
Abstract
Background
Incubation period, disease progression, pathology and clinical presentation of classical scrapie in sheep are highly dependent on PRNP genotype, time and route of inoculation and prion strain. Our experimental model with pre-colostrum inoculation of homozygous VRQ lambs has shown to be an effective model with extensive PrPSc dissemination in lymphatic tissue and a short incubation period with severe clinical disease. Serum protein analysis has shown an elevation of acute phase proteins in the clinical stages of this experimental model, and here, we investigate changes in gene expression in whole blood, liver and brain.
Results
The animals in the scrapie group showed severe signs of illness 22 weeks post inoculation necessitating euthanasia at 23 weeks post inoculation. This severe clinical presentation was accompanied by changes in expression of several genes. The following genes were differentially expressed in whole blood: TLR2, TLR4, C3, IL1B, LF and SAA, in liver tissue, the following genes differentially expressed: TNF-α, SAA, HP, CP, AAT, TTR and TF, and in the brain tissue, the following genes were differentially expressed: HP, CP, ALB and TTR.
Conclusions
We report a strong and evident transcriptional innate immune response in the terminal stage of classical scrapie in these animals. The PRNP genotype and time of inoculation are believed to contribute to the clinical presentation, including the extensive dissemination of PrPSc throughout the lymphatic tissue.
Similar content being viewed by others


Background
Prion diseases are a group of diseases with common pathology of the central nervous system including neuronal degeneration, vacuolation and gliosis [1]. The causative agent is an infectious protein, called the prion (PrPSc), which is an abnormal isoform of the cellular prion protein, PrPC, found in many cell types in the body [2]. Prion diseases are also named Transmissible Spongiform Encephalopathies (TSEs), and scrapie is the TSE that affects sheep and goats. Sheep can become infected early in life, around and soon after birth, from infectious prions in foetal fluids and membranes and bodily secretions like colostrum/milk and faeces [3,4,5,6].
Susceptibility to classical scrapie in sheep is highly dependent on polymorphism at codons 136, 154 and 171 of the PrP gene (PRNP). The five possible alleles, ARR, AHQ, ARH, ARQ and VRQ, give rise to 15 combinations found in sheep, and these are also linked to the different presentations of classical scrapie [7]. These genotypes differ widely in their susceptibility to scrapie, from extreme susceptibility in homozygous V136R154Q171 genotype, to very low susceptibility in homozygous A136R154R171 [8,9,10]. The PrP genotype not only influences the degree of susceptibility, but also the course of disease, incubation period and clinical picture [7, 11]. This genetic susceptibility is the basis behind the breeding for resistance program successfully implemented in the EU as per EC Regulation 999/2001, where only ewes carrying at least one ARR allele and no VRQ allele, and rams of ARR/ARR genotype, are bred from [12]. Several member states have implemented breeding programs to select for resistance to TSEs in their ovine population, resulting in a marked increase in ARR frequency and a decreased VRQ frequency. There are now reports of a shift in PRNP genotypes in national flocks after years of breeding from low susceptibility genotypes [13,14,15,16]. Thus, the prevalence of classical scrapie at population level decreased as highly susceptible genotypes are eliminated.
Presentation and progression of disease are influenced by PRNP genotype, PrPSc strain, and route of inoculation. Lateral transmission via the oral route is the most important and commonest route of infection, and in a flock where scrapie is endemic, new-born lambs become infected at birth from their infected dam and/or from the contaminated environment [17]. After oral infection, prions seem to be taken up across the gastrointestinal mucosa by M-cells, and dendritic cells (DCs) and possibly macrophages are involved in the spread to the lymphoreticular system (LRS). Follicular dendritic cells (FDCs) within the LRS, are the site of prion replication, but the extent and speed of lymphatic involvement is dependent on PRNP genotype [18]. Sheep carrying the least susceptible ARR allele show minimal PrPSc deposition within the LRS, while carriers of the VRQ allele are associated with extensive PrPSc deposition [19,20,21,22,23,24]. When lambs with the most susceptible genotype are orally challenged with scrapie infection, PrPSc is detected very early in lymphoid tissues associated with the gastrointestinal tract, and is followed by dissemination of PrPSc to other, non-gastrointestinal, lymphoid tissues [24,25,26]. Due to this early PrPSc accumulation in lymphatic tissue in certain genotypes, the European Food Safety Authority considers young stock, less than six months of age and from TSE infected flocks, as potentially highly infectious [27].
The pathogenesis and progression of TSE in different species is still not fully understood, and sheep with classical scrapie is a good model to study the disease mechanisms. Pre-colostrum inoculation of lambs at birth is an effective model that has shown to produce severe clinical disease after a relatively short incubation period [28]. One can say, our model produces a “worse-case scenario” of prion disease, where the genotype, donor material and time of infection are optimal. Ersdal and co-workers reported a study of PrPSc uptake and dissemination in an earlier experimental model, where older lambs where orally inoculated, with the same VRQ-prion donor material, between 46 and 61 days of age [22]. Later, this experimental model was repeated, only this time the lambs where inoculated at birth, and produced a much shorter incubation period and more severe clinical disease [28]. Both these models revealed an early and strong dissemination of PrPSc in lymphatic tissue in combination with relatively moderate cytopathogenic changes in the CNS in VRQ/VRQ animals. We have earlier published our findings of an acute phase response in lambs inoculated at birth, evident at serum protein level [29]. In the present study, transcriptional changes of the above lambs were (further) studied. This article describes the expression of a selection of genes in RNA isolated from whole blood, liver and posterior obex of healthy and scrapie affected lambs.
Many genes are expressed by leukocytes in the blood as a systemic response to pathology in peripheral tissues. Injured and diseased tissues and organs will alter the gene expression profile in blood leukocytes and in the liver, which is the main producer of acute phase proteins associated with an innate immune response. Several studies of gene expression in circulating blood cells have revealed expression profiles characteristic for a wide variety of diseases [30,31,32]. Gene expression studies of whole blood can thus improve our knowledge of disease related processes regardless of which tissue is primarily affected. White blood cells have an important role in the immune system, and, although scrapie is known to not produce a specific immune response per se, cells of the monocyte line seem to play an important role in the uptake from the gut and dissemination of PrPSc to lymphoid organs and further into the CNS resulting in gliosis and astrocytosis [20, 22].
Collection and stabilization of total RNA from whole blood using PAXgene Blood RNA Tubes (BD Biosciences) offer a robust and simple method that minimize sampling-to-sampling differences. PAXgene technology capture the RNA profiles at the time of collection of all cell types in whole blood, including peripheral blood monocytes, lymphocytes, erythrocytes/reticulocytes, granulocytes and platelets [33].
The aim of this study was to investigate how scrapie infection influences the gene expression profile of selected genes in whole blood during the preclinical incubation period and through to the terminal clinical end stage. Further, the gene expression patterns were studied in liver and posterior obex at the terminal end stage.
Methods
Animals
The animals and experimental model used in this project were described in detail previously, and all animal procedures were approved by the Norwegian Animal Research Authority [28, 29]. Briefly, nine lambs were inoculated by stomach tube, at birth and before colostrum intake, with pooled brain material from either healthy, scrapie free sheep (control group) or from confirmed cases of classical scrapie (scrapie group). All the lambs originated from the commercial sheep flock belonging to the Norwegian University of Life Sciences, Faculty of Veterinary Medicine, Department of Production Animal Clinical Sciences, Section of Small Ruminant Research, 4325 Sandnes, Norway. Within this flock, a number of animals have selectively been bred to maintain the homozygous V136R154Q171 PRNP genotype. The control group consisted of two twins (four lambs) and the scrapie group consisted of triplets and twins (five lambs). The lambs and their dams were housed individually until euthanasia at 23 weeks of age. All the animals were inspected daily, and the scrapie group was under video surveillance. Euthanasia was performed by an intravenous injection of sodium pentobarbital, using the jugular vein.
Blood and tissue collection and RNA isolation
Blood samples were collected at regular intervals from the jugular vein into appropriate blood tubes, depending on the requirement for the different analyses. Whole blood (2.5 mL) was collected at 12, 14, 16, 20, 22 and 23 weeks post inoculation (wpi) in PAXgene Blood RNA Tubes (Qiagen/BD Company). All the samples were handled according to manufacturer’s instructions and stored at minus 70 °C until analysis.
Brain and liver tissues were removed immediately post mortem and placed in RNAlater (Qiagen) prior to storage at minus 70 °C. Total RNA was extracted and purified from stabilized blood samples manually, using PAXgene® Blood miRNA Kit (Qiagen/BD Company) according to manufacturer’s instructions using the protocol for manual purification. The RNA eluate was immediately stored at minus70°C.
Total RNA was isolated from 20 to 25 mg tissue sections, lysed and homogenized, from the posterior obex region in the brain and from the liver using RNeasy® Lipid Tissue Midi Kit and (Qiagen) and RNeasy® Lipid Tissue Mini Kit (Qiagen), respectively, including digestion of DNA using RNase-Free DNase kit (Qiagen).
Total RNA was quantified by UV spectrometry at 260 nm using a NanoDrop ND-1000 spectrophotometer (Saveen and Werner AB, Limhamn, Sweden). In addition, for each sample 260/280 and 260/230 ratios were provided, enabling RNA sample purity estimations. RNA integrity was evaluated using the Agilent® 2100 Bioanalyzer with Agilent RNA 6000 Nano Kit (Agilent Technologies, Waldbronn, Germany), according to manufacturer’s instructions. Each total RNA sample was assigned an RNA Integrity Number (RIN) ranging from 1 to 10. The mean and standard deviation of all the RIN values were calculated and are shown in Additional file 1.
Target genes
Two different primer pairs were designed to amplify different regions of each gene to ensure high quality results. Sequences used for primer design were obtained from public database (GenBank, NCBI) and are listed together with reaction conditions in Additional file 2. Primers were designed using Primer3 (http://bioinfo.ut.ee/primer3-0.4.0/primer3/) and synthesized at TAG Copenhagen A/S (Copenhagen, Denmark). BLAST searches were performed to confirm gene specificity of the primer sequence, and to show the absence of polymorphisms at the primer site. Whenever possible, only sequences from Ovis aries were used, if not, sequences specific for Bos taurus or Capra ibex were used. Further, NetPrimer software available at http://www.premierbiosoft.com was used to obtain a rating value for each primer pair.
High-throughput qPCR
Two separate technical replicates of complementary DNA (cDNA) were synthesised from each RNA sample using 300 ng of total RNA using the QuantiTect reverse-transcription kit (Qiagen®/BD Company), according to the manufacturer’s instructions. All samples were diluted, with a dilution factor of 0.13, in low EDTA TE-buffer (VWR, APLIA8569.0500) in a total solution of 20 μl.
Custom-designed primers (TAG Copenhagen, Copenhagen, Denmark) were diluted to 100 μM. For preamplification and qPCR, each primer pair, consisting of forward and reverse primers was further diluted in low EDTA TE-buffer (VWR, APLIA8569.0500) to reach a final concentration of 20 μM. Then a primer mix was created from 5 μl of each 20 μM primer pair and low EDTA TE-buffer in a 500 μl, 200 nM solution. A PreAmp mix, consisting of 5 μl TaqMan PreAmp Master Mix (Applied Biosystems, PN 4391128) and 2.5 μl 200 nM primer mix, was added to 2.5 μl of each cDNA sample. This mix was incubated under thermal cycling conditions as follows; initially held at 95 °C for 10 min, followed by 16 pre-amplification cycles of 15 s at 95 °C and 4 min at 60 °C, before returning to 4 °C. Before storage at − 20 °C, each sample was diluted 1:4 in low EDTA TE-buffer. The liver samples were optimised by a second run, L2; using a 1:10 dilution.
High-throughput qPCR was performed in 48.48 Dynamic Arrays using the BioMark thermocycler (Fluidigm Corporation, CA, USA) following the manufacturer’s protocol. The 48.48 dynamic array enables 2304 separate and simultaneous qPCR reactions from 48 samples and 48 primers in one operation. Each 48.48 dynamic array was prepared with control line fluid and primed in an IFC Controller MX (Fluidigm Corporation, CA, USA) while assays and samples were prepared. The Assay master mix (2.75 μl), consisted of 2.5 μl loading reagent (85,000,800 Sample and Assay Loading kit, Fluidigm) and 0.25 μl low EDTA TE-buffer, were combined with 2.3 μl 20 μM of each specific primer pair before loading each corresponding assay inlets on the 48.48 dynamic array. The Pre-sample mix (4.5 μl), consisting of 3 μl TaqMan Gene Expression Master mix (Applied Biosystems, PN 4369016), 0.3 μl DNA binding dye (100–0388, Fluidigm), 0.3 μl EvaGreen (VWR, BTIU31000) and 0.9 μl low EDTA TE-buffer, and 1.5 μl preamplified cDNA sample were added to each corresponding sample inlets on the 48.48 dynamic array. The 48.48 dynamic array was then returned to the IFC Controller to load samples and assays into the Integrated Fluidic Circuit of the dynamic array. The 48.48 dynamic array was placed in the BioMark HD instrument for qPCR with a ten-minute hot start phase at 95 °C followed by 35 cycles of denaturing at 95 °C for 15 s and annealing/elongation at 60 °C for 1 min. Melting curves were generated after each run to confirm the presence of a single PCR product (from 60 °C to 95 °C, increasing 1 °C/3 s). Non-template controls, non-reverse transcriptase controls and three interplate calibrators were included on each dynamic array if data from multiple dynamic arrays were analysed.
Data collection and pre-processing
RT-qPCR data was collected using Fluidigm Real-Time PCR Analysis Software Version 3.0.2 (Fluidigm Corporation, CA, USA). Melting curves and non-template control (NTC) were used to monitor non-specific amplification and sample contamination. Non-reverse transcriptase controls were used to assess potential genomic DNA contamination. Each sample was examined to confirm a single PCR product, and lack of such product resulted in the rejection of that quantification cycle (Cq) value.
Expression data were pre-processed and analysed using GenEx Pro Version 5.3.6 (MultiD Analyses AB, Goteborg, Sweden). Pre-processed data were separated into four data subsets (blood, obex, liver1 and liver2) according to type of sample and time of analysis, and data analysis was performed with GenEx Pro Version 5.3.6 in the following order: 1. Initial visual evaluation of melting curves and amplification curves; 2. Efficiency correction. PCR amplification efficiency was established by the means of calibration curves and determination of the slope (S) of the log-linear portion. The calibration curve was constructed from dilution series (4 × 4) using a pool consisting of 1 μl of each sample tested, and diluted into following relative concentrations: F1 0.333, F2 0.067, F3 0.0133 and F4 0.00267. Both slope value (m) and correlation coefficient R2 for each assay was determined using the above. Efficiency value (E) was calculated as follows: E = 101/m – 1, and used for the efficiency correction of each primer assay individually in GenEx. Any assay with E values less than 0.8 or higher than 1.1, and R2 values less than 0.95, were discarded and excluded for further analysis; 3. Inter-plate calibration (IPC): based on the three interplate calibrators; 4. Normalization to reference genes. Six different genes (RPLP0, PPIA, UBC, HPRT1, GAPDH and SDHA) were evaluated using GeNorm and NormFinder incorporated in GenEx; 5. Averaging technical cDNA replicates. Before cDNA technical replicates were averaged, the data set was inspected for level of variation between replicates. A maximum of 20% of difference in Cq between the two replicates was accepted.; 6. The relative expression of each sample analysed, was calculated using the mean Cq value of the control croup as a base line. Any sample with higher Cq (lower expression) than mean Cq of control group, get a value below 1, and any sample with lower Cq (higher expression) than mean Cq of control group, get a value above 1. This was done for each primer assay individually; 7. Finally, the data was converted into logarithmic scale, using log2 transformation for all statistical analysis. Fold change was calculated by scaling the relative expression data. A scaling factor was calculated based on the average relative expression (fold change) in the control group was scaled to 1. Individual expression data was multiplied by the scaling factor, giving individual fold change. Average fold change and standard error of the mean (SEM) was calculated for each group/primer assay. When relative expression (RE) was below 1, it was translated to a negative fold change value using − 2-(log2RE), i.e. a relative expression of 0.8, gave a fold change of − 1.2.
Statistical analysis
The data were analysed separately using statistical features of GenEx software (MultiD). The pre-processed data from the qPCR analyses was separated into subsets of data, based on time and type of sampling. The non-parametric statistical model, Mann-Whitney U Test, was used to calculate p-values between scrapie and control groups in each of the data subsets, and significant p-value was set to p < 0.05. Due to the small group size, p-values should be interpreted with some caution. Genes were considered as differentially expressed if they showed a fold change of ≥1.5 and reached significant p-values.
Results
Animals
Animals in the control group remained healthy without any clinical symptoms for the full experimental period. No obvious clinical signs of illness were visible in the scrapie group until 22 wpi, although subtle signs of unspecific ill-thrift were observed in two animals from 19 wpi. By the end of 22 wpi., all the animals in the scrapie group showed signs of disease, such as weakness, dullness and lethargy. Two animals in the infected group displayed an accelerated worsening of clinical signs to terminal recumbency and severe depression. This sudden change, over a few days, made euthanasia necessary. Brain tissue from all the animals in both groups was examined for PrPSc by immunohistochemistry (IHC) and Western Blot (WB), and only the animals in the scrapie group were found PrPSc-positive (Fig. 1). There were moderate vacuolation of neuropil and neurons with marked distribution of PrPSc deposits in the CNS, and PrPSc was found to be widely distributed in lymphoid organs (spleen and retropharyngeal lymph nodes), consistent with previous findings [28].

Western Blot image. Western immunoblot using P4 antibody for the detection of PrPSc in equal amount of homogenated brain tissue from animals and inoculation material used in the experiment (TeSeE Western Blot, Bio-Rad). Lanes 1–5 represent the five animals in the scrapie group, and lanes 6 and 7 represent two animals in the control group. Lane 8 represents inoculation material (donor) used in the scrapie group and a molecular marker was placed in lane 9. PrPSc was detected in inoculation material and in all the animals from the scrapie group
Gene expression analysis in blood
Statistically differentially expressed genes, fold change and p-value are summarized in Table 1. The magnitude of the fold change at each time points, reflects the gene expression alterations relative to the mean level of the control group scaled to 1. Of the 10 target genes analysed after pre-processing, only six were significantly differently expressed in the scrapie group compared to the control group at any time point. No genes were differentially expressed at 12 nor 14 wpi. IL1B was differentially expressed at 16 and 22 wpi, and TLR2, TLR4, C3 and SAA were differentially expressed at 22 and 23 wpi. LF was only differentially expressed at 23 wpi. Apart from IL-1B which was downregulated at 16 wpi, all the others were upregulated. Expression levels of TLR9, IL1RN, IL8 and LR/LAMR1 were not significantly altered at any of the time points. (Table 1).
Gene expression analysis in liver at 23 wpi
Due to the small group size, only descriptive analyses were performed. Differentially expressed genes of the scrapie group relative to the control group with a fold change of ≥1.5 can be seen in Table 2. The fold change value is relative to the mean expression level in the control group, set as value 1. Of the 12 target genes analysed after pre-processing, seven genes were expressed with fold change ≥1.5 (Table 2). Three genes were upregulated (SAA, Hp and Cp) and four were downregulated (TNFa, AAT, TF and TTR).
Gene expression analysis in brain at 23 wpi
Gene expression differences in brain tissue are listed in Table 3. Out of 12 target genes analysed after pre-processing, only four were expressed with fold change ≥1.5. Hp and Cp were upregulated, and ALB and TTR were downregulated.
Discussion
Classical scrapie is known to present with a variety of clinical signs, and clinical presentation is dependent on PRNP genotype, PrPSc strain, infective dose and age of host at infection. Our experimental model resembles natural infection and illustrates the “worst-case” scenario which can occur naturally when the right conditions are present. Clinical signs of disease in the scrapie group was first observed at 19 wpi. The symptoms aggravated quickly, and by 23 wpi, two lambs had reached the terminal stages. The very short incubation period was dominated by sudden progressive weakness, followed by rapid worsening and necessitating euthanasia at an early age. Hence, the observed clinical presentation is not typical for classical scrapie known to present in older animals, which is characterised by a slow and long incubation period with signs of progressive neurodegeneration associated with dissemination and accumulation of PrPSc within the CNS. Although this experimental scrapie model attempts to eliminate many of the factors that generate disease variation, individual variation still occurred. Individual progression of clinical disease and expression profiles in blood and tissues differs between animals. The APR is fast, and the level of inflammatory mediators, such as cytokines, changes continuously. Cytokines are multifunctional (both pro-inflammatory and anti-inflammatory) intra-cellular and inter-cellular signalling molecules, operating in a very complex signalling network to initiate and fine-tune the immune response [34,35,36,37,38]. Their levels are tightly controlled within a narrow range by inhibitory mechanisms and antagonists, whilst APPs can increase quite dramatically depending on type of protein [39, 40].
In the present study, we have investigated the expression of a selection of genes in blood approximately half-way through the incubation period, at 12–16 wpi, and at the clinical end-stage, at 22–23 wpi. Expression levels of selected genes were also investigated in liver and brain tissues, harvested at post mortem examination. In summary, the mean expression of six genes were significantly altered in the scrapie group compared to the control group. We were not able to calculate statistical significance in mean expression levels in brain and liver tissues, but we believe that a difference in mean expression levels greater than 1.5 fold change is of biological relevance and will be discussed. Despite our best efforts, only four genes in the brain tissue were differently expressed in the scrapie group, compared to the control group. These findings were unexpected as all the animals in the scrapie group were clinically affected and were positive for PrPSc in the brain on both IHC examination and WB (Fig. 1).
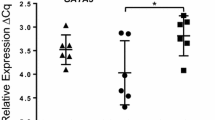
Our overall results, presented in Fig. 2, indicate an innate immune response, where genes involved in pattern recognition, complement system and acute phase proteins, are significantly altered in blood at the clinical stages (22 and 23 wpi). Mean gene expression levels of brain and liver tissues in the scrapie group show ≥1.5 fold change of several APPs. These findings coincide well with our earlier reports on increased serum levels of APPs, and identification of SAA as being a discriminating protein by proteomic analysis of serum from the scrapie group [29, 41].

Bar diagram presenting average fold change and SEM in the scrapie group of selected genes. The fold change value is scaled, in each data set, to the mean fold change value of the control group equals 1 (one)
Prion disease and the innate immune system have previously been reviewed by Bradford and Mabbott, and activation of cells within the innate immune system is critical to peripheral and central prion pathogenesis [42]. Gene network analyses of brain tissue from prion infected mice show that increased expression of C3, PRRs (including TLR2), and other receptors involved with PrPSc recognition and uptake are one of the first transcriptome changes [43, 44]. TLR2 is one of the, at least, ten members of the ovine TLRs family which are membrane-bound PRRs [45]. Our results show significantly increased TLR2 and TLR4 expression in blood from 22 wpi, thus, indicating an increased transcription of these receptors in circulating blood cells. Both these TLRs are situated on the cell surface and recognize a variety of microbial components, and activation will trigger expression of several genes and signalling pathways involved in the immune response [37, 46, 47]. Activation of different TLRs leads to specific gene expression profile patterns, and the specific signalling pathways in prion diseases are not yet fully revealed. TLR2 and TLR4 signalling pathways share a common MyD88-dependent pathway which results in production of inflammatory cytokines. In addition, TLR4 stimulation can also initiate a MyD88-independent pathway [46]. Prinz et al. demonstrated that MyD88−/− mice were highly susceptible to prion infection, and thus pathogenesis and neuroinvasion are not solely dependent on the MyD88-dependent pathway [48]. Spinner et al. showed that scrapie pathogenesis occurs more rapidly in mice mutant of TLR4 signalling and concludes that this may be due to a reduced innate response to PrPSc [49]. The TLRs may be involved in binding PrPSc and initiating the innate immune response [42, 48,49,50,51,52]. Alongside the TLRs, there is a significant increased expression of C3 in blood at both 22 and 23 wpi. Binding of PrPSc to complement components plays an important role in getting the agent to lymphoid tissue and early accumulation in FDCs [53]. PrPSc interacts directly with C1q and thereby activates C3 through the classical pathway [54,55,56]. The complement system, in general, sense and react quickly to danger signals and can potentially initiate a strong inflammatory response, which needs to be tightly balanced between activation and inhibition [54].
Binding to and activation of PRRs lead to the production of pro-inflammatory cytokines that function both protective and degenerative. Increased levels of pro-inflammatory cytokines are associated with the systemic inflammatory response seen in prion infection and other neurodegenerative diseases [57, 58]. Cytokine antibody array analysis of scrapie-infected Tg338 mice shows significant alteration in expression of interleukins in spleen, mesenteric lymph node, brain and serum at both preclinical and clinical stages of infection [59]. We analysed several different interleukins, but only the expression IL1B was significantly changed in blood in the scrapie group; first downregulated at 16 wpi and then upregulated at 22 wpi. IL-1 beta is a powerful pro inflammatory mediator, and increased levels initiate a negative feedback loop [35, 39].
Translation and release of proinflammatory cytokines initiate a wide range of systemic inflammatory effects, including the synthesis and release of APPs mainly from the liver, but also extrahepatically [36, 38, 47, 60]. The APPs are classified as positive or negative depending whether their serum concentration increases or decreases during the APR [36, 47, 61, 62]. Our results show changes at transcription levels of both negative and positive APPs at the end stage. ALB, TTR and TF are negative APPs, and these have decreased expression in brain and liver. ALB is the major negative APP and synthesis is down-regulated during the APR. Transthyretin is a transport protein in serum and CSF, and one of its functions is inhibition of interleukin-1 production by monocytes and endothelial cells, and a decrease may, thus be pro-inflammatory [36]. TTR expression is reduced during the APR and the protein has been seen to decrease in serum in late stage scrapie in sheep [63, 64].
Of the positive APPs, LF, Cp, SAA and Hp have increased expression at clinical end stage, while AAT was down regulated. Alpha-1 antitrypsin (AAT) is a circulating serine protease inhibitor (serpin) and an acute phase protease, and it is classified as a minor positive acute phase protein in ruminants [64]. It is synthesised and secreted from the liver, and it plays an important role in the control of the inflammatory response [47, 65]. Serpinopathies and AAT deficiency have been associated with protein misfolding diseases [65, 66]. We found a decreased transcription of AAT mRNA in the liver in the scrapie group compared to the control group.
Gene expression of lactoferrin (LF), another minor positive APP, was increased by a 13.7-fold in blood in the scrapie group at 23 wpi. LF is a multifunctional glycoprotein with great iron-binding affinity and it is produced by mucosal epithelial cells [67]. It is involved in regulation of iron absorption and immune responses, and it has been suggested that LF has multifunctional antiprion activities [68]. Neutrophils are the source of lactoferrin in serum [69].
Ceruloplasmin (Cp) is a moderate APP with important biological functions. It is the major copper-carrying protein in blood, it plays a role in iron homeostasis and has oxidase activity in the CNS [47, 70,71,72]. Cp is mainly produced in the liver, and we detected an increased transcription in liver tissue in the scrapie group at 23 wpi. This coincides well with our previous finding of increased serum levels of ceruloplasmin at 23 wpi [29].
Serum amyloid A (SAA) and haptoglobin (Hp) are major APPs in sheep and increased serum levels are associated with a number of diseases. These proteins are primarily synthesised by hepatocytes, but other tissues and cell types are able to produce these two locally, though at a much lower level [47]. Our results show an increased transcription of both genes in the scrapie group at the clinical end stage. SAA gene expression in blood was significantly increased at both 22 and 23 wpi, with an average fold change of 5.00 and 21.99, respectively. In liver tissue, the mean expression level was increased by an average 89.98 fold change in the scrapie group. These results indicate that the increased level of SAA in in serum, reported earlier, originates both from increased synthesis in the liver and in the blood.
Hp showed increased gene transcription in both liver and brain tissue, with a mean fold change of 542.86 and 7.91, respectively. This indicates an increased synthesis of Hp hepatically and extrahepatically within the brain in the scrapie group at 23 wpi.
There was considerable variation in relative expression of individual gene expression within the scrapie group, indicated by the SEM values (Tables 1-3). One animal stands out in gene expression fold change in liver and blood, and on clinical presentation (individual data not shown). We believe there is relationship between the magnitude of the systemic acute phase response and clinical presentation in these animals. The acute phase response leads to a number of behavioural, physiological, biochemical and nutritional changes [36]. The brain recognizes proinflammatory cytokines as molecular signals of sickness, and metabolic and behavioural changes are initiated. These changes are collectively called “sickness behaviour” [73]. These peripherally released cytokines communicate and act on the brain via a fast neural pathway through afferent neurons (especially the vagus nerve), and via a slow humoral pathway [37]. In this sense, the immune- and nervous systems are in close contact. The afferent branches of the vagus nerve are involved in transmitting signals of inflammation to the brain via cytokine receptors [37]. Dendritic cells and macrophages are present in close association to vagal nerve fibres and they are important in the immune-brain communication [74]. Dendritic cells and macrophages express membrane TLRs and can produce proinflammatory cytokines upon activation, and at the same time, these cell types play a role in peripheral prion pathogenesis by interacting with PrPSc [42, 75]. Immune signals and PrPSc neuroinvasion may share common neural pathway transmitting immune messages and PrPSc infectivity, respectively, from periphery to the brain, where DCs have an important role [37, 76]. The innate immune system and the nervous system are, thus closely linked through common hormonal and neuronal routes. The ANS and CNS enhance and dampen the inflammatory response, and regulation through negative feedback loops are important. Any interference may have deleterious consequences. Recently, Salvesen et al. suggested that PrPC is one of the modulators of the innate immune response, and loss of PrPC prolonged sickness behaviour and stimulated pro-inflammatory activity [77].
Similar experimental scrapie models in sheep show PrPSc accumulation in peripheral lymphatic tissue and gene expression changes about half-way through the incubation period, while the animals appear asymptomatic [20, 22, 24, 43]. We believe the situation in the scrapie group was similar, an asymptomatic incubation period was followed be a short clinical end stage characterised by progressive apathy, dirty wool and passiveness. By 23 wpi two sheep were recumbent, weak, incapacitated and responded poorly to external stimuli. Sheep are very good at hiding behaviour changes as indicators of disease, thus presence of clinical signs cannot always be relied upon [78]. Sickness behaviour can be both beneficial and disadvantageous to the animal, and it can, to some extent, be overruled by fear and mothering behaviour [73, 79]. Why the long asymptomatic incubation period, despite the ongoing accumulation of PrPSc in LRS and CNS and gene expressions alterations, is not fully understood. Many of the genes involved are part of the innate immune system, and it is known that a prolonged and excessive inflammatory response can have negative or even lethal effects [34, 37, 80].
The particular disease phenotype seen in present study, is the result of the interaction between donor material, dosage and time of inoculation and recipient genotype, also described by González et al. [81]. The inoculation at birth before access to colostrum results in an effective and fast uptake of PrPSc across the susceptible new-born gut, similarly described by Tabouret et al. [24, 28]. These lambs have a characteristic dissemination of PrPSc throughout peripheral lymphatic tissues well in advance of PrPSc detection in the brain. The early and progressive development of clinical signs is unusual and have only been described earlier in a few rare cases of natural scrapie by Ulvund, although not at such a young age [82]. The histopathological examinations of the brain from these pre-colostrum inoculated lambs revealed only moderate cytopathological changes, with a mild to moderate gliosis with few vacuolated neurons. This is in somewhat in contrast to an earlier experimental model, described by Ersdal et al., where lambs of the same genotype (VRQ) were orally inoculated with brain tissue homogenate at ages between 46 and 61 days of age (mean 55.6 days).
Conclusion
We believe our experimental model produces a “worst-case” scenario which can occur in natural scrapie settings. Homozygous VRQ offspring from subclinically infected ewes, can become infected at birth and develop clinical scrapie similar to the cases described here. In naturally occurring scrapie, such animals will pose a threat to other susceptible sheep through environmental contamination due to high infectivity. The young age and atypical clinical presentation of these cases can result in misdiagnosis and not be recognisable as scrapie. Further work is needed to investigate the connection between the innate immune system and prion diseases, and whether the severe response we observed in our model can be linked to loss of modulatory function of PrPC.
Abbreviations
- AAT:
-
Alfa-1 antitrypsin
- ALB:
-
albumin
- ANS:
-
Autonomic nervous system
- APP:
-
Acute Phase Protein
- APR:
-
acute phase response
- C3:
-
complement component 3
- cDNA:
-
complementary DNA
- CNS:
-
Central nervous system
- Cp:
-
Ceruloplasmin
- CSF:
-
cerebrospinal fluid
- DCs:
-
Dendritic cells
- DNA:
-
Deoxyribonucleic acid
- FC:
-
fold change
- FDCs:
-
follicular dendritic cells
- Hp:
-
Haptoglobin
- IHC:
-
immunohistochemistry
- IL1B:
-
Interleukin-1-beta
- LF:
-
Lactoferrin
- LRS:
-
Lymphoreticular system
- mRNA:
-
Messenger ribonucleic acid
- NTC:
-
No template control
- PNS:
-
Peripheral nervous system
- PRNP:
-
Prion protein gene
- PrPC :
-
Normal cellular prion protein
- PrPSc :
-
Abnormal isoform of prion protein
- PRRs:
-
pattern recognition receptors
- RIN:
-
RNA integrity number
- RNA:
-
Ribonucleic acid
- RT-qPCR:
-
Reverse transcription quantitative real-time polymerase chain reaction
- SAA:
-
Serum amyloid A
- SEM:
-
Standard errors of mean
- TF:
-
Transferrin
- TLR2:
-
Toll-like receptor 2
- TLR4:
-
Toll-like receptor 4
- TLRs:
-
Toll-like receptors
- TNFa:
-
Tumor necrosis factor alfa
- TSE:
-
Transmissible spongiform encephalopathy
- TTR:
-
Transthyretin
- WB:
-
western blot
- wpi:
-
weeks post inoculation
References
Prusiner SB. Molecular biology of prion diseases. Science. 1991;252:1515–22.
Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–44.
Detwiler LA, Baylis M. The epidemiology of scrapie. Rev Sci Tech Int Off Epizoot. 2003;22:121–43.
Hoinville LJ, Tongue SC, Wilesmith JW. Evidence for maternal transmission of scrapie in naturally affected flocks. Prev Vet Med. 2010;93:121–8.
Lantier F, Schelcher F, Lafond-Benestad S, Chabert A, Monnereau L, Andréoletti O, et al. PrPSc accumulation in placentas of ewes exposed to natural scrapie: influence of foetal PrP genotype and effect on ewe-to-lamb transmission. J Gen Virol. 2002;83:2607–16.
Konold T, Moore S, Bellworthy SJ, Terry LA, Thorne L, Ramsay A, et al. Evidence of effective scrapie transmission via colostrum and milk in sheep. BMC Vet Res. 2013;9:99.
Goldmann W, Hunter N, Smith G, Foster J, Hope J. PrP genotype and agent effects in scrapie: change in allelic interaction with different isolates of agent in sheep, a natural host of scrapie. J Gen Virol. 1994;75:989–95.
Dawson M, Hoinville LJ, Hosie BD, Hunter N. Guidance on the use of PrP genotyping as an aid to the control of clinical scrapie. Scrapie Information Group Vet Rec. 1998;142:623–5.
Baylis M, Chihota C, Stevenson E, Goldmann W, Smith A, Sivam K, et al. Risk of scrapie in British sheep of different prion protein genotype. J Gen Virol. 2004;85(Pt 9):2735–40.
Ortiz-Pelaez A, Bianchini J. The impact of the genotype on the prevalence of classical scrapie at population level. Vet Res. 2011;42:31.
Tranulis MA. Influence of the prion protein gene, Prnp, on scrapie susceptibility in sheep. APMIS. 2002;110:33–43.
European Parliament, Council of the European Union. REGULATION (EC) No 999/2001 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 22 May 2001 laying down rules for the prevention, control and eradication of certain transmissible spongiform encephalopathies. 2001. http://eur-lex.europa.eu/legal-content/EN/ALL/?uri=CELEX:02001R0999-20170701.
Hagenaars TJ, Melchior MB, Bossers A, Davidse A, Engel B, van Zijderveld FG. Scrapie prevalence in sheep of susceptible genotype is declining in a population subject to breeding for resistance. BMC Vet Res. 2010;6:25.
Dobly A, Van der Heyden S, Roels S. Trends in genotype frequency resulting from breeding for resistance to classical scrapie in Belgium (2006~2011). J Vet Sci. 2013;14:45.
Ortiz-Pelaez A, Thompson CE, Dawson M. The impact of the National Scrapie Plan on the PRNP genotype distribution of the British national flock, 2002–2012. Vet Rec 2014;174:530–530.
Stepanek O, Horin P. Genetic diversity of the prion protein gene (PRNP) coding sequence in Czech sheep and evaluation of the national breeding programme for resistance to scrapie in the Czech Republic. J Appl Genet. 2017;58:111–21.
Touzeau S, Chase-Topping ME, Matthews L, Lajous D, Eychenne F, Hunter N, et al. Modelling the spread of scrapie in a sheep flock: evidence for increased transmission during lambing seasons. Arch Virol. 2006;151:735–51.
Weissmann C, Raeber AJ, Montrasio F, Hegyi I, Frigg R, Klein MA, et al. Prions and the lymphoreticular system. Philos Trans R Soc B Biol Sci. 2001;356:177–84.
Bossers A, Schreuder BE, Muileman IH, Belt PB, Smits MA. PrP genotype contributes to determining survival times of sheep with natural scrapie. J Gen Virol. 1996;77(Pt 10):2669–73.
Ersdal C, Ulvund MJ, Benestad SL, Tranulis MA. Accumulation of pathogenic prion protein (PrPSc) in nervous and lymphoid tissues of sheep with subclinical scrapie. VetPathol. 2003;40:164–74.
Heggebø R, Press CM, Gunnes G, Ulvund MJ, Tranulis MA, Lsverk T. Detection of PrPSc in lymphoid tissues of lambs experimentally exposed to the scrapie agent. J Comp Pathol. 2003;128:172–81.
Ersdal C, Ulvund MJ, Espenes A, Benestad SL, Sarradin P, Landsverk T. Mapping PrPSc propagation in experimental and natural scrapie in sheep with different PrP genotypes. VetPathol. 2005;42:258–74.
Georgsson G, Adolfsdottir JA, Palsdottir A, Jorundsson E, Sigurdarson S, Thorgeirsdottir S. High incidence of subclinical infection of lymphoid tissues in scrapie-affected sheep flocks. Arch Virol. 2008;153:637–44.
Tabouret G, Lacroux C, Lugan S, Costes P, Corbiere F, Weisbecker JL, et al. Relevance of oral experimental challenge with classical scrapie in sheep. J Gen Virol. 2010;91:2139–44.
Ryder SJ, Dexter GE, Heasman L, Warner R, Moore SJ. Accumulation and dissemination of prion protein in experimental sheep scrapie in the natural host. BMC Vet Res. 2009;5:9.
Gossner AG, Hopkins J. The effect of PrP Sc accumulation on inflammatory gene expression within sheep peripheral lymphoid tissue. Vet Microbiol. 2015;181:204–11.
European Food Safety Authority (EFSA). TSE risk assessment from carcasses of ovine and caprine animals below 6 months of age from TSE infected flocks intended for human consumption - Scientific Opinion of the Panel on Biological Hazards. EFSA J. 2008;6. doi:https://doi.org/10.2903/j.efsa.2008.719.
Ulvund MJ, Brundtland E, Bårdsen K, Meling S, Ersdal C, Espenes A, et al. A new experimental model for studying scrapie and prion diseases in sheep. Edinburgh: Conference abstract; 2007.
Meling S, Bardsen K, Ulvund MJ. Presence of an acute phase response in sheep with clinical classical scrapie. BMCVetRes. 2012;8:113.
Tang Y, Lu A, Aronow BJ, Sharp FR. Blood genomic responses differ after stroke, seizures, hypoglycemia, and hypoxia: blood genomic fingerprints of disease. AnnNeurol. 2001;50:699–707.
Tang Y, Nee AC, Lu A, Ran R, Sharp FR. Blood genomic expression profile for neuronal injury. JCerebBlood Flow Metab. 2003;23:310–9.
Liew CC, Ma J, Tang HC, Zheng R, Dempsey AA. The peripheral blood transcriptome dynamically reflects system wide biology: a potential diagnostic tool. JLab ClinMed. 2006;147:126–32.
Debey S, Schoenbeck U, Hellmich M, Gathof BS, Pillai R, Zander T, et al. Comparison of different isolation techniques prior gene expression profiling of blood derived cells: impact on physiological responses, on overall expression and the role of different cell types. PharmacogenomicsJ. 2004;4:193–207.
Koj A. Initiation of acute phase response and synthesis of cytokines. Biochim Biophys Acta. 1996;1317:84–94.
Moshage H. Cytokines and the hepatic acute phase response. J Pathol. 1997;181:257–66.
Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med. 1999;340:448–54.
Dantzer R. Cytokine, sickness behavior, and depression. Immunol Allergy Clin N Am. 2009;29:247–64.
Ceciliani F, Giordano A, Spagnolo V. The systemic reaction during inflammation: the acute-phase proteins. Protein Pept Lett. 2002;9:211–23.
Baumann H, Gauldie J. The acute phase response. Immunol Today. 1994;15:74–80.
Cray C, Zaias J, Altman NH. Acute phase response in animals: a review. Comp Med. 2009;59:517–26.
Meling S, Kvalheim OM, Arneberg R, Bårdsen K, Hjelle A, Ulvund MJ. Investigation of serum protein profiles in scrapie infected sheep by means of SELDI-TOF-MS and multivariate data analysis. BMC Res Notes. 2013;6:466.
Bradford BM, Mabbott NA. Prion disease and the innate immune system. Viruses. 2012;4:3389–419.
Hwang D, Lee IY, Yoo H, Gehlenborg N, Cho J-H, Petritis B, et al. A systems approach to prion disease. Mol Syst Biol. 2009;5 https://doi.org/10.1038/msb.2009.10.
Crespo I, Roomp K, Jurkowski W. Kitano H, del SA. Gene regulatory network analysis supports inflammation as a key neurodegeneration process in prion disease BMCSystBiol. 2012;6:132.
Chang J-S, Russell GC, Jann O, Glass EJ, Werling D, Haig DM. Molecular cloning and characterization of toll-like receptors 1-10 in sheep. Vet Immunol Immunopathol. 2009;127:94–105.
Takeda K, Akira S. Toll-like receptors in innate immunity. IntImmunol. 2005;17:1–14.
Ceciliani F, Ceron JJ, Eckersall PD, Sauerwein H. Acute phase proteins in ruminants. J Proteome. 2012;75:4207–31.
Prinz M, Heikenwalder M, Schwarz P, Takeda K, Akira S, Aguzzi A. Prion pathogenesis in the absence of toll-like receptor signalling. EMBO Rep. 2003;4:195–9.
Spinner DS, Cho IS, Park SY, Kim JI, Meeker HC, Ye X, et al. Accelerated prion disease pathogenesis in toll-like receptor 4 signaling-mutant mice. J Virol. 2008;82:10701–8.
Kang S-G, Kim C, Cortez LM, Carmen Garza M, Yang J, Wille H, et al. Toll-like receptor-mediated immune response inhibits prion propagation: Innate Immune Defense Against Prions. Glia. 2016;:n/a-n/a.
Aguzzi A, Zhu C. Microglia in prion diseases. J Clin Invest. 2017;127:3230–9.
Aguzzi A, Nuvolone M, Zhu C. The immunobiology of prion diseases. NatRevImmunol. 2013;13:888–902.
Mabbott NA. The complement system in prion diseases. Curr Opin Immunol. 2004;16:587–93.
Sjöberg AP, Trouw LA, Blom AM. Complement activation and inhibition: a delicate balance. Trends Immunol. 2009;30:83–90.
Blanquet-Grossard F, Thielens NM, Vendrely C, Jamin M, Arlaud GJ. Complement Protein C1q Recognizes a Conformationally Modified Form of the Prion Protein †. Biochemistry (Mosc). 2005;44:4349–56.
Mitchell DA, Kirby L, Paulin SM, Villiers CL, Sim RB. Prion protein activates and fixes complement directly via the classical pathway: implications for the mechanism of scrapie agent propagation in lymphoid tissue. Mol Immunol. 2007;44:2997–3004.
McGeer PL, McGeer EG. The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res Brain Res Rev. 1995;21:195–218.
Członkowska A, Kurkowska-Jastrzębska I. Inflammation and gliosis in neurological diseases – clinical implications. J Neuroimmunol. 2011;231:78–85.
Newsom DM, Liggitt HD, O’Rourke K, Zhuang D, Schneider DA, Harrington RD. Cytokine antibody array analysis in brain and periphery of scrapie-infected Tg338 mice. Comp Immunol Microbiol Infect Dis. 2011;34:387–97.
Gruys E, Toussaint MJM, Niewold TA, Koopmans SJ. Acute phase reaction and acute phase proteins. J Zhejiang Univ Sci. 2005;6B:1045–56.
Ritchie RF, Palomaki GE, Neveux LM, Navolotskaia O, Ledue TB, Craig WY. Reference distributions for the negative acute-phase serum proteins, albumin, transferrin and transthyretin: a practical, simple and clinically relevant approach in a large cohort. J Clin Lab Anal. 1999;13:273–9.
Petersen HH, Nielsen JP, Heegaard PMH. Application of acute phase protein measurements in veterinary clinical chemistry. Vet Res. 2004;35:163–87.
Batxelli-Molina I, Salvetat N, Andréoletti O, Guerrier L, Vicat G, Molina F, et al. Ovine serum biomarkers of early and late phase scrapie. BMC Vet Res. 2010;6:49.
Tothova C, Nagy O, Kovac G. Acute phase proteins and their use in the diagnosis of diseases in ruminants: a review. Veterinární Medicína. 2014;59 No. 4:163–80.
Belorgey D, Hägglöf P, Karlsson-Li S, Lomas DA. Protein misfolding and the serpinopathies. Prion. 2007;1:15–20.
Carrell RW, Lomas DA. Alpha 1 -antitrypsin deficiency — a model for conformational diseases. N Engl J Med. 2002;346:45–53.
García-Montoya IA, Cendón TS, Arévalo-Gallegos S, Rascón-Cruz Q. Lactoferrin a multiple bioactive protein: an overview. Biochim Biophys Acta BBA - Gen Subj. 1820;2012:226–36.
Iwamaru Y, Shimizu Y, Imamura M, Murayama Y, Endo R, Tagawa Y, et al. Lactoferrin induces cell surface retention of prion protein and inhibits prion accumulation. J Neurochem. 2008;107:636–46.
Adlerova L, Bartoskova A, Faldyna M. Lactoferrin: a review. Veterinární Medicína. 2008;53 No. 9:457–68.
Eckersall PD, Saini PK, McComb C. The acute phase response of acid soluble glycoprotein, alpha(1)-acid glycoprotein, ceruloplasmin, haptoglobin and C-reactive protein, in the pig. Vet Immunol Immunopathol. 1996;51:377–85.
Patel BN, Dunn RJ, Jeong SY, Zhu Q, Julien J-P, David S. Ceruloplasmin regulates iron levels in the CNS and prevents free radical injury. J Neurosci. 2002;22:6578–86.
Frieden E, Hsieh HS. Ceruloplasmin: the copper transport protein with essential oxidase activity. Adv Enzymol Relat Areas Mol Biol. 1976;44:187–236.
Tizard I. Sickness behavior, its mechanisms and significance. Anim Health Res Rev. 2008;9:87–99.
Goehler LE, Gaykema RP, Nguyen KT, Lee JE, Tilders FJ, Maier SF, et al. Interleukin-1beta in immune cells of the abdominal vagus nerve: a link between the immune and nervous systems? J Neurosci. 1999;19:2799–806.
Beekes M, McBride PA. The spread of prions through the body in naturally acquired transmissible spongiform encephalopathies: spread of prions through the body. FEBS J. 2007;274:588–605.
Langevin C, Gousset K, Costanzo M, Richard-Le Goff O, Zurzolo C. Characterization of the role of dendritic cells in prion transfer to primary neurons. Biochem J. 2010;431:189–98.
Salvesen ø., Reiten MR, Espenes A, Bakkebø MK, Tranulis MA, Ersdal C. LPS-induced systemic inflammation reveals an immunomodulatory role for the prion protein at the blood-brain interface. J Neuroinflammation. 2017;14. doi:https://doi.org/10.1186/s12974-017-0879-5.
Hindson JC, Winter AC. Manual of sheep diseases. 2. ed. Oxford: Blackwell Science; 2002.
Aubert A. Sickness and behaviour in animals: a motivational perspective. Neurosci Biobehav Rev. 1999;23:1029–36.
Sternberg EM. Neural regulation of innate immunity: a coordinated nonspecific host response to pathogens. Nat Rev Immunol. 2006;6:318–28.
Gonzalez L, Jeffrey M, Dagleish MP, Goldmann W, Siso S, Eaton SL, et al. Susceptibility to scrapie and disease phenotype in sheep: cross-PRNP genotype experimental transmissions with natural sources. VetRes. 2012;43:55.
Ulvund MJ. Scrapie: a clinical Challenge. Norwegian School of Veterinary Science: Oslo; 1998.
Acknowledgements
The authors gratefully thank Eli Brundtland for assisting in sampling of material and PRNP-genotyping, and Karin Tarp for excellent help and guidance on sample preparation and RT-qPCR analysis.
Consent to publication
Not applicable.
Funding
This study was supported by grant NRC 178268 (2006–2010) funded by the Research Council of Norway, and Johan Haaland’s Legat.
Availability of data and materials
Data and materials supporting the conclusions are contained within the manuscript. Any additional data are available upon request. RIN values and calculated are shown in Additional file 1. Primer sequences are listed together with reaction conditions in Additional file 2.
Author information
Authors and Affiliations
Contributions
MJU, SM and KB designed the study, and SM, MJU and KB performed the experiments. KS participated in designing primers and laboratory procedures and pre-processing of the expression data. SM designed primers, carried out laboratory procedures, performed the statistical analyses and drafted the manuscript. MJU participated in the interpretation of analytical data and in drafting the manuscript. PMHH coordinated the analyses in Denmark. All authors have critically read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interest
The authors declare that they have no competing interests.
Ethics approval
The animal experiment was performed in compliance with ethical guidelines and approved by the Norwegian Animal Research Authority (FOTS id852) with reference to the Norwegian regulation on animal experimentation (FOR-1996-01-15-23).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional information
§Corresponding author
Additional files
Additional file 1:
Quantity and quality calculations of extracted RNA. Descriptive statistical presentation of quantitative and qualitative properties of the extracted RNA from blood, liver tissue and brain tissue. (DOCX 13 kb)
Additional file 2:
Primers and reaction conditions. Gene functional class, gene abbreviation, gene name, gene bank entry and primer sequences used in the real time qPCR analysis. Calculated PCR efficiency and correlation are also listed. (XLSX 28 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Meling, S., Skovgaard, K., Bårdsen, K. et al. Expression of selected genes isolated from whole blood, liver and obex in lambs with experimental classical scrapie and healthy controls, showing a systemic innate immune response at the clinical end-stage. BMC Vet Res 14, 281 (2018). https://doi.org/10.1186/s12917-018-1607-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12917-018-1607-9