
Abstract
Background
Extreme panting under heat stress threatens dairy cattle milk production. Previous research has revealed that the gas exchange-mediated respiratory drive in critically ill dairy cattle with low O2 saturation induces panting. Vascular endothelial growth factor (VEGF) signaling may play important roles in immunosuppression and oxidative stress during severe respiratory stress responses in heat-stressed cattle. The objectives of this study were to transcriptomically analyze mRNA expression mediating heat-induced respiratory stress-associated panting, evaluate gas exchange, screen hub genes, and verify the expression of proteins encoded by differentially expressed genes in lymphocyte pathways.
Results
Jersey cattle were naturally heat-exposed. Physiological data were collected for response evaluation, and blood was collected for gas exchange and gene expression assays at 06:00, 10:00 and 14:00 continuously for 1 week. Lymphocytes were isolated from whole-blood samples for mRNA-seq and expression analysis of key pathway genes/proteins. The cattle respiration rates differed with time, averaging 51 bpm at 06:00, 76 bpm at 10:00, and 121 bpm at 14:00 (p < 0.05). Gas exchange analysis showed that both pH and pCO2 differed with time: they were 7.41 and 41 mmHg at 06:00, 7.45 and 37.5 mmHg at 10:00, and 7.49 and 33 mmHg at 14:00, respectively (p < 0.01). Sixteen heat-related differentially expressed genes (DEGs; 13 upregulated and 3 downregulated) were screened between 212 DEGs and 1370 heat stress-affected genes. Kyoto Encyclopedia of Genes and Genomes (KEGG) hub gene functional analysis annotated eleven genes to signal transduction, six genes to the immune response, and five genes to the endocrine response, including both prostaglandin-endoperoxide synthase 2 (PTGS2) and VEGF. Gene Ontology (GO) functional enrichment analysis revealed that oxygen regulation was associated with the phosphorus metabolic process, response to oxygen levels, response to decreased oxygen levels, response to hypoxia and cytokine activity terms. The main signaling pathways were the VEGF, hypoxia inducible factor-1(HIF-1), cytokine-cytokine receptor interaction and TNF pathways. Four genes involved Integrin beta 3 (ITBG3), PTGS2, VEGF, and myosin light chain 9 (MYL9) among the 16 genes related to immunosuppression, oxidative stress, and endocrine dysfunction were identified as participants in the VEGF signaling pathway and oxygenation.
Conclusion
These findings help elucidate the underlying immune and oxygen regulation mechanisms associated with the VEGF signaling pathway in heat-stressed dairy cattle.
Similar content being viewed by others

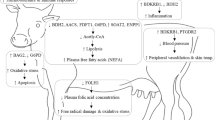

Background
The specific physiological responses of homeothermal animals under heat stress have been found to involve hypoxia, oxidative stress, decreased immunity, respiratory acidosis, and electrolyte disturbances. A previous study has shown that the physiological panting response of dairy cattle is affected by interactions between breed and the environment [1, 2]. Panting related to the chemical and metabolic acid-base balance of animals that occurs under a range of environmental stressors affecting respiratory condition is based on the mechanism of H+ equivalent exchange. For instance, panting reduces systemic arterial pO2 and Hb O2 (HbO2) saturation but increases the partial pressure of CO2, leading to increased H+ accumulation [3]. Furthermore, under heat stress, dairy cattle attempt to dissipate heat by panting. The mechanism of oxygenation is insufficient for immune cell metabolism, although quick breathing increases gas exchange between the environment and lungs. In addition, a previous transcriptomic analysis has revealed that 2'-5'-oligoadenylate synthetase 2 (OAS2), MX dynamin like GTPase 2 (MX2), interferon induced protein with tetratricopeptide repeats 5 (IFIT5) and TGFB2 are potential regulatory genes related to heat tolerance in Holstein dairy cattle [4]. However, transcriptomic analysis of messenger RNA (mRNA) sequences with a focus on the immune response and oxidative stress in dairy cattle under heat stress has not been performed.
Immune responses assist dairy cattle in maintaining a healthy microenvironment by releasing cytokines and activating genes to protect tissue and cells from damage caused by heat stress [5]. In line with the dynamic nature of heat stress, two phases are recognized: an early inflammatory phase and a late immunosuppressive phase [6]. Shalova et al. [7] found that the early phase is characterized by leukocyte activation, a cytokine storm, and a systemic inflammatory response, while the late phase is characterized by immunosuppression involving leukocyte deactivation, increased risk of secondary infection, and high mortality during heat stress. For example, it would effected in the early inflammation on immune function due to increased white blood cell counts when dairy cattle are under heat stress [8]. Furthermore, previous studies have reported that the effects of hypoxia on cytokine gene expression, inflammation and immunosuppression involve VEGF [9], PTGS2 [9, 10], endothelin-1 (EDN1) [11], nuclear receptor subfamily 4 group A member 2 (NR4A2) [12,13,14], C-X-C motif chemokine ligand 8 (CXCL8) [15, 16], interleukin 1 alpha (IL1A) [17] and MYL9 [18]. It is possible for heat to induce both hypoxia and immune responses to affect the expression of common genes with related functions.
In addition, oxidative stress, a physiological response to heat stress, is provoked in dairy cattle by oxygen metabolism-related enzymes [19]. Several studies have concluded that exposure of dairy cattle to heat stress induces oxidative stress, which can lead to cytotoxicity [20]. It is well known that biological macromolecules can be damaged by oxidative stress, which interferes with metabolic and physiological pathways [21]. Moreover, the symptoms of heat stress have been suggested to be similar to those of oxidative stress because of correspondences in the expressed genes, including genes encoding heat-shock proteins and antioxidant enzymes [22, 23]. It is possible that the VEGF signaling pathway plays an important role in regulating oxygen levels.
Therefore, it is interesting to explore whether the effects of immune-mediated destruction of cells under heat stress on gene expression help to maintain homeostasis in dairy cattle. Thus far, the gene regulation mechanism of heat-induced panting with regard to the mRNA expression levels of hub genes that regulate immunosuppression and oxygen stress in blood immune cells in dairy cattle under heat stress has remained unknown. It has been hypothesized that the heat stress-induced panting response of dairy cattle may be regulated by hub genes within the VEGF signaling pathway that play important roles in immunosuppression, oxidative stress, and the endocrine system.
Results
Meteorological data, physiological responses, and gas exchange
Meteorological data, namely, the dry-bulb temperature, wet-bulb temperature, and relative humidity, significantly differed with the temperature humidity index (THI) (p < 0.01) (Table 1). All physiological responses, including respiratory rate (RR), rectal temperature (RT), and skin temperature (ST), increased as the THI increased; the strongest response appeared at 14:00 when THI was highest (THI = 88) (p < 0.01) (Table 1). Significant differences in were observed; the RR was 51 bpm at 06:00(THI77), 76 bpm at 10:00(THI82) and 121 bpm at 14:00(THI88) (p < 0.01). The gas exchange data showed that both pH and pCO2 significantly differed and were affected by the THI (p < 0.01). The pH values increased as the THI increased, while the pCO2 values decreased (Table 1).
RNA-seq results
The transcriptome sequencing results are shown in Table 2. From the samples in the THI = 77 (THI77) group, 61,562,712 (THI = 77_1), 59,409,952 (THI = 77_2), 67,050,696 (THI = 77_3), 58,325,238 (THI = 77_4) and 69,485,932 (THI = 77_5) raw reads were obtained. From the samples in the THI = 82 (THI82) group, 50,837,238 (THI = 82_1), 58,619,002 (THI = 82_2), 50,511,366 (THI = 82_3), 59,646,870 (THI = 82_4), and 63,367,910 (THI = 82_5) raw reads were obtained. From the samples in the THI = 88 (THI88) group, 62,085,400 (THI = 88_1), 59,070,634 (THI = 88_2), 59,661,384 (THI = 88_3), 69,221,370 (THI = 88_4), and 61,949,988 (THI = 88_5) raw reads were obtained. Clean reads were obtained by removing reads containing adapters and undetermined bases and low-quality reads from the group of raw reads. From the samples in the THI77 group, 61,069,186 (THI = 77_1), 58,867,260 (THI = 77_2), 66,516,354 (THI = 77_3), and 57,799,350 (THI = 77_4) clean reads were obtained. From the samples in the THI82 group, 50,474,676 (THI = 82_1), 58,178,126 (THI = 82_2), 50,049,474 (THI = 82_3), 59,215,152 (THI = 82_4), and 62,924,034 (THI = 82_5) clean reads were obtained. From the samples in the THI88 group, 61,551,498 (THI = 88_1), 58,633,654 (THI = 88_2), 59,176,084 (THI = 88_3), 68,728,564 (THI = 88_4), and 61,437,404 (THI = 88_5) clean reads were obtained. All obtained Q20 and Q30 values were greater than 95%, and the error rates were less than 0.05% in the THI77, THI82 and THI88 groups.
DEG analysis and screening of 16 heat-related DEGs in lymphocytes of Jersey dairy cattle affected by the THI under heat stress
The DEGs between each pair of groups were revealed via differential expression analysis using normalized read counts and are shown in Fig. 1A. The upregulated DEGs between each pair of groups are shown in Fig. 1B, while the downregulated DEGs are shown in Fig. 1C. RNA-seq analysis screened 170 DEGs in the THI82 group compared to the control group of THI77 cells, of which 26 genes were downregulated and 144 were upregulated. Additionally, RNA-seq analysis identified 30 DEGs in the THI88 group compared to the control group of THI82 cells, of which 2 were upregulated and 28 were downregulated. Finally, RNA-seq analysis identified 65 DEGs in the THI77 group compared to the control group of THI88 cells, of which 58 were upregulated and 5 were downregulated (Fig. 1D). Subsequently, 16 heat-related DEGs were screened by Venn analysis between 1370 heat-related genes and 212 DEGs (Fig. 1E). Furthermore, 16 heat-related DEGs (including 13 upregulated DEGs and 3 downregulated DEGs) were obtained using the DESeq2 package (version 1.10.1) according to the values of a |log2 fold change| ≥ 1 and a p < 0.05 (Fig. 1F, Table 3).

DEGs were subjected to Venn analysis using normalized read counts from three pairs of groups: THI77 vs THI82, THI82 vs THI88 and THI77 vs THI88. The 16 heat-related DEGs were screened, and a heatmap was created. a. Venn statistics showing the total DEGs in each pair of THI groups. b. Venn statistics showing the upregulated DEGs in each pair of THI groups. c. Venn statistics showing the downregulated DEGs in each pair of THI groups. d. Diagram showing the total numbers of DEGs and the numbers of upregulated and downregulated DEGs in each pair of THI groups. e. Venn diagram showing the 16 heat-related DEGs screened between 212 DEGs and 1370 heat-related genes. f. Heatmap of the 16heat-related DEGs
Functional annotation and enrichment of the 16 heat-related DEGs by KEGG analysis
Functional annotation and enrichment of the 16 heat-related DEGs revealed that immunosuppression, oxidative stress, and endocrine disorder are effected by heat stress. The KEGG functional annotations focused mainly on three major functions: signal transduction for eleven genes (AF356445.1(DUP6), intercellular adhesion molecule 1(ICAM1), CP027091.1, IL1A, ITGB3, PTGS2, VEGF, MYL9, HNHc-like endonuclease (END1), TGFB2, and CXCL8), the immune system for six genes (VEGF, IL1A, ITGB3, PTGS2, MYL9, and CXCL8) and the endocrine system for five genes (PTGS2, NR4A2, MYL9, EDN1, and ITBG3) (Fig. 2A). Some of the 16 heat-related DEGs were enriched in the following KEGG pathways: the AGE-RAGE signaling pathway in diabetic complications (enriched genes: PTGS2, VEGF, IL1A, EDN1, TGFB2, CXCL8, and ICAM1), the VEGF signaling pathway (map04370) (enriched genes: vascular endothelial growth factor A [VEGFA], PTGS2 and CP027091.1), cytokine-cytokine receptor interactions (enriched genes: IL1A, VEGFA, CXCL8 and CP027091.1), the TNF signaling pathway (map04668) (enriched genes: EDN1, ICAM1 and PTGS2), the hypoxia inducible factor-1 (HIF-1) signaling pathway (map04066) (enriched genes: VEGFA, EDN1 and CP027091.1), the MAPK signaling pathway (map04010) (enriched genes: IL1A and DUSP6), and EGFR tyrosine kinase inhibitor resistance (map01521) (enriched gene: CP027091.1) (Fig. 2C).

GO and KEGG functional enrichment of 16 heat-related DEGs. a The main annotated functions were associated with the endocrine system (PTGS2, NR4A2, MYL9, EDN1and ITGB3), the immunesystem (VEGF, IL1A, ITGB3, PTGS2, MYL9 and CXCL8) and signaling molecules and interaction(11 genes). b The enriched GO terms included the regulation of phosphorus metabolic process, response to oxygen levels, response to decreased oxygen levels, response to hypoxia and cytokine activity terms. c The enriched KEGG pathways included the cytokine-cytokine receptor interaction pathway, the VEGF signaling pathway, the TNF signaling pathway and the HIF-1 signaling pathway
Functional annotation and enrichment of the 16 heat-related DEGs by GO analysis
The top five enriched process terms in GO analysis were the phosphorus metabolic process term, the response to oxygen levels term, the response to decreased oxygen levels term, the response to hypoxia term and the cytokine activation term (Fig. 2B). The genes enriched for phosphorus metabolic process-related GO functions included SAM domain, SH3 domain and nuclear localization signals 1(SAMSN1)(GO:0050732), DUSP6(GO:0004725) and CP027091.1 (GO:0050731); those enriched for oxygen balance-related GO functions included VEGFA (GO:0043117), EDN1 (GO:0007589 and GO:0019229), NR4A2 (GO:0034599 and GO:0043576), and PTGS2 (GO:0006954, GO:0071347 and GO:0006979); those enriched for hypoxia-related GO functions included VEGFA (GO:0071456), EDN1 (GO:0001666), NR4A2 (GO:0001666) and CP027091.1 (GO:0071456); and those enriched for cytokine activation-related GO terms included IL1A (GO:0005125), VEGFA (GO:0005125), EDN1 (GO:0005125), CXCL8 (GO:0008009), and CP027091.1 (GO:0005125) (Addtional 1).
Q-PCR validation
To verify the reliability of the transcriptome sequencing results in this study, Q-PCR was used to detect the expression of heat-related DEGs. Compared to the control group, the experimental group exhibited increased mRNA expression of VEGF, PR/SET domain 1 (PRDM1), SAMSN1, basic helix-loop-helix family member e40 (BHLHE40), NR4A2, DUSP6, EDN1, MYL9, prostaglandin-endoperoxide synthase 2 (PTGS2), CXCL8 and IL1A. These results were consistent with the RNA-seq results (Fig. 3).

Heat-related DEG expression determined by RNA-seq and Q-PCR
Strong heat stress-mediated activation of heat-related DEG coexpression and upregulation of the gene expression of VEGF and PTGS2 in the VEGF pathway in blood-derived lymphocytes
A coexpression network was constructed for the 16 heat-related DEGs in the blood-derived lymphocytes of Jersey dairy cattle affected by heat stress. This network included TGFB2, ITBG3, VEGF, PTGS2, NR4A2, EDN1, DUSP6, IL1A, MYL9, PRDM1, SAMSN1, BHLHE40 and ICAM1. The results showed that the VEGF gene was strongly related to other genes in the VEGF signaling pathway, including the PTGS2 gene (Fig. 4A). The results also showed that the VEGF signaling pathway was activated in the blood-derived lymphocytes of Jersey dairy cattle (Bos taurus). VEGFR-2 is the major mediator of VEGF (VEGFA)-driven responses in lymphocytes. Binding of VEGFA to VEGFR-2 leads to dimerization of the receptor, triggering intracellular activation of the PLCy and IP3-Ca + kinase- cyclooxygenase 2 (COX2) pathways of arachidonic acid metabolism. Subsequently, the initiation of COX2/PTGS2 gene expression increases permeability (Fig. 4B). The levels of both the VEGF and PTGS2 proteins are shown (Fig. 4C). The relative mRNA expression levels determined using Q-PCR were significantly different (p < 0.001) (Fig. 4c). Furthermore, both the gene and protein expression of VEGF and PTGS2 increased significantly in association with high values of THI (p < 0.001) (Fig. 4D, d, E, e).

Heat-related DEG coexpression network and the expression of VEGF and PTGS2 in the VEGF pathway. a Coexpression network diagram of the 16 heat-related DEGs, with a positive relationship between PTGS2 and VEGF. b Diagram of the VEGF signaling pathway showing that VEGFA binds VEGFR2 at the membrane before triggering PLCy-CNLP-NFAT signaling to activate expression of the target gene COX2/PTGS2. c Verification of VEGF and PTGS2 expression. Scanned images of the protein levels of both VEGF and PTGS2 under different THIs (THI77, THI82, and THI88). c) Changes in relative mRNA expression determined using Q-PCR. d Changes in VEGF expression in lymphocte caused by different THIs (THI77, THI82, and THI88). d) Changes in relative mRNA expression determined using Q-PCR. e Changes in PTGS2 expression in lymphocytes caused by different THIs (THI77, THI82, and THI88). e) Changes in relative mRNA expression determined using Q-PCR
Discussion
The effects of heat stress on the heat-tolerant Jersey cattle breed (Bos taurus) have been investigated by Smith et al. [24]. In this study, an animal model of heat stress was successfully induced by an average temperature of 30.3 °C and an average humidity of 68% from July to August in Chongqing (29.4316°N, 106.9123°E). It was confirmed that the Jersey dairy cattle initiated panting for evaporative heat dissipation. The RR was significantly affected by THI: it was 51 bpm at THI77, 76 bpm at THI82, and 121 bpm at THI88. In addition, the gas exchange parameters of pH and pCO2 in the blood of Jersey dairy cattle were significantly affected: they were 7.41 and 41 mmHg at THI77, 7.45 and 37.5 mmHg at THI82, and 7.49 and 33 mmHg at THI88, respectively. Furthermore, the findings were consistent with the following standard categories: THI < 72, no stress; 72 < THI < 79, vasodilation and increased RR; and 80 < THI < 90, increased RR or even panting [2].
In this study, transcriptome sequencing analysis screened 212 DEGs and 1370 heat-related genes in the lymphocytes of Jersey dairy cattle naturally exposed to hot environmental conditions of THI77, THI82 and THI88. Ultimately, 16 heat-related DEGs were screened from the 212 DEGs and 1370 heat-related genes, including transforming growth factor beta 2 (TGFB2), ITBG3, VEGF, CP027091.1, PTGS2, NR4A2, EDN1, DUSP6, IL1A, CXCL8, ICAM1, MYL9, DnaJ heat shock protein family (Hsp40) member B13(DNAJB13), PRDM1, SAMSN1 and BHLHE40. The functional annotation results revealed that the 16 heat-related DEGs were associated with the response to hypoxia, oxidative stress, the immune response, inflammation and endocrine disorder. Likewise, six genes were annotated to the immune system, including VEGF, IL1A, ITBG3, PTGS2, MYL9 and CXCL8; five genes were annotated to the endocrine system, including PTGS2, NR4A2, MYL9, EDN1, and ITBG3; and eleven genes were annotated to signal transduction, including DUP6, ICAM1, CP027091.1, IL1A, ITBG3, PTGS2, VEGF, MYL9, END1, TGFB2 and CXCL8.
In a previous study, hypoxia was found to affect cytokine gene expression, inflammation and immunosuppression in immune cells in a manner that involved VEGF, PTGS2, EDN1, CXCL8, IL1A and MYL9. Hypoxia has also been found to affect the expression of the gene encoding EDN1 in U87 glioma cells [11] and the genes encoding PTGS2 and VEGFA in breast cancer and melanoma cells [9]. Overproduction of PTGS2 has also been observed in lung cancer for immunosuppression [10]. Furthermore, HIF-1 is activated in response to hypoxia to modulate the expression of genes such as VEGF and COX-2 via cytokines and chemokines. For example, endothelin-2 (EDN2) has been investigated in macrophages and cancer cells [9]. An investigation has also proven that MYL9 is a ligand activated by CD69 on leukocytes that is strongly detected inside blood vessels in inflamed lungs [18]. In addition, the IL1A gene plays a role in vascular regulation and hematopoiesis induced by hypoxia, but factor 1α (HIF-1α) induces the cytokine CXCL8, which activates heat shock transcription via AKT/mTOR/STAT3 pathways [15,16,17]. Moreover, ITGB3 (αVβ3), a type of integrin that consists of two components, integrin alpha V and integrin beta 3 (CD61), is a receptor for phagocytosis on macrophages. ITBG3 is physically and functionally associated with important therapeutic targets [25]. The findings in this study show that ITBG3, PTGS2, VEGF and MYL9 are hub genes that are possible biological markers of the immune response in dairy cattle under heat stress.
Oxidative stress is provoked in dairy cattle as a physiological response to heat stress [19]. Several studies have investigated whether exposure of dairy cattle to heat stress can lead to enhanced ROS production, oxidative stress and cytotoxicity [20]. Oxidative stress can interfere with metabolic and physiological pathways by damaging biological macromolecules [21]. Moreover, heat stress and oxidative stress induce very similar gene expression patterns in dairy cattle; for example, they induce heat-shock proteins and antioxidant enzymes [22]. The results of this study show that the gene expression of VEGF and PTGS2 in lymphocytes significantly increases in order to defend against oxidative stress in Jersey dairy cattle when the THI increases. Furthermore, the results show that the PTGS2 gene plays important roles in oxidative stress, oxidation-reduction processes, cellular oxidant detoxification, peroxidase activity, prostaglandin-endoperoxide synthase activity, and dioxygenase activity. It is possible that VEGF and PTGS2 expression in the VEGF signaling pathway promotes the production of antioxidants and enzymes.
Moreover, the results of this study show that the main signaling pathways are the AGE-RAGE pathway, the VEGF pathway, cytokine-cytokine receptor interactions, and TNF and HIF-1 pathways. These pathways mediate hypoxia responses and immunosuppression and are enriched for the VEGF and PTGS2 genes as well. Lee et al. [26] found that VEGF plays vital roles in hypoxia responses by controlling the expression of numerous hypoxia-responsive genes functioning in diverse processes of oxygen delivery. In addition, an investigation has shown that VEGF not only regulates oxygen supply and growth but also acts as a mediator of inflammatory cytokines [27].
Moreover, Ferrara et al. [28] have proven that in low-oxygen environments, VEGF can bind to the VEGF receptor on the endothelial cell membrane, causing receptor autophosphorylation. Therefore, it is possible that heat stress may cause immunosuppression, oxidative stress and endocrine disorder by significantly increasing VEGF and PTGS2 gene expression when the THI increases. In addition, heat may induce the VEGF signaling pathway to increase PTGS2 release. Camacho et al. [29] found that PTGS2 increases PGI2 release in human vascular cells. During exposure to inflammatory stimuli, blood vessels are also induced to contract by PGI2. Given that this study revealed that both the VEGF and PTGS2 genes are upregulated by THIs of 77, 82 and 88, it is possible that blood vessels in the lung exhibit changes in resistance during panting in dairy cattle.
Conclusions
The present study shows that RR, pH, and pCO2 are significantly affected by the THI and that Jersey dairy cattle (Bos taurus) employ panting-type respiration to dissipate excess body heat when the THI is close to 90. A total of 212 DEGs were screened from different pairs of THI groups (THI77, THI82 and THI88). Furthermore, 16 heat-related DEGs were screened between 212 DEGs and 1370 heat-related genes. Importantly, four hub genes (ITBG3, PTGS2, VEGF, and MYL9) related to immunosuppression, oxidative stress, and endocrine disorder were identified from the 16 genes. It is possible that VEGF and PTGS2 upregulate the VEGF signaling pathway to improve oxygenation in dairy cattle under heat stress. These findings will help elucidate the underlying immune and oxygen regulation mechanisms of the VEGF signaling pathway in dairy cattle under heat stress.
Methods
This experiment was conducted with a random complete design (RCD) involving environmental temperature evaluation, identification of the physiological panting response, screening and analysis of heat-related differentially expressed genes, and gene verification (Fig. 5).

Diagram of the experimental design
Environment and experimental animals
Randomly selected healthy and dry Jersey dairy cattle (n = 5, 340 ± 10 kg, 4.5 years old) naturally exposed at hot environment in Chongqing (29.4316° N, 106.9123° E) were sampled continuously for 1 week from July to August of 2019 in this study.
Physiological data for the physiological response test, blood sample collection for the gas exchange test and lymphocyte isolation
Physiological data including rectal temperature (RT), respiration rate (RR) and skin temperature (ST), as well as blood samples, were collected at 06:00, 10:00 and 14:00. Simultaneously, meteorological data were recorded using dry- and wet-bulb thermometers. The temperature and humidity index (THI) was calculated as described by Bohmanova et al. [30]. THI was calculated by the equation:
Where Tdb is dry-bulb temperature and Twb is wet- bulb temperature.
RRs were determined by observing and counting the number of flank movements for 60 s. RT was measured using a digital clinical thermometer, whereas ST on skin with and without hair was measured with an infrared surface thermometer (Everest Interscience Inc., Fullerton, CA, USA). Blood pH and the partial pressure of carbon dioxide (pCO2) were measured with a Roche Omni C blood gas analyzer (MedWOW, UK). Whole-blood samples (10 ml) were obtained from the juguglar veins of Jersey dairy cattle and by coccygeal vein puncture with permission and were keptc with K2 EDTA anticoagulant to isolate lymphocytes. Briefly, blood samples were added to a tube (227,290, Greiner) and centrifuged at 1500 g at room temperature (18 °C) in a horizontal rotor (swing-out head) for a minimum of 20 min. After centrifugation, mononuclear cells and platelets were collected immediately. Mononuclear cells and cell pellets were resuspended by gentle vortexing with PBS and centrifuged at 1500 g for 10 min. The supernatant was aspirated as much as possible without disturbing the cell pellet, and then the lymphocytes were resuspended in PBS to isolate the cell pellet successfully for the subsequent procedure [31].
RNA isolation, cDNA library construction and sequencing
Total RNA was isolated using TRIzol® Reagent according to the manufacturer’s protocol (Invitrogen, USA), and genomic DNA was removed using DNase I (Takara, Japan). RNA concentrations, purity and integrity were determined with a NanoDrop ND-2000 (NanoDrop Technologies, Inc., Wilmington, DE, USA) and with an RNA Nano 6000 Assay Kit on a Bioanalyzer 2100 system (Agilent Technologies, CA, USA). An RNA-seq transcriptome library was prepared using a TruSeq™ RNA sample preparation kit (Illumina, San Diego, CA, USA). Briefly, mRNA was isolated according to the polyA selection method with oligo (dT) beads and then fragmented with fragmentation buffer. Double-stranded cDNA was synthesized using a SuperScript Double-Stranded cDNA Synthesis Kit (Invitrogen, CA, USA) with random hexamer primers (Illumina, San Diego, CA, USA). Then, the synthesized cDNA was subjected to end repair, phosphorylation and ‘A’ base addition according to Illumina’s library construction protocol. The libraries were size-selected for cDNA target fragments of 200–300 bp on 2% Low Range Ultra Agarose and then subjected to PCR amplification using Phusion DNA polymerase (NEB, China) for 15 PCR cycles. After quantification with a TBS-380 instrument, the paired-end RNA-seq sequencing library was sequenced with the Illumina HiSeq X Ten (2 × 150 bp read length).
Functional annotation and pathway analysis
Functional enrichment analyses including Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses were performed to identify the significantly enriched GO terms and pathways for the DEGs. GO functional enrichment and KEGG pathway analysis were carried out with the GO R package GOATOOLS and KOBAS, respectively [32].
Library data analysis
The raw data were first filtered. The obtained clean data were compared with the reference genome of the species. To identify DEGs between two different samples, the expression level of each transcript was calculated according to the fragments per kilobase of exon per million mapped reads (FPKM) method. RSEM was used to quantify gene abundances, and the expression of each gene was calculated. Then, differential expression analysis and KEGG and GO [33] enrichment analyses were performed with the Database for Annotation, Visualization and Integrated Discovery (DAVID) bioinformatics software (version 2.5.0) [34].
Quantitative real-time polymerase chain reaction validation (Q-PCR)
To ensure the reliability of the mRNA-seq data, an RT Reagent Kit with gDNA Eraser (Takara, Chengdu, China) was used according to the manufacturer’s protocol. Q-PCR was performed on a StepOnePlus Real-Time PCR System (Life Technologies, Gaithersburg, MD, USA) according to standard methods using Fast Start Universal SYBR Green Master (ROX) (Roche, Mannheim, Germany). The relative expression of target genes was normalized to the expression of β-actin. The primers used for Q-PCR are given in Table 4. The relative gene expression values were calculated by the 2-ΔΔCt method [35].
Heat-related DEG coexpression analysis and VEGF pathway visualization
Sixteen heat-related DEGs were identified by coexpression analysis with a bioinformatics software package in DAVID (version 2.0). The VEGF pathway was drawn with BioRender (https://biorender.com/).
Immunoblotting
The samples were lysed and analyzed by immunoblotting according to a previously reported protocol [36]. Denatured protein samples (20 to 50 μg) in Laemmli buffer (Alfa Aesar, Heysham, UK) were separated on 4 to 20% sodium dodecyl sulfate-polyacrylamide gels and transferred by electroblotting onto nitrocellulose membranes (Bio-Rad, Watford, UK). The membranes were then incubated overnight at 4 °C with primary antibodies (diluted 1:1000 in PBS). The primary antibodies included anti-VEGF (catalog no. ab46154, Abcam), anti-PTGS2 (catalog no. ab169782, Abcam) and anti-β-Actin (clone AC-74, Sigma-Aldrich, Gillingham, UK). The secondary antibody was HRP-linked anti-rabbit IgG (polyclonal, Cell Signaling). The blots were incubated with SuperSignal West Dura substrate (Thermo), developed with Amersham Hyperfilm (GE Healthcare, Chalfont St. Giles, UK) or imaged with a Bio-Rad gel imager and analyzed using ImageJ or Image Lab (Bio-Rad). Semiquantitative densitometric analysis, the results of which are dependent on the detection method, exposure time and substrate reaction kinetics, does not imply linearity of the measurements. The resulting expression values supported the protein representation of the bands, increasing the reliability of the results.
Statistical analysis
All data were statistically analyzed in SAS software [37] using one-way ANOVA. Analyses were conducted on both environmental and physiological data. In addition, the GO and KEGG pathway enrichment statistics were analyzed by Fisher’s exact test with the following cutoffs: a p < 0.05 was considered to indicate a significant difference, while a p < 0.01 was considered to indicate a highly significant difference. Furthermore, the Q-PCR validation and immunoblotting data were statistically analyzed using one-way ANOVA with Tukey post hoc testing. The experimental error is shown as the standard deviation, standard error of the mean or 95% confidence interval depending on the experiment. The significance levels are shown as follows: *, p ≤ 0.05; **, p ≤ 0.01; and ***, p ≤ 0.001.
Availability of data and materials
The data of transcriptome analysis would show with excel involved total DEGs, up-regulation DEGs and down-regulation DEGs among THI77, THI82 and THI88; The 16 genes of heatmap, GO analysis, KEGG analysis, gene analysis, relative analysis.
Abbreviations
- VEGF:
-
Vascular endothelial growth factor
- DEGs:
-
Differentially expressed genes
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- GO:
-
Gene ontology
- HIF-1:
-
Hypoxia inducible factor-1
- ITBG3:
-
Integrin beta 3
- OAS2:
-
2'-5'-oligoadenylate synthetase 2
- MX2:
-
MX dynamin like GTPase 2
- IFIT5:
-
Interferon induced protein with tetratricopeptide repeats 5
- NR4A2:
-
Nuclear receptor subfamily 4 group A member 2
- CXCL8:
-
C-X-C motif chemokine ligand 8
- IL1A:
-
Interleukin 1 alpha
- DUP6:
-
AF356445.1(Dual specificity phosphatase 6)
- ICAM1:
-
Intercellular adhesion molecule 1
- END1:
-
HNHc-like endonuclease
- SAMSN1:
-
SAM domain, SH3 domain and nuclear localization signals 1
- BHLHE40:
-
Basic helix-loop-helix family member e40
- PTGS2:
-
Prostaglandin-endoperoxide synthase 2
- TGFB2:
-
Transforming growth factor beta 2
- DNAJB13:
-
DnaJ heat shock protein family (Hsp40) member B13
- TNF:
-
Tumor necrosis factor
- THI:
-
Temperature humidity index
- Hb:
-
Hemoglobin
- HbO2 :
-
Oxyhemoglobin
- pO2 :
-
Partial pressure of oxygen
- ANOVA:
-
Analysis of variance
- PPI:
-
Protein–protein interaction
- RT:
-
Rectal temperature
- RR:
-
Respiratory rate
- ST:
-
Skin temperature
- FRKM:
-
Fragments per kilobase of exon per million mapped reads
- MF:
-
Molecular function
- BP:
-
Biological process
- CC:
-
Cellular component
- CO2 :
-
Carbon dioxide
- Bpm:
-
Respiration rate unite:breath per minute
- ml:
-
Millilitre
- °C:
-
Degree Celsius
References
Wang J, Duangjinda M, Vajrabukka C, Katawatin S. Differences of skin morphology in Bos indicus, Bos taurus, and their crossbreds. Int J Biometeorol. 2014;58(6):1087–94. https://doi.org/10.1007/s00484-013-0700-9.
Wang J, Ke Y, Cheng L. Physiological responses and lactation to cutaneous evaporative heat loss in Bos indicus, Bos taurus, and their crossbreds. Asian Austral J Anim. 2015;8(11):1558–64. https://doi.org/10.5713/ajas.14.0526.
Wideman RF, Fedde MR, Tackett CD, Weigle GE. Cardio-pulmonary function in preascitic (hypoxemic) or normal broilers inhaling ambient air or 100% oxygen. Poult Sci. 2000;79(3):415–25. https://doi.org/10.1093/ps/79.3.415.
Liu S, Yue T, Ahmad MJ, Hu X, Zhang X, Deng YH, et al. Transcriptome analysis reveals potential regulatory genes related to heat tolerance in holstein dairy cattle. Genes (Basel). 2020;11(1):68. Published 2020 Jan 7. https://doi.org/10.3390/genes11010068.
Salak-Johnson JL, McGlone JJ. Making sense of apparently conflicting data: stress and immunity in swine and cattle. J Anim Sci. 2007;85(13 Suppl):E81–8. https://doi.org/10.2527/jas.2006-538.
Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. 2009;30(10):475–87. https://doi.org/10.1016/j.it.2009.07.009.
Shalova IN, Lim JY, Chittezhath M, Zinkernagel AS, Beasley F, Hernández-Jiménez E, et al. Human monocytes undergo functional re-programming during sepsis mediated by hypoxia-inducible factor-1α. Immunity. 2015;42(3):484–98. https://doi.org/10.1016/j.immuni.2015.02.001.
Min L, Cheng J, Zhao S, Tian H, Zhang Y, Li S, et al. Plasma-based proteomics reveals immune response, complement and coagulation cascades pathway shifts in heat-stressed lactating dairy cows. Proteomics. 2016;146:99–108. https://doi.org/10.1016/j.jprot.2016.06.008.
Campillo N, Falcones B, Otero J, Gozal D, Naniel N, Famon F, et al. Differential oxygenation in tumor microenvironment modulates macrophage and cancer cell crosstalk: novel experimental setting and proof of concept. Front Oncol. 2019;9:43. Published 2019 Feb 6. https://doi.org/10.3389/fonc.2019.00043.
Lin XM, Luo W, Wang H, Li RZ, Huang YS, Chen LK, et al. The role of prostaglandin-endoperoxide synthase-2 in chemoresistance of non-small cell lung cancer. Front Pharmacol. 2019;10:836. https://doi.org/10.3389/fphar.2019.00836.
Minchenko DO, Tsymbal DO, Riabovol OO, Viletska YM, Lahanovska YO, Sliusar MY, et al. Hypoxic regulation of EDN1, EDNRA, EDNRB, and ECE1 gene expressions in ERN1 knockdown U87 glioma cells. Endocr Regul. 2019;53(4):250–62. https://doi.org/10.2478/enr-2019-0025.
Herring JA, Elison WS, Tessem JS. Function of Nr4a orphan nuclear receptors in proliferation, apoptosis and fuel utilization across tissues. Cells. 2019;8(11):1373. https://doi.org/10.3390/cells8111373.
McCoy JM, Walkenhorst DE, McCauley KS, Elaasar H, Everett JR, Mix KS. Orphan nuclear receptor NR4A2 induces transcription of the immunomodulatory peptide hormone prolactin. J Inflamm-Lond. 2015;12(1):13. https://doi.org/10.1186/s12950-015-0059-2.
Doi Y, Oki S, Ozawa T, Hohjoh H, Miyake S, Yamamura T. Orphan nuclear receptor NR4A2 expressed in T cells from multiple sclerosis mediates production of inflammatory cytokines. PNAS. 2008;105(24):8381–6. https://doi.org/10.1073/pnas.0803454105.
Li XP, Yang XY, Biskup E, Zhou J, Li HL, Wu YF, et al. Co-expression of CXCL8 and HIF-1α is associated with metastasis and poor prognosis in hepatocellular carcinoma. Oncotarget. 2015;6(26):22880–9. https://doi.org/10.18632/oncotarget.4412.
Singh IS, Gupta A, Nagarsekar A, Cooper Z, Manka C, Hester L, et al. Heat shock co-activates interleukin-8 transcription. Am J Resp Cell Mol. 2008;39(2):235–42. https://doi.org/10.1165/rcmb.2007-0294OC.
Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nat Rev J Cancer. 2008;8(12):967–75. https://doi.org/10.1038/nrc2540.
Kimura MY, Koyama-Nasu R, Yagi R, Nakayama T. A new therapeutic target: the CD69-Myl9 system in immune responses. Semin Immunopathol. 2019;41(3):349–58. https://doi.org/10.1007/s00281-019-00734-7.
Chen S, Wang J, Peng D, Li G, Chen J, Gu X. Exposure to heat-stress environment affects the physiology, circulation levels of cytokines, and microbiome in dairy cows. Rep Sci. 2008;8(1):14606. https://doi.org/10.1038/s41598-018-32886-1.
Bernabucci U, Ronchi B, Lacetera N, Nardone A. Markers of oxidative status in plasma and erythrocytes of transition dairy cows during hot season. J Dairy Sci. 2002;85(9):2173–9. https://doi.org/10.3168/jds.S0022-0302(02)74296-3.
Zhang H, Yin M, Huang L, Wang J, Gong L, Liu J, et al. Evaluation of the cellular and animal models for the study of antioxidant activity: a review. J Food Sci. 2017;82(2):278–88. https://doi.org/10.1111/1750-3841.13605.
Slimen IB, Najar T, Ghram A, Abdrrabba M. Heat stress effects on livestock: molecular, cellular and metabolic aspects, a review. J Anim Physiol Anim Nutr. 2016;100(3):401–12. https://doi.org/10.1111/jpn.12379.
Liu S, Wang X, Sun F, Zhang J, Feng J, Liu H, et al. RNA-Seq reveals expression signatures of genes involved in oxygen transport, protein synthesis, folding, and degradation in response to heat stress in catfish. Physiol Genomics. 2013;45(12):462–76. https://doi.org/10.1152/physiolgenomics.00026.2013.
Smith DL, Smith T, Rude BJ, Ward SH. Short communication: comparison of the effects of heat stress on milk and component yields and somatic cell score in Holstein and Jersey cows. J Dairy Sci. 2013;96(5):3028–33. https://doi.org/10.3168/jds.2012-5737.
Wang Q, Onuma K, Liu C, Wong H, Bloom MS, Elliott EE, et al. Dysregulated integrin αvβ3 and cd47 signaling promotes joint inflammation, cartilage breakdown, and progression of osteoarthritis. JCI Insight. 2019;4(18):e128616. Published 2019 Sep 19. https://doi.org/10.1172/jci.insight.128616.
Lee DC, Sohn HA, Park ZY, Oh S, Kang YK, Lee KM, et al. A lactate-induced response to hypoxia. Cell. 2015;161(3):595–609. https://doi.org/10.1016/j.cell.2015.03.011.
Kasai N, Kojima C, Sumi D, Ikutomo A, Goto K. Inflammatory, oxidative stress, and Angiogenic growth factor responses to repeated-Sprint exercise in hypoxia. Front Physiol. 2019;10:844. https://doi.org/10.3389/fphys.2019.00844.
Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9(6):669–76. https://doi.org/10.1038/nm0603-669.
Camacho M, Rodríguez C, Guadall A, Alcolea S, Orriols M, Escudero JR, et al. Hypoxia upregulates PGI-synthase and increases PGI2 release in human vascular cells exposed to inflammatory stimuli. J Lipid Res. 2011;52(4):720–31. https://doi.org/10.1194/jlr.M011007.
Bohmanova J, Misztal I, Cole JB. Temperature-humidity indices as indicators of milk production losses due to heat stress. J Dairy Sci. 2007;90(4):1947–56. https://doi.org/10.3168/jds.2006-513.
Feezor RJ, Baker HV, Mindrinos M, Hayden D, Tannahill CL, Brownstein BH, et al. Inflammation and host response to injury, large-scale collaborative research program. Whole blood and leukocyte RNA isolation for gene expression analyses. Physiol Genomics. 2004;19(3):247–54. https://doi.org/10.1152/physiolgenomics.00020.
Xie C, Mao X, Huang J, Ding Y, Wu J, Dong S, et al. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011;39(Web Server issue):W316–22. https://doi.org/10.1093/nar/gkr483.
Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011;12(1):323. https://doi.org/10.1186/1471-2105-12-323.
Huang D, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. https://doi.org/10.1038/nprot.2008.211.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods (San Diego, Calif.). 2001;25(4):402–8. https://doi.org/10.1006/meth.2001.1262.
Mahmood T, Yang PC. Western blot: technique, theory, and trouble shooting. N Am J Med Sci. 2012;4(9):429–34. https://doi.org/10.4103/1947-2714.100998.
SAS. SAS/STATTM. Guide for personal computers version, 6th edn. Cary: SAS Institute; 1988.
Acknowledgments
The authors thank the Guangda Dairy Farm (Xiaohui Xu) for providing the experimental Jersey dairy cattle for physiological data collection and blood sample collection. The authors would like to acknowledge the animal medicine workshop (Meilan Jin) at Southwest University for advice on high-throughput sequencing by liquid chromatography-mass spectrometry, Q-PCR and immunoblotting. The authors would like to acknowledge the following scientific research facility funded by the Department of Oncology at the University of Oxford for advice related to immunity and hypoxia, statistical analysis and manuscript preparation: the mechanical workshop (Len Seymour group).
Funding
This research was funded by the National Natural Science Foundation of China (NSFC) grant 31501991 to support J. W for the collection. Additionally, J.W. was supported by the Chongqing People’s Livelihood Project (cstc2017shms-xdny80002) for the analysis, and the Key Research and Development Projects of the Chongqing Social and Livelihood Sectors (cst2019jscx-gksb0321) for the interpretation of data, Hongchang Li was supported by the University Student Innovation Project (S201010635045) for writing the manuscript.
Author information
Authors and Affiliations
Contributions
Conceptualization: J.W. Investigation, methodology, and formal analysis: J.W., Y.X., H.C.L and C.F. Statistical analysis: M.D. and S.K. Writing—whole draft preparation: J.W. Writing – review and editing: J.W., S.S.J., H.C.L and C.F. Visualization: J.W. Supervision: S.S.J. and J.W. Funding acquisition: J.W. All authors have read and agreed to the published version of the manuscript.
Authors’ information
Experimental design and reporting, Jian Wang from southwest University in Chongqing, China; Experimental was done by Yang Xiang and Hong Chang Li from Southwest University in Chongqing China. Data was analysis by Monchai Duangjingda and Suporn Katawatin from Khon Kaen University at Khon Kaen in Thailand; mRNA transcriptome analysis and visualization by Shisong Jiang and Flurin Caviezel from Oxford University at Oxford in UK.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
It was approved IACUC No.IACUC2019004 from veterinary faculty southwest university. Project named Involvement of the VEGF Signaling Pathway in Immunosuppression and Hypoxia Stress: Analysis of mRNA Expression in Lymphocytes Mediating Panting in Jersey Cattle under Heat Stress. Animal Experiment Protocol was investigated by principle investigator Dr. Jian Wang. This Project has been supervised and approved by Institutional Animal Care and Use Committee (IACUC) of Southwest University.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
The 16 genes enriched at GO and KEGG related to immunosuppression, oxidative stress, and endocrine disorder in lymphocytes under heat stress.
Additional file 2.
Complemary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, J., Xiang, Y., Jiang, S. et al. Involvement of the VEGF signaling pathway in immunosuppression and hypoxia stress: analysis of mRNA expression in lymphocytes mediating panting in Jersey cattle under heat stress. BMC Vet Res 17, 209 (2021). https://doi.org/10.1186/s12917-021-02912-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12917-021-02912-y