondii Antibodies [30]. This technique was employed using the BIO-RAD PASTOREX™ TOXO 100 antibody test kit (BIO-RAD, California, USA) according to the manufacturer ‘s instructions. Positive and negative controls were supplied with the test kit. Briefly, a drop of positive control, negative control sera, and 15 µL of the sera to be examined were applied to different fields of the agglutination card without touching each other. A diluent drop was then applied to each area on the card, followed by the addition of the latex solution. The cards were then agitated for 5 min, and the results read. The formation of a green background with red aggregates indicated that the serum contained T. gondii antibodies while a brown homogenous suspension showed a negative result [31].
Statistical analysis
The data were collected and filtered using Microsoft Excel version 2016 and analyzed using Stata 15 (StataCorp, College Station, diagnostic laboratory data, USA). We assessed univariate associations of species, province of origin and year of sampling with Q fever and toxoplasmosis seropositivity using a two-tailed Fisher’s exact test. The same three variables were then included in multiple logistic regression models to adjust for confounding; however, multivariable analysis was not possible for Q fever seropositivity due to extensive collinearity. Model fit was assessed using the Hosmer–Lemeshow goodness-of-fit test.
PCR detection of C. burnetii
DNA extraction and PCR for detection of C. burnetii
PCR confirmation was conducted for all diagnostic tissue samples from 11 animals. The diagnostic tissue samples consisted of various tissue specimens from different species originating from other provinces of South Africa, as shown in Table 1. Tissue samples were cut into small pieces and 10 g from each sample in placed 10 mL ice-cold buffered phosphate saline (PBS) pH 7.4 in 50 mL bead ruptor homogenizing tubes containing 2.8 mm ceramic beads. The tissue samples were then homogenized using the automated BEAD RUPTOR ELITE Bead Mill homogenizer (Omni International, Georgia, USA). The tissues were homogenized at a speed of 3 m/s for 90 s. DNA extraction from the homogenates was conducted using the Qiagen DNeasy® blood and tissue kit as previously described [17]. The homogenates were centrifuged for 15 min at 4000 rpm, and 200 µL of the supernatant was transferred to 2 mL centrifuge tubes. To the tubes, 180 µL of tissue lysis (ATL) buffer and 20 µL of proteinase K were added, suspension vortexed, and incubated at 56 °C overnight. After overnight incubation, 200 µL of lysis (AL) buffer was added, and the suspension vortexed for 15 sand incubated at 70 °C for 10 min. Absolute ethanol (200 µL) was added to the mixture, vortexed, and transferred to DNeasy® spin columns. The columns were washed twice with wash buffers; AW1 and AW2, respectively. DNA was eluted from the columns with 200 µl of elution buffer (AE).
PCR for detection of C. burnetii in tissues (liver, spleen, kidney, placenta) was conducted in a 50 µL reaction targeting the multi-copy transposase gene in insertion element; IS1111 using primers 5’CGCAGCACGTCAAACCG3’ and 5’TATCTTTAACAGCGCTTGAACGTC3’ [4, 30]. The Coxiella gene fragment (gblock) from Integrated DNA Technologies (IDT, Iowa, USA) was used as template positive control while distilled water used as template negative control in the reaction. The reaction mixture contained 400 nM of each primer (IS1111F and IS1111R), 25 µL of the Amplicon 2 × Taq DNA polymerase Master Mix Red (Amplicon A/S, Odense, Denmark), and 10 µL of the extracted DNA. PCR amplification was conducted using BIO-RAD T100™ thermal cycler (BIO-RAD, California, USA). Cycling conditions consisted of initial denaturation at 95 °C for 15 min, 35 cycles of denaturation at 95 °C for 30 s, annealing at 60 °C for 30 s, and extension at 72 °C for 60 s for 35 cycles. The final extension was carried out at 72 °C for 10 min, and amplicons were visualized on a 1.5% w/v ethidium bromide-stained agarose gel with an expected size of 146 bp [32] estimated using Quick-Load® 100 bp DNA Ladder (New England Biolabs, Massachusetts, USA).
Sequence confirmation of C. burnetii
PCR confirmation of 16 tissues from 10/11 animals; 2271(a), 2271(b), 5030, 5269, 12,072(a), 12,072(b), 4243, 4322(a), 4322(b), 4322(c), 4460(a), 4460(b), 6450, 6451(a), 6451(b) and 2208 was conducted using Sanger sequencing. The IS1111 PCR products of the 16 tissues were sent to Inqaba Biotechnical Industries (Pty) Ltd (Pretoria, South Africa) for sequencing by Sanger. Both reverse and forward PCR primers were also used as sequencing primers Sequences were manually edited using the BioEdit Sequence alignment editor (version 7.2.5) and analyzed using the BLAST search online tool (http://www.ncbi.nlm.nih.gov/blast).
Study limitations
-
1.
Factors such as the age of animals, sex, and breed could not be determined as this information was also missing from the databases and sample submission forms.
-
2.
We did not investigate subclinical infections of C. burnetii and histopathological changes. This is because clients only submitted tissues for PCR detection of C. burnetii, and only pathogen DNA and not the disease was detected. However we confirmed the bacterium by sequencing. Some of the tissues are the ones that tested negative for other abortifacient pathogens such as brucellosis and Chlamydiosis.
-
3.
We did not rule out co-infections because we did not investigate or confirm the cause of abortions, only focusing on C. burnetii detection and confirmation by sequencing.
-
4.
Information about the reasons for submission of the animal samples for testing was not available in the database, and sample submission forms resulted in difficulties in establishing whether sampling was random or biased. This is because animal samples can be submitted for testing for various purposes such as symptoms, export, import, diagnostic, and screening, resulting in sample bias which would not be considered representative samples. Therefore, the results do not reflect true seropositivity to C. burnetii and T. gondii antibodies.
Availability of data and materials
In order to protect the privacy and confidentiality of clients who submitted the sera and tissues for Q fever and toxoplasmosis testing, has been de-identified.
Abbreviations
- ARC-OVR:
-
Onderstepoort Veterinary Research
- PCR:
-
Polymerase chain reaction
- IS1111 :
-
Insertion element of the transposase gene
- DALY:
-
Disability-adjusted life year
- C. burnetii :
-
Coxiella burnetii
- CFT:
-
Complement fixation test
- ELISA:
-
Enzyme-linked immunosorbent assay
- IFA:
-
Immunofluorescence assay
- T. gondii:
-
Toxoplasma gondii
- US $:
-
United States Dollar
- LAT:
-
Latex agglutination test
- CAT:
-
Card agglutination test
- PBS:
-
phosphate- buffered saline
- DLD:
-
Diagnostic laboratory data
- EPV:
-
Epidemiology, Parasites, and Vectors
- USA:
-
United States of America
- TX:
-
Texas
- DNA:
-
Deoxyribonucleic acid
- DALRRD:
-
Department of Agriculture, Land Reform and Rural Development
- AEC:
-
Animal Use Ethics Committee
- SA:
-
South Africa
References
Kittelberger R, Mars J, Wibberley G, Sting R, Henning K, Horner GW, et al. Comparison of the q-fever complement fixation test and two commercial enzyme-linked immunosorbent assays for the detection of serum antibodies against Coxiella burnetii (Q-fever) in ruminants: Recommendations for use of serological tests on imported animals in New Zealand. N Z Vet J. 2009;57(5):262–8.
Angelakis E, Raoult D. Q fever. Vet Microbiol. 2010;40:(3-4):297–309.
van Asseldonk MAPM, Prins J, Bergevoet RHM. Economic assessment of Q fever in the Netherlands. Prev Vet Med. 2013;112(1–2):27–34.
Arricau-Bouvery N, Rodolakis A. Is Q fever an emerging or re-emerging zoonosis? Vet Res. 2005;36(3):327–49.
Rahman MA, Alam MM, Islam MA, Bhuiyan AKFH, Rahman AKMA. Serological and Molecular Evidence of Q fever in Domestic Ruminants in Bangladesh. Vet Med Int. 2016;2016. https://doi.org/10.1155/2016/9098416.
Rousset E, Durand B, Berri M, Dufour P, Prigent M, Russo P, et al. Comparative diagnostic potential of three serological tests for abortive Q fever in goat herds. Vet Microbiol. 2007;124(3–4):286–97.
Wyrosdick HM, Schaefer JJ. Toxoplasma gondii: History and diagnostic test development. Anim Health Res Rev. 2015;16(2):150–62.
Cook AJC. Sources of toxoplasma infection in pregnant women: European multicentre case-control study Commentary: Congenital toxoplasmosis–-further thought for food. BMJ. 2000;321(7254):142–7.
Stelzer S, Basso W, Benavides Silván J, Ortega-Mora LM, Maksimov P, Gethmann J, et al. Toxoplasma gondii infection and toxoplasmosis in farm animals: Risk factors and economic impact, vol. 15. Food and Waterborne Parasitology: Elsevier Inc; 2019.
Maenz M, Schlüter D, Liesenfeld O, Schares G, Gross U, Pleyer U. Ocular toxoplasmosis past, present and new aspects of an old disease. Prog Retin Eye Res. 2014;39:77–106.
Liu Q, das Singla L, Zhou H. Vaccines against Toxoplasma gondii: Status, challenges and future directions. Hum Vaccin Immuno Ther. 2012;8(9):1305–8.
MacRì G, Sala M, Linder AM, Pettirossi N, Scarpulla M. Comparison of indirect fluorescent antibody test and modified agglutination test for detecting Toxoplasma gondii immunoglobulin G antibodies in dog and cat. Parasitol Res. 2009;105(1):35–40.
Su C, Khan A, Zhou P, Majumdar D, Ajzenberg D, Dardé ML, et al. Globally diverse Toxoplasma gondii isolates comprise six major clades originating from a small number of distinct ancestral lineages. Proc Natl Acad Sci U S A. 2012;109(15):5844–9.
Liu Q, Wang ZD, Huang SY, Zhu XQ. Diagnosis of toxoplasmosis and typing of Toxoplasma gondii. Parasit Vectors. 2015;8:1–4.
Blignaut B, van Heerden J, Reininghaus B, Fosgate GT, Heath L. Characterization of SAT2 foot-and-mouth disease 2013/2014 outbreak viruses at the wildlife–livestock interface in South Africa. Transbound Emerg Dis. 2020;67(4):1595–606.
Pavlin BI, Schloegel LM, Daszak P. Risk of importing zoonotic diseases through wildlife trade, United States. Emerg Infect Dis. 2009;15(11):1721–6.
Mangena M, Gcebe N, Pierneef R, Thompson PN, Adesiyun AA. Q fever: Seroprevalence, risk factors in slaughter livestock and genotypes of coxiella burnetii in South Africa. Pathogens. 2021;10(3):1–14.
van der Hoek W, Dijkstra F, Schimmer B, Schneeberger PM, Vellema P, Wijkmans C, et al. Deventer, the Netherlands 4. Municipal Health Service "Hart voor Brabant. Vol. 3, Municipal Health Service Brabant-Southeast. Available from: www.eurosurveillance.orghttp://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=19520
Honarmand H. Q fever: An old but still a poorly understood disease. Vol Interdiscip Perspect Infect Dis. 2012;2012. https://doi.org/10.1155/2012/131932.
Gear JHS, Wolstenholme B.* & Cort A. Q fever: Serological evidence of the occurrence of a case in South Africa. SAMJ. 1950;24(22):409–10.
de Boni L, Mall S, Msimang V, de Voux A, Rossouw J, Frean J. Exposure of South African Abattoir Workers to Coxiella burnetii. Trop Med Infect Dis. 2022;7(2):1–12.
Frean J. Q fever – an underappreciated occupational disease in South Africa. Occup Health South Afric. 2022;28(3):101–2.
Samra NA, Mccrindle ME, Penzhorn BL, Cenci-Goga B. Seroprevalence of toxoplasmosis in sheep in South Africa. J S Afr Vet Assoc. 2007;78(3):116–20.
Hammond-Aryee K, van Helden LS, van Helden PD. The prevalence of antibodies to Toxoplasma gondii in sheep in the Western Cape, South Africa. Onderstepoort J Vet Res. 2015;82(1):1–5.
Adesiyun AA, Knobel DL, Thompson PN, Wentzel J, Kolo FB, Kolo AO, et al. Sero-Epidemiological Study of Selected Zoonotic and Abortifacient Pathogens in Cattle at a Wildlife-Livestock Interface in South Africa. Vector Borne Zoonotic Dis. 2020;20(4):258–67.
Webster JP. Review of 'Toxoplasmosis of Animals and Humans' by J.P Dubey, J.P. (second edition). Parasit Vectors. 2010;3(1):112.
Mobarez AM, Khalili M, Mostafavi E, Esmaeili S. Molecular detection of Coxiella burnetii infection in aborted samples of domestic ruminants in Iran. PLoS One. 2021;16(4):e0250116
Pritchard GC, Smith RP, Errington J, Hannon S, Jones RM, Mearns R. Prevalence of Coxiella burnetii in livestock abortion material using PCR. Veterinary Record. 2011;169(15):391.
Herr S, Huchzermeyer HF, te Brugge LA, Williamson CC, Roos JA, Schiele GJ. The use of a single complement fixation test technique in bovine brucellosis, Johne’s disease, dourine, equine piroplasmosis and Q fever serology. Onderstepoort J Vet Res. 1985;52(4):279–82.
Jenum PA, Stray-Pedersen B, Melby KK, Kapperud G, Whitelaw A, Eskild A, et al. Incidence of Toxoplasma gondii infection in 35,940 pregnant women in Norway and pregnancy outcome for infected women. J Clin Microbiol. 1998;36(10):2900–6.
Parker SP, Cubitt WD. Modified latex agglutination test for antibodies to Toxoplasma gondii in eluates from Guthrie cards. J Clin Pathol. 1992;45(10):907–9.
de Bruin A, Tw Van Alphen P, Qj Van Der Plaats R, Nd De Heer L, Reusken CB, van Rotterdam BJ, et al. Molecular typing of Coxiella burnetii from animal and environmental matrices during Q fever epidemics in the Netherlands. 2012. Available from: http://www.biomedcentral.com/1746-6148/8/165
Acknowledgements
We thank Dr. Andrew Potts from ARC-OVR Serology and Mr. Brian Peba at the ARC-OVR EPV laboratories for providing access to Q fever and toxoplasmosis DLD records, respectively. We also thank Dr Rian Pierneef from the University of Pretoria for assistance with drawing of the map.
Funding
Financial support to conduct the study was obtained from the National Research Foundation (Grant # SFH150702122812), the Red Meat Research and Development in South Africa (RMRD-SA), CSIR IBS-HCD Bursary, and the Department of Trade, Industry and Competition (DTIC)-THRIP.
Author information
Authors and Affiliations
Contributions
Maruping Mangena conducted the experiments, wrote, and edited the manuscript. Nomakorinte Gcebe , Peter Thompson, and Abiodun Adesiyun equally conceptualized, wrote, edited, and reviewed the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Permission to conduct the study was obtained from the Department of Agriculture, Land Reform and Rural Development (DALRRD) in terms of the Animal Diseases Act 35 of 1984, Reference number 12/11/1/1/6. The study was approved by the University of Pretoria Animal Ethics Committee (V085-17). Ethical clearance to conduct the study was also obtained from the Agricultural Research Council-Onderstepoort Veterinary Research, Animal Use Ethics Committee (AEC 12.16). All experiments for Q fever and toxoplasmosis testing were carried out under Biosafety Level III conditions at the ARC-OVR campus. The experiments were conducted in a South Africa National Accreditations System (SANAS) accredited and Department of Agriculture, Rural Development and Land Reform (DALRDD), South Africa approved laboratories by SANAS accredited personnel. In addition, all methods were performed in accordance with the relevant guidelines and regulations. Consent for participation is not applicable in study.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Mangena, M.L., Gcebe, N., Thompson, P.N. et al. Q fever and toxoplasmosis in South African livestock and wildlife: a retrospective study on seropositivity, sporadic abortion, and stillbirth cases in livestock caused by Coxiella burnetii. BMC Vet Res 19, 168 (2023). https://doi.org/10.1186/s12917-023-03645-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12917-023-03645-w