
Abstract
Background
Canine morbillivirus (canine distemper virus, CDV) is a member of the Paramyxoviridae family. Canine distemper is a serious viral disease that affects many mammalian species, including members of the Mustelidae family. These animals have an elusive nature, which makes related virological studies extremely challenging. There is a significant knowledge gap about the evolution of their viruses and about the possible effects of these viruses to the population dynamics of the host animals. Spleen and lung tissue samples of 170 road-killed mustelids belonging to six species were collected between 1997 and 2022 throughout Hungary and tested for CDV with real-time RT-PCR.
Results
Three species were positive for viral RNA, 2 out of 64 Steppe polecats (Mustela eversmanii), 1 out of 36 European polecats (Mustela putorius) and 2 out of 36 stone martens (Martes foina); all 18 pine martens (Martes martes), 10 least weasels (Mustela nivalis) and 6 stoats (Mustela erminea) tested negative. The complete CDV genome was sequenced in five samples using pan-genotype CDV-specific, amplicon-based Nanopore sequencing. Based on the phylogenetic analysis, all five viral sequences were grouped to the Europe/South America 1 lineage and the distribution of one sequence among trees indicated recombination of the Hemagglutinin gene. We verified the recombination with SimPlot analysis.
Conclusions
This paper provides the first CDV genome sequences from Steppe polecats and additional complete genomes from European polecats and stone martens. The infected specimens of various species originated from distinct parts of the country over a long time, indicating a wide circulation of CDV among mustelids throughout Hungary. Considering the high virulence of CDV and the presence of the virus in these animals, we highlight the importance of conservation efforts for wild mustelids. In addition, we emphasize the importance of full genomic data acquisition and analysis to better understand the evolution of the virus. Since CDV is prone to recombination, specific genomic segment analyses may provide less representative evolutionary traits than using complete genome sequences.
Similar content being viewed by others
Background
Canine morbillivirus (canine distemper virus, CDV) is a single-stranded, negative-sense RNA virus that belongs to the Morbillivirus genus of the Paramyxoviridae family [1,2,3]. The length of the CDV genome is 15,690 nucleotides, and the genome encodes six structural proteins; two glycoproteins: hemagglutinin (H) and fusion (F) proteins, one envelope-associated matrix (M) protein, one nucleocapsid (N) protein, and two transcriptase-associated proteins: phosphoprotein (P) and a large polymerase (L) protein [3, 4]. Several distinct genotypes are known and classified according to different hosts and geographical areas based on nucleotide sequence analysis of the H gene [3, 5,6,7,8]. In Hungary, three different CDV genotypes (Europe, Arctic-like and European wildlife lineages) was described so far based on the H gene nucleotide sequences. In addition to dogs, CDV infection was detected in other carnivores, including the red fox and raccoon (Procyon lotor), Eurasian otter and ferret (Mustela furo) [9,10,11,12,13]. These genotypes have significantly different history, geographic distribution and known host range. Notably, all these CDV is a significant viral pathogen among wild and domesticated animals with a high mortality rate [14, 15]. The virus is primarily transmitted through bodily fluids, e.g., saliva, respiratory droplets, urine, and feces, including transmission due to direct contact [16]. Cross-species transmission occurs frequently, which may lead to conservation problems regarding vulnerable species [17].
Fatal CDV outbreaks are known to occur in wild populations of endangered species. In Africa, CDV caused outbreaks in a diverse range of wild mammals such as the lion (Panthera leo), African wild dog (Lycaon pictus) and Ethiopian wolf (Canis simensis) [14, 18,19,20]. In Asia, the virus poses a serious threat to the vulnerable Giant panda (Ailuropoda melanoleuca) and the endangered Amur tiger (Panthera tigris altaica) [21,22,23]. Additionally, in Europe, CDV infection was also reported in one of the most endangered felid species, the Iberian lynx (Lynx pardinus) [24]. In the case of mustelids, CDV infection was previously associated with a high mortality rate approaching 100% [25]. The most remarkable CDV outbreak in black-footed ferret (Mustela nigripes) population occurred in Wyoming, Western USA, seriously affecting a captive breeding program and leading to the extirpation of the species from the wild [26, 27]. A recent report from Spain investigated the CDV seroprevalence trends in association to the population size of the Critically Endangered European mink (Mustela lutreola). They found that CDV seroprevalence is an indicator for the population trend of these animals, supporting our hypothesis that CDV may be an important wildlife disease [28]. In Europe, CDV has been reported among multiple species to date, including the stone marten (Martes foina), pine marten (Martes martes), Eurasian badger (Meles meles), Eurasian otter (Lutra lutra), European mink (Mustela lutreola), European polecat (Mustela putorius) and the American mink (Mustela vison) [13, 29,30,31,32,33,34].
In Hungary, the Steppe polecat (Mustela eversmanii), least weasel (Mustela nivalis), stoat (Mustela erminea) and pine marten are protected species, the European polecat is periodically considered, and the stone marten is a legally hunted species throughout the year. The stone marten and the European polecat are common, habitat generalists [35, 36]; the least weasel and pine marten are relatively common, whereas the stoat and the Steppe polecat are rare species [37,38,39]. These mustelids belong to small mammal consumers and omnivorous trophic guilds. Frequent coexistence of up to 5–6 carnivore species and known killings among smaller related species [40] indicate interspecific encounters. These direct contacts may result in cross-infection.
Next generation sequencing (NGS) technologies are increasingly being used to detect and characterize pathogens in wildlife [41,42,43,44]. MinION (Oxford Nanopore Technologies, Oxford, UK) has been used in many areas of virology, for instance, metagenomics or sequencing of complete genomes [45,46,47,48]. Amplicon-based NGS sequencing of specific pathogens is a method for rapid detection and genomic characterization of target pathogens which may yield high-coverage genomic sequence information [12, 49,50,51,52]. With the aid of this technology, we can gain more knowledge about the complete viral genomes, like detection of recombination events.
Detection and investigation of viral diseases are important factors for conserving protected and rare species; however, the elusive nature of several mustelids hampers our understanding of their viruses. Monitoring road-killed animals is a general practice for population genetic studies on rare species [53, 54], but it also gives a good opportunity to get data about pathogens of these animals [13, 55]. Herein we present the results of a post-mortem retrospective surveillance study to detect CDV RNA among road-killed mustelids and perform complete genomic sequencing, phylogenetic and recombination analyses on these virus sequences.
Results
PCR screening
Canine morbillivirus RNA was detected in three out of the six investigated species: 2 positives out of 64 Steppe polecats, 1 positive out of 36 European polecats and 2 positives out of 36 stone martens. Samples screened from 18 pine martens, 10 least weasels and 6 stoats were negative. The European polecat detected in 2019 and the stone marten in 2020 originated from Western Hungary, both Steppe polecats (collected in 2018 and 2021) originated in Eastern Hungary, and the stone marten (sampled in 2017) was collected in Southern Hungary (Table 1). Two CDV test positive animals (a stone marten and a Steppe polecat) showed signs of bites on their bodies, which indicates combat with another carnivore (Table 1). As the sample collection efforts were not evenly distributed during the study period, CDV prevalence could not be estimated.
Sequencing and phylogenetic analysis
Complete genomes were successfully retrieved from all positive samples. Sequences were deposited in GenBank (accession numbers OP209185-OP209189). Based on the phylogenetic analysis of complete genomes, all these sequences belong to the Europe/South America 1 lineage (Fig. 1). The Hemagglutinin (H) gene sequence-based analysis confirmed this result (Fig. 2).

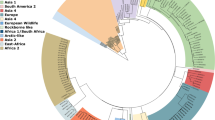
A Maximum likelihood phylogenetic tree based on 221 CDV complete genomes. Phocine distemper virus (PDV) (GenBank accession number: KY629928) was used as an outgroup to root the phylogenetic tree. The Europe/South America 1 lineage of interest is highlighted in blue. B Expanded portion of Europe/S Am 1 lineage. Dots represent sequences obtained in this study

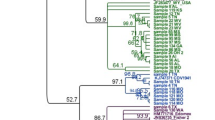
A Maximum Likelihood phylogenetic tree based on 969 complete Hemagglutinin (H) nucleotide sequences. Phocine distemper virus (PDV) (GenBank accession number: KY629928) was used as an outgroup to root the phylogenetic tree. The Europe/South America 1 lineage of interest is highlighted in blue. B Expanded portion of Europe/S Am 1 lineage. Dots represent sequences obtained in this study
Sequences are dispersed among two clusters within the Europe/South America 1 lineage, and both clusters are composed of sequences from Hungary. Based on complete genomes (Fig. 1), one cluster contains only mustelid sequences, whereas one Steppe polecat sample was grouped with red fox (Vulpes vulpes) samples in a separate clade. Based on the H gene phylogenetic tree, both Steppe polecat samples (OP209186) grouped with red fox samples on a distant clade (Fig. 2). The secondary analysis with RAxML plugin for Geneious supported the primary phylogenetic patter of the sequences, all main lineages and the novel sequences were positioned similarly (Supplementary material; Supplementary Figs. 2 and 3).
The distinct clustering pattern of OP209185 from a Steppe polecat on the phylogenetic trees (Fig. 1 and 2) indicates a recombination event in association with the Hemagglutinin genomic region. The SimPlot analysis confirmed the recombination of the Hemagglutinin gene with a closely related, Europe/South America 1 lineage strain. Also, it confirmed multiple additional recombination points in the genome (Fig. 3). The secondary analysis with DualBrothers plugin in Geneious also detected multiple recombination points throughout the genome with the same crossing-points (Supplementary material; Supplementary Fig. 4).

Recombination analysis of the canine distemper virus (OP209185): (A) Similarity Plot analysis of the complete genome sequences of OP209185 (Steppe polecat, Hungary, 2021) and its putative parents OP209186 (Steppe polecat, Hungary, 2018) and OP209187 (European polecat, Hungary, 2019). The OP209185 was used as the query. (B) Boot Scan analysis of OP209185 and its parent sequences. (C) Similarity Plot analysis of the H gene sequences of OP209185 and its putative parents. (D) Boot Scan analysis of OP209185 and its parent sequences in the H gene. A CDV isolate, HM046486 (Caspian lineage), was used as an outgroup in all analyses. The red vertical line represents the H gene segment region. The y-axis indicates the percentage of identity with a window size of 600 bp and a step size of 20 bp. The comparison was performed using 50% consensus sequences with 1000 bootstrap replicates
Discussion
We present the circulation of CDV throughout the country over several years, supporting the endemic nature of this virus among mustelids. An important finding of the current study is the detection of CDV in wild-living Steppe polecats. It is a rare and protected mammal species of our region and by using retrospective virus surveillance methods (i.e. without disturbance and invasive sampling of the animals), we were able to indicate the role of these animals in CDV transmission. Steppe polecat was already a suspected host for CDV [56]; however, due to its rareness and elusive nature, only a few molecular biological investigations have been performed on this species without presenting viral genomic data [30]. In the current study we present the first two complete CDV genomes from the Steppe polecat, enriching the diversity of available CDV genomes. By revealing the presence of a recombinant CDV strain in these animals we demonstrated the importance of generating complete genomic data. This approach may ultimately lead to better understanding CDV evolution, since partial genome fragments are not suitable to understand the impact of recombination events in CDV evolution or the role of coding regions other than H. Furthermore, the presence of CDV was confirmed in two additional species in this study. The European polecat is also at risk of infection by CDV; for instance, the virus was detected with RT-PCR (qPCR) from the Asturias region of Atlantic Spain in 2021 [57]. The stone marten is a well-known host of CDV, and in recent decades, many cases have been detected in nearby countries including such as Austria, the Czech Republic, Germany, Switzerland and Italy [29,30,31, 58,59,60,61].
According to our findings and previous literature data, CDV is present in 4 out of the 8 species of the Mustelidae family in our region [9, 10, 13, 40]. Considering the relevance of these animals in conservation biology, vaccination in wildlife rescue centers may be an important tool in the conservation of rare and protected mustelids [62]. For instance, the black-footed ferret, which population has almost been extinct due to CDV infection, is a close relative of the Steppe polecat. The vaccination of black-footed ferret × Steppe polecat hybrids was reported as surrogates for endangered black-footed ferrets [63, 64]. In Europe, CDV was detected in Spain in four carnivore species collected in 2020–2021, including the Eurasian badger, pine marten, European polecat and the red fox [57]. In the Czech Republic, CDV was detected between 2012–2020 in the red fox, stone marten, raccoon, pine marten and the European badger [59]. Similar outbreaks were observed among red foxes across Europe due to this strain [12, 60, 65,66,67]. Europe/South America 1 lineage was also detected in many other species such as Iberian wolves (Canis lupus signatus), an Asian marmot (Marmota caudata) kept in a zoo, a stone marten, pine marten, Eurasian lynx (Lynx lynx), Iberian lynx and a domestic dog [24, 31, 42, 68,69,70].
For effective transmission of CDV, close contact among infected and susceptible animals is necessary. Bites on two positive animals (stone marten and Steppe polecat) were observed as a direct indication of contact with other carnivores. Aggressive intra- and interspecific behavior are relatively common in the mustelid species, and competition for territory [71], food, or mating partner can effectively facilitate the spread of the disease. Nonetheless, according to published literature, skin contact, feces or urine are less important means of transmission [22, 51, 52]. However, the primary method of transmission in CDV infection is theorized to be via the respiratory tract droplets [72, 73], which may have relevance under fighting conditions. More studies and observational data are necessary to better understand the natural transmission and circulation patterns of CDV.
Based on literature data, the Europe/South America 1 lineage of CDV, which circulates among mustelids throughout Hungary, is also present in surrounding countries [31, 59, 60, 66, 67]. Similar to most of the CDV surveillance studies, H-gene phylogeny was a useful tool for lineage categorization. However as a major limitation, H-gene based analysis is not adequate to reveal genome-scale recombination patterns and understand fine-scale evolutionary patterns. Based on literature data these viruses are prone to recombine in several genomic regions, most frequently in the H gene [74, 75]. We support this with our observation and presentation of multiple recombination points in our recombinant CDV strain. More complete genomic sequence data in the future can reveal a more accurate evolutionary scenario for our sequence. In addition the dispersive pattern among these two phylogenetic clades, composed by different CDV strains from other animal species raises the possibility for cross-species transmission events. This was already known from literature data as an important feature of CDV transmission [76, 77].
A limitation of our study is the lack of autopsy or histology data to better understand the pathogenicity of the CDV infection in these animals. Further studies are needed to discuss the pathogenic nature of these different CDV strains. However, our study highlighted the importance of genome-scale monitoring of CDV evolution, which may serve as a first step to understand genomic evolution in relation to pathogenesis. In addition to these, our study demonstrated that road-killed carcasses are a valuable source of CDV surveillance in wildlife species.
Conclusion
Understanding the long-term presence of CDV in free-living mammals is of great importance, especially among mustelids, which are particularly sensitive to CDV. As we demonstrated in our study, retrospective sample surveillance coupled with complete genomic sequencing are useful tools to understand the host range of CDV and describe a more detailed evolutionary picture of the virus. Amplicon-based NGS methods are ideal tools to gain complete genomes even from organ samples stored over a long time and most importantly from samples with low viral titers.
Methods
Sample collection
Road-killed mustelids (n = 170) were collected in Hungary between 1997 and 2022 by the staff of National Park Directorates and volunteers and stored at -20 °C until processing. Tissue samples from spleen and lung via general dissection procedures were collected from the Steppe polecat (n = 64), European polecat (n = 36), stone marten (n = 36), pine marten (n = 18), least weasel (n = 10) and stoat (n = 6) [see Supplementary material; Table 1, Fig. 1]. The post-mortem examination was carried out by the Carnivore Ecology Research Group at the Kaposvár Campus of the Hungarian University of Agriculture and Life Sciences [78] and by the Hungarian Natural History Museum, Budapest [36, 37, 79]. We scored the body condition based on fat deposit over flanks between 1 (poor), 2 (average) and 3 (good) [80]. Tissue samples were stored at -20 °C in the Kaposvár Campus. A few months before nucleic acid extraction, they were deposited in the National Laboratory of Virology at -80 °C.
Research and sample collection permits were issued by the relevant authorities to the Kaposvár Campus (SO-04Z/TO/392–2/2019) and to the Hungarian Natural History Museum (14/6156/7/2011, OKTF-KP/6903–21/2015, PE-KTF/736–6/2017, PE-KTFO/329–16/2019, PE-KTFO/1568–18/2020, PE-KTFO/1403–3/2022).
Nucleic acid extraction and PCR reactions
For most animals, nucleic acids were extracted from the spleen, but lung was substituted when spleen was not available. Tissue samples were homogenized in 500 μl phosphate buffered saline (PBS), using a TissueLyser LT device (Qiagen, Hilden, Germany) at maximum speed for three minutes, supplemented with two glass beads per sample to facilitate tissue disruption. The total RNA was extracted using the Monarch Total RNA Miniprep Kit (NEB, USA) in full adherence to the manufacturer’s recommended guidance. The samples were screened with a CDV-specific real-time RT-PCR method [3] using OneStep RT-PCR Kit (Qiagen, Germany). RNA was added to each tube and the cycling was adjusted to one cycle of 50 °C for 30 min for the reverse transcription of RNA to cDNA, followed by one cycle at 95 °C for 15 min. The cDNA was amplified by PCR for 50 cycles, each cycle consisting of denaturation at 94 °C for20 sec, annealing at 46 °C for 30 s, extension at 72 °C for 30 s and final extension at 72 °C for 10 min. RT-PCRs were performed immediately following RNA extraction without freeze-thawing the nucleic acid to avoid RNA degradation.
Nanopore sequencing and data analysis
The complete genome sequencing was performed with MinION Nanopore sequencing technology (Oxford Nanopore Technologies, UK). We used a previously published universal amplicon-based sequencing method designed for CDV [12, 13]. The detailed protocol and the primers are available at our laboratory protocols.io page [81]. In brief, the CDV RNA positive nucleic acids were used for cDNA preparation with Superscript IV Reverse Transcriptase (Invitrogen, USA) using random hexamers. Two sets of primers were used to generate overlapping genome fragments that differ in the length of amplicons (1000 bp, 2000 bp). These multiplex PCRs were conducted directly from the cDNA with the usage of Q5 Hot Start HF Polymerase (New England Biolabs, USA). For the cleanup step, we used AMPure XP beads (Beckman Coulter, USA), and the PCR products were end-prepped with NEBNext Ultra II End Repair/dA-Tailing Module (New England Biolabs, USA). Barcodes from EXP-NBD196 (Nanopore Technologies, UK) were ligated to generate amplicons with NEBNext Ultra II Ligation Module (NEB, USA). The sequencing runs were performed on a R9.4.1. (FLO-MIN106D) flow cell with the AMX-F motor protein from SQK-LSK110 kit (Nanopore Technologies, UK). Sequencing raw data was processed by regular methods for Oxford Nanopore sequencing. Base-calling and demultiplexing of the raw data was performed with Guppy software (version 6.0.1.) using the super accurate base-calling model and default parameters with the “barcode_both_end” option. The generated reads were further processed, as 50 bases pairs were trimmed from both ends and the dataset was filtered to eliminate the short and chimeric sequence reads. Following the previously mentioned processes, all generated reads from a sample were mapped to the MN267060 reference sequence using Geneious Prime (version 1.6.0.). The preconsensus sequences were polished with Medaka (version 2022.1.1) to generate final consensus sequences.
Phylogenetic and recombinant analysis
Prior to the phylogenetic reconstruction, sequences of interest were retrieved from GenBank (NCBI, Bethesda, USA) and aligned with our obtained sequences in MUSCLE alignment webserver. Two datasets were used for phylogenetic tree analysis comprising 221 complete genomes and 969 complete hemagglutinin gene sequences, respectively. Subsequently, the Maximum Likelihood phylogenetic tree was constructed under the General Time Reversible Model, Gamma Distributed with Invariant Sites (GTR + G + I) substitution model with best model selection in MEGA X (MEGA, Pennsylvania, USA) [82]. The clustering of the sequences was verified with and additional method, using the RAxML (Randomized Axelerated Maximum Likelihood) plugin for Geneious Prime® 2022.2.2 [83]. The resultant tree was edited in iTOL (iTOL, Heidelberg, Germany) [84]. Phocine distemper virus (PDV) was used as an outgroup for all phylogenies.
The potential recombinant CDV genomes were tested through recombination analysis using similarity plot and bootscan analyses in SimPlot software package (version 3.5.1.) [85]. The recombination analysis was modeled with Kimura 2-parameter distance model using a window size of 600 bp and step size of 20 bp in the case of complete genomes and H gene sequences. To support our observation, we used a secondary recombination analysis method with the DualBrothers plugin in Geneious Prime® 2022.2.2 [86].
Availability of data and materials
Sequence data are deposited under GenBank accession numbers OP209185, OP209186, OP209187, OP209188 and OP209189. All data analyzed during this study are included in the results section.
Abbreviations
- CDV:
-
Canine distemper virus
- PDV:
-
Phocine distemper virus
- PBS:
-
Phosphate buffered saline
References
de Vries R, Duprex W, de Swart R. Morbillivirus Infections: An Introduction Viruses. 2015;7:699–706.
Elia G, Decaro N, Martella V, Cirone F, Lucente MS, Lorusso E, et al. Detection of canine distemper virus in dogs by real-time RT-PCR. J Virol Methods. 2006;136:171–6.
Martella V, Cirone F, Elia G, Lorusso E, Decaro N, Campolo M, et al. Heterogeneity within the hemagglutinin genes of canine distemper virus (CDV) strains detected in Italy. Vet Microbiol. 2006;116:301–9.
Martella V, Elia G, Buonavoglia C. Canine Distemper Virus. Vet Clin North Am - Small Anim Pract. 2008;38:787–97.
Bolt G, Jensen TD, Gottschalck E, Arctander P, Appel MJG, Buckland R, et al. Genetic diversity of the attachment (H) protein gene of current field isolates of canine distemper virus. J Gen Virol. 1997;78:367–72.
Duque-Valencia J, Forero-Muñoz NR, Díaz FJ, Martins E, Barato P, Ruiz-Saenz J. Phylogenetic evidence of the intercontinental circulation of a Canine distemper virus lineage in the Americas. Sci Rep. 2019;9:1–15.
McCarthy AJ, Shaw M-A, Goodman SJ. Pathogen evolution and disease emergence in carnivores. Proc R Soc B Biol Sci. 2007;274:3165–74.
Duque-Valencia J, Sarute N, Olarte-Castillo XA, Ruíz-Sáenz J. Evolution and Interspecies Transmission of Canine Distemper Virus—An Outlook of the Diverse Evolutionary Landscapes of a Multi-Host Virus. Viruses. 2019;11:582.
Demeter Z, Lakatos B, Palade EA, Kozma T, Forgách P, Rusvai M. Genetic diversity of Hungarian canine distemper virus strains. Vet Microbiol. 2007;122:258–69.
Demeter Z, Palade EA, Rusvai M. Canine Distemper : Still a Major Concern in Central Europe. Lucr Stiint - Univ Stiint Agric a Banat Timisoara, Med Vet. 2009;42:136–50.
Lanszki Z, Zana B, Zeghbib S, Jakab F, Szabó N, Kemenesi G. Prolonged Infection of Canine Distemper Virus in a Mixed-Breed Dog. Vet Sci. 2021;8:61.
Lanszki Z, Tóth GE, Schütz É, Zeghbib S, Rusvai M, Jakab F, et al. Complete genomic sequencing of canine distemper virus with nanopore technology during an epizootic event. Sci Rep. 2022;12:4116.
Lanszki Z, Lanszki J, Tóth GE, Zeghbib S, Jakab F, Kemenesi G. Retrospective Detection and Complete Genomic Sequencing of Canine morbillivirus in Eurasian Otter (Lutra lutra) Using Nanopore Technology. Viruses. 2022;14:1433.
Viana M, Cleaveland S, Matthiopoulos J, Halliday J, Packer C, Craft ME, et al. Dynamics of a morbillivirus at the domestic–wildlife interface: Canine distemper virus in domestic dogs and lions. Proc Natl Acad Sci. 2015;112:1464–9.
Martinez-Gutierrez M, Ruiz-Saenz J. Diversity of susceptible hosts in canine distemper virus infection: A systematic review and data synthesis. BMC Vet Res. 2016;12:1–11.
Greene CE, Vandevelde M, Canine Distemper In: Greene CE, editor. Infectious diseases of the dog and cat. 4th ed. Philadelphia: Saunders Elsevier Co; 2012. p. 25–42.
Beineke A, Baumgärtner W, Wohlsein P. Cross-species transmission of canine distemper virus—an update. One Heal. 2015;1:49–59.
Roelke-Parker ME, Munson L, Packer C, Kock R, Cleaveland S, Carpenter M, et al. A canine distemper virus epidemic in Serengeti lions (Panthera leo). Nature. 1996;379:441–5.
van de Bildt MWG. Distemper Outbreak and Its Effect on African Wild Dog Conservation. Emerg Infect Dis. 2002;8:212–3.
Gordon CH, Banyard AC, Hussein A, Laurenson MK, Malcolm JR, Marino J, et al. Canine Distemper in Endangered Ethiopian Wolves. Emerg Infect Dis. 2015;21:824–32.
Feng N, Yu Y, Wang T, Wilker P, Wang J, Li Y, et al. Fatal canine distemper virus infection of giant pandas in China. Sci Rep. 2016;6:27518.
Wang R, Wang X, Zhai J, Zhang P, Irwin DM, Shen X, et al. A new canine distemper virus lineage identified from red pandas in China. Transbound Emerg Dis. 2021. https://doi.org/10.1111/tbed.14370.
Seimon TA, Miquelle DG, Chang TY, Newton AL, Korotkova I, Ivanchuk G, et al. Canine Distemper Virus: an Emerging Disease in Wild Endangered Amur Tigers (Panthera tigris altaica). MBio. 2013;4:e00410–13.
Meli ML, Simmler P, Cattori V, Martínez F, Vargas A, Palomares F, et al. Importance of canine distemper virus (CDV) infection in free-ranging Iberian lynxes (Lynx pardinus). Vet Microbiol. 2010;146:132–7.
Kiupel M, Perpiñán D. Viral Diseases of Ferrets. In: Fox JG, Marini RP, editor. Biology and Diseases of the Ferret. 3rd ed. Ames: John Wiley & Sons, Inc.; 2014. p. 439–517.
Thorne ET, Williams ES. Disease and Endangered Species: The Black-footed Ferret as a Recent Example. Conserv Biol. 1988;2:66–74.
Williams ES, Thome ET, Appel MJG, Belitsky DW. Canine distemper in black-footed ferrets (Mustela nigripes) from Wyoming. J Wildl Dis. 1988;24:385–98.
Fournier‐Chambrillon C, Ceña J, Urra‐Maya F, van de Bildt M, Ferreras MC, Giralda‐Carrera G, et al. A 9‐Year Demographic and Health Survey of a European Mink Population in Navarre (Spain). In: San EDL, Sato J, Belant JL, Somers MJ editor. Small Carnivores. 1st ed. Wiley. 2022. p. 231–47.
Frölich K, Czupalla O, Haas L, Hentschke J, Dedek J, Fickel J. Epizootiological investigations of canine distemper virus in free-ranging carnivores from Germany. Vet Microbiol. 2000;74:283–92.
Pavlacik L, Celer V, Koubek PL, I. Prevalence of canine distemper virus in wild mustelids in the Czech Republic and a case of canine distemper in young stone martens. Vet Med (Praha). 2007;52:69–73.
Origgi FC, Plattet P, Sattler U, Robert N, Casaubon J, Mavrot F, et al. Emergence of Canine Distemper Virus Strains With Modified Molecular Signature and Enhanced Neuronal Tropism Leading to High Mortality in Wild Carnivores. Vet Pathol. 2012;49:913–29.
Di Sabatino D, Di Francesco G, Zaccaria G, Malatesta D, Brugnola L, Marcacci M, et al. Lethal distemper in badgers (Meles meles) following epidemic in dogs and wolves. Infect Genet Evol. 2016;46:130–7.
Akdesir E, Origgi FC, Wimmershoff J, Frey J, Frey CF, Ryser-Degiorgis M-P. Causes of mortality and morbidity in free-ranging mustelids in Switzerland: necropsy data from over 50 years of general health surveillance. BMC Vet Res. 2018;14:195.
Philippa J, Fournier-Chambrillon C, Fournier P, Schaftenaar W, van de Bildt M, van Herweijnen R, et al. Serologic survey for selected viral pathogens in free-ranging endangered European mink (Mustela lutreola) and other mustelids from south-western France. J Wildl Dis. 2008;44:791–801.
Blandford PRS. Biology of the polecat Mustela putorius: a literature review. Mamm Rev. 1987;17:155–98.
Virgos E, Zalewski A, Rosalino LM, Mergey M. Habitat ecology of Martens species in Europe. A Review of the Evidence: Cornell University Press; 2012.
Zalewski A, Jędrzejewski W. Spatial organisation and dynamics of the pine marten Martes martes population in Białowieza Forest (E Poland) compared with other European woodlands. Ecography (Cop). 2006;29:31–43.
King CM, Powell RA. The natural history of weasels and stoats: ecology, behavior, and management. Second: Oxford University Press; 2006.
Šálek M, Spassov N, Anděra M, Enzinger K, Ottlecz B, Hegyeli Z. Population status, habitat associations, and distribution of the steppe polecat Mustela eversmanii in Europe. Acta Theriol (Warsz). 2013;58:233–44.
Lanszki J, Heltai M, Kövér G, Zalewski A. Non-linear relationship between body size of terrestrial carnivores and their trophic niche breadth and overlap. Basic Appl Ecol. 2019;38:36–46.
Arumugam R, Uli JE, Annavi G. A Review of the Application of Next Generation Sequencing (NGS) in Wild Terrestrial Vertebrate Research. Annu Res Rev Biol. 2019;31:1–9.
Conceição-neto N, Godinho R, Álvares F, Yinda CK, Deboutte W, Zeller M, et al. Viral gut metagenomics of sympatric wild and domestic canids , and monitoring of viruses : Insights from an endangered wolf population. 2017;12:4135–46.
Jo WK, Peters M, Kydyrmanov A, van de Bildt MWG, Kuiken T, Osterhaus A, et al. The Canine Morbillivirus Strain Associated with An Epizootic in Caspian Seals Provides New Insights into the Evolutionary History of this Virus. Viruses. 2019;11:894.
da Costa VG, Saivish MV, de Oliveira PG, Silva-Júnior A, Moreli ML, Krüger RH. First complete genome sequence and molecular characterization of Canine morbillivirus isolated in Central Brazil. Sci Rep. 2021;11:13039.
Kilianski A, Haas JL, Corriveau EJ, Liem AT, Willis KL, Kadavy DR, et al. Bacterial and viral identification and differentiation by amplicon sequencing on the MinION nanopore sequencer. Gigascience. 2015;4:12.
Batista FM, Stapleton T, Lowther JA, Fonseca VG, Shaw R, Pond C, et al. Whole Genome Sequencing of Hepatitis A Virus Using a PCR-Free Single-Molecule Nanopore Sequencing Approach. Front Microbiol. 2020;11:874.
Peserico A, Marcacci M, Malatesta D, Di Domenico M, Pratelli A, Mangone I, et al. Diagnosis and characterization of canine distemper virus through sequencing by MinION nanopore technology. Sci Rep. 2019;9:1714.
Young KT, Lahmers KK, Sellers HS, Stallknecht DE, Poulson RL, Saliki JT, et al. Randomly primed, strand-switching, MinION-based sequencing for the detection and characterization of cultured RNA viruses. J Vet Diagnostic Investig. 2021;33:202–15.
Freed NE, Vlková M, Faisal MB, Silander OK. Rapid and inexpensive whole-genome sequencing of SARS-CoV-2 using 1200 bp tiled amplicons and Oxford Nanopore Rapid Barcoding. Biol Methods Protoc. 2020;5:bpaa014.
Kemenesi G, Tóth GE, Mayora-Neto M, Scott S, Temperton N, Wright E, et al. Isolation of infectious Lloviu virus from Schreiber’s bats in Hungary. Nat Commun. 2022;13:1706.
Park K, Lee S-H, Kim J, Lee J, Lee G-Y, Cho S, et al. Multiplex PCR-Based Nanopore Sequencing and Epidemiological Surveillance of Hantaan orthohantavirus in Apodemus agrarius. Republic of Korea Viruses. 2021;13:847.
Quick J, Loman NJ, Duraffour S, Simpson JT, Severi E, Cowley L, et al. Real-time, portable genome sequencing for Ebola surveillance. Nature. 2016;530:228–32.
Nussberger B, Wandeler P, Weber D, Keller LF. Monitoring introgression in European wildcats in the Swiss Jura. Conserv Genet. 2014;15:1219–30.
Szatmári L, Cserkész T, Laczkó L, Lanszki J, Pertoldi C, Abramov A V., et al. A comparison of microsatellites and genome‐wide SNPs for the detection of admixture brings the first molecular evidence for hybridization between Mustela eversmanii and M. putorius (Mustelidae, Carnivora). Evol Appl. 2021;14:2286–304.
Maas M, Tatem-Dokter R, Rijks JM, Dam-Deisz C, Franssen F, van Bolhuis H, et al. Population genetics, invasion pathways and public health risks of the raccoon and its roundworm Baylisascaris procyonis in northwestern Europe. Transbound Emerg Dis. 2022;69:2191–200.
Heptner VG. Mammals of the Soviet Union, vol. II. Moscow: Vysshaya Shkola; 1967.
Oleaga Á, Vázquez CB, Royo LJ, Barral TD, Bonnaire D, Armenteros JÁ, et al. Canine distemper virus in wildlife in south-western Europe. Transbound Emerg Dis. 2021. https://doi.org/10.1111/tbed.14323.
Benetka V, Leschnik M, Affenzeller N, Mösti K. Phylogenetic analysis of Austrian canine distemper virus strains from clinical samples from dogs and wild carnivores. Vet Rec. 2011;168:377.
Kličková E, Černíková L, Dumondin A, Bártová E, Budíková M, Sedlák K. Canine Distemper Virus in Wild Carnivore Populations from the Czech Republic (2012–2020): Occurrence, Geographical Distribution, and Phylogenetic Analysis. Life. 2022;12:289.
Trogu T, Canziani S, Salvato S, Bianchi A, Bertoletti I, Gibelli LR, et al. Canine Distemper Outbreaks in Wild Carnivores in Northern Italy. Viruses. 2021;13:99.
Balboni A, Savini F, Scagliarini A, Berti E, Naldi M, Urbani L, et al. Natural distemper infection in stone martens (Martes foina): From infection to neutralizing antibodies. Res Vet Sci. 2021;138:196–200.
Wright ML, Livieri TM, Santymire RM. Recombitek canine distemper vaccine as an alternative for purevax distemper vaccine in endangered black-footed ferrets (mustela nigripes). J Zoo Wildl Med. 2022;53:194–99.
Williams ES, Anderson SL, Cavender J, Lynn C, List K, Hearn C, et al. Vaccination of black-footed ferret (Mustela nigripes)× Siberian polecat (M. eversmanni) hybrids and domestic ferrets (M. putorius furo) against canine distemper. J Wildl Dis. 1996;32:417–23.
Wimsatt J, Biggins D, Innes K, Taylor B, Garell D. Evaluation of oral and subcutaneous delivery of an experimental canarypox recombinant canine distemper vaccine in the Siberian polecat (Mustela eversmanni). J Zoo Wildl Med. 2003;34:25–35.
Nikolin VM, Wibbelt G, Michler FUF, Wolf P, East ML. Susceptibility of carnivore hosts to strains of canine distemper virus from distinct genetic lineages. Vet Microbiol. 2012;156:45–53.
Sekulin K, Hafner-Marx A, Kolodziejek J, Janik D, Schmidt P, Nowotny N. Emergence of canine distemper in Bavarian wildlife associated with a specific amino acid exchange in the haemagglutinin protein. Vet J. 2011;187:399–401.
Martella V, Bianchi A, Bertoletti I, Pedrotti L, Gugiatti A, Catella A, et al. Canine Distemper Epizootic among Red Foxes, Italy, 2009. Emerg Infect Dis. 2010;16:2007–9.
Maganga GD, Labouba I, Ngoubangoye B, Nkili-Meyong AA, Obame Ondo D, Leroy EM, et al. Molecular characterization of complete genome of a canine distemper virus associated with fatal infection in dogs in Gabon. Central Africa Virus Res. 2018;247:21–5.
Origgi FC, Sattler U, Pilo P, Waldvogel AS. Fatal Combined Infection With Canine Distemper Virus and Orthopoxvirus in a Group of Asian Marmots (Marmota caudata). Vet Pathol. 2013;50:914–20.
Müller A, Silva E, Santos N, Thompson G. Domestic Dog Origin of Canine Distemper Virus in Free-ranging Wolves in Portugal as Revealed by Hemagglutinin Gene Characterization. J Wildl Dis. 2011;47:725–9.
Palomares F, Caro TM. Interspecific Killing among Mammalian Carnivores. Am Nat. 1999;153:492–508.
Shin D-L, Chludzinski E, Wu N-H, Peng J-Y, Ciurkiewicz M, Sawatsky B, et al. Overcoming the Barrier of the Respiratory Epithelium during Canine Distemper Virus Infection. MBio. 2022;13:e03043–21.
Rendon-Marin S, da Fontoura BR, Canal CW, Ruiz-Saenz J. Tropism and molecular pathogenesis of canine distemper virus. Virol J. 2019;16:30.
Piewbang C, Radtanakatikanon A, Puenpa J, Poovorawan Y, Techangamsuwan S. Genetic and evolutionary analysis of a new Asia-4 lineage and naturally recombinant canine distemper virus strains from Thailand. Sci Rep. 2019;9:3198.
Ke G-M, Ho C-H, Chiang M-J, Sanno-Duanda B, Chung C-S, Lin M-Y, et al. Phylodynamic analysis of the canine distemper virus hemagglutinin gene. BMC Vet Res. 2015;11:164.
Yuan C, Liu W, Wang Y, Hou J, Zhang L, Wang G. Homologous recombination is a force in the evolution of canine distemper virus. PLoS ONE. 2017;12: e0175416.
Nikolin VM, Olarte-Castillo XA, Osterrieder N, Hofer H, Dubovi E, Mazzoni CJ, et al. Canine distemper virus in the Serengeti ecosystem: molecular adaptation to different carnivore species. Mol Ecol. 2017;26:2111–30.
Wereszczuk A, Hofmeester TR, Csanády A, Dumić T, Elmeros M, Lanszki J, et al. Different increase rate in body mass of two marten species due to climate warming potentially reinforces interspecific competition. Sci Rep. 2021;11:24164.
Cserkész T, Kiss C, Barkaszi Z, Görföl T, Zagorodniuk I, Sramkó G, et al. Intra- and interspecific morphological variation in sympatric and allopatric populations of Mustela putorius and M. eversmanii (Carnivora: Mustelidae) and detection of potential hybrids. Mammal Res. 2021;66:103–14.
Simpson VR. Post mortem protocol for otters. In: JWH Conroy, P Yoxon AG, editor. Proceedings of the First Otter Toxicology Conference. Scotland: Isle of Skye; 2000. p. 159–64.
2021. https://www.protocols.io/view/universal-amplicon-based-sequencing-method-for-can-bykwpuxe
Tamura K, Stecher G, Kumar S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol Biol Evol. 2021;38:3022–7.
Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–90.
Letunic I, Bork P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49:W293–6.
Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, et al. Full-Length Human Immunodeficiency Virus Type 1 Genomes from Subtype C-Infected Seroconverters in India, with Evidence of Intersubtype Recombination. J Virol. 1999;73:152–60.
Minin VN, Dorman KS, Fang F, Suchard MA. Dual multiple change-point model leads to more accurate recombination detection. Bioinformatics. 2005;21:3034–42.
Acknowledgements
We sincerely thank the volunteers and staff of the National Park Directorates and other volunteers for collecting the carcasses. Collection of polecat road-kills was supported by the GRASSLAND-HU LIFE project (LIFE17 IPE/HU/000018).
Funding
This work was supported by the National Laboratory of Virology (RRF-2.3.1–21-2022–00010) and Z.L. was supported by the Biological and Sportbiological Doctoral School of the University of Pécs, Hungary.
Author information
Authors and Affiliations
Contributions
J.L., T.C., G.C. and A.I.C. sample collection. Z.L. and G.E.T. laboratory work and data analysis. Z.L. and T.G. phylogenetic analysis. G.E.T. and Z.L. performed NGS experiments. G.E.T. bioinformatic analysis of NGS data. Z.L. and J.L. drafted the manuscript. G.E.T., T.C., G.C., T.G., A.I.C., J.F. and G.K. finalized the manuscript. Z.L., G.E.T., G.K. conceptualization. G.K., J.F. supervision. All authors have read and agreed to the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lanszki, Z., Lanszki, J., Tóth, G.E. et al. Detection and sequence analysis of Canine morbillivirus in multiple species of the Mustelidae family. BMC Vet Res 18, 450 (2022). https://doi.org/10.1186/s12917-022-03551-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12917-022-03551-7