
Abstract
Background
The timing of puberty may have an important impact on adolescent mental health. In particular, earlier age at menarche has been associated with elevated rates of depression in adolescents. Previous research suggests that this relationship may be causal, but replication and an investigation of whether this effect extends to other mental health domains is warranted.
Methods
In this Registered Report, we triangulated evidence from different causal inference methods using a new wave of data (N = 13,398) from the Norwegian Mother, Father, and Child Cohort Study. We combined multiple regression, one- and two-sample Mendelian randomisation (MR), and negative control analyses (using pre-pubertal symptoms as outcomes) to assess the causal links between age at menarche and different domains of adolescent mental health.
Results
Our results supported the hypothesis that earlier age at menarche is associated with elevated depressive symptoms in early adolescence based on multiple regression (β = − 0.11, 95% CI [− 0.12, − 0.09], pone-tailed < 0.01). One-sample MR analyses suggested that this relationship may be causal (β = − 0.07, 95% CI [− 0.13, 0.00], pone-tailed = 0.03), but the effect was small, corresponding to just a 0.06 standard deviation increase in depressive symptoms with each earlier year of menarche. There was also some evidence of a causal relationship with depression diagnoses during adolescence based on one-sample MR (OR = 0.74, 95% CI [0.54, 1.01], pone-tailed = 0.03), corresponding to a 29% increase in the odds of receiving a depression diagnosis with each earlier year of menarche. Negative control and two-sample MR sensitivity analyses were broadly consistent with this pattern of results. Multivariable MR analyses accounting for the genetic overlap between age at menarche and childhood body size provided some evidence of confounding. Meanwhile, we found little consistent evidence of effects on other domains of mental health after accounting for co-occurring depression and other confounding.
Conclusions
We found evidence that age at menarche affected diagnoses of adolescent depression, but not other domains of mental health. Our findings suggest that earlier age at menarche is linked to problems in specific domains rather than adolescent mental health in general.
Similar content being viewed by others

Background
Early pubertal timing has been associated with problems in a wide range of adolescent mental health domains (e.g., depression [1,2,3,4,5,6,7,8,9,10,11,12,13], anxiety [7, 14, 15], conduct disorders [3, 7, 16,17,18,19], and attention-deficit hyperactivity disorder (ADHD) [6]) across different indicators of pubertal development and across sexes [20]. The consistency of associations between early timing and adolescent mental health has led to the hypothesis that early pubertal timing is a transdiagnostic risk factor for psychopathology in adolescents [21].
Despite the apparent generality of associations between early pubertal timing and adolescent mental health, the prominent rise in rates of female depression beginning during puberty [22] has led to this outcome receiving particular empirical focus [23]. The timing of puberty in females is commonly indexed using the onset of menses (menarche). Earlier age at menarche has been associated with elevated depressive symptoms in adolescents in several observational studies [4, 24,25,26,27,28,29,30,31,32], but not in all [33,34,35,36,37], and also higher rates of clinical depression during adolescence [31, 37]. However, although early pubertal maturation in females has been associated with a wide range of problems in adolescence, these associations may dissipate by adulthood [3, 38]. A notable exception in a large prospective study was that heightened risk of depression persisted into young adulthood for early maturers, in particular, for those with a history of conduct disorder [3].
The association between pubertal timing and depression in adolescent females may be due to the biological underpinnings of reproductive maturation. The female sex hormone estradiol increases with puberty and is associated with depression [39, 40], and hormonal contraceptive use has been associated with higher levels of depressive symptoms, especially in adolescence [41]. In fact, it has been found that the stage of breast development (governed primarily by estradiol) was associated with depression independently of the timing of menarche in adolescent females from the Avon Longitudinal Study of Parents and Children (ALSPAC) [42]. Interestingly, a recent study in the same sample found that a polygenic score for age at menarche showed a potential indirect association with adolescent depressive symptoms through the stage of breast development [43]. Alongside psychosocial pathways (i.e., visible breast development leading to unwanted sexual attention at a younger age), increases in estradiol represent a plausible biological mechanism for the link between early pubertal timing and depression in females.
Despite the series of observational studies, it is unknown whether the link between age at menarche and depression represents a truly causal relationship. This is important because several robust observational associations in epidemiology have turned out not to be causal and may instead have been the result of confounding (i.e., vitamin E supplement use and cardiovascular disease [44]). In the case of associations between age at menarche and depression, body mass index (BMI) is a particularly likely candidate for confounding given the robust (and plausibly causal) links between BMI and age at menarche [45] and between BMI and depression [46,47,48]. Failure to appropriately account for potential confounding, especially by BMI [4, 25, 27,28,29,30, 32,33,34,35,36,37], has been a relatively common shortcoming of the literature on this topic to date. In previous studies that explicitly controlled for BMI, the relationship was somewhat attenuated [26, 49]. Another study found BMI to be a partial mediator of the relationship between earlier menarche and depression [31].
Mendelian randomisation (MR) is a causal inference method that can be implemented in instrumental variable analyses [50, 51], which is particularly useful when experimental manipulation of the variable of interest is not ethical or feasible. Since hundreds of genetic variants are strongly linked with age at menarche [52], single nucleotide polymorphisms (SNPs) that are independently associated with this phenotype can be used as genetic instruments in MR analyses. The logic of MR is analogous to that of a randomised controlled trial (RCT). Unlike in an RCT design where individuals are randomly assigned to experimental groups, in MR, we use random “assignment” to genotype (ensured by the random transmission of one of two possible alleles at each genetic locus from each parent to their child at conception [53]). Specifically, these genetic variants are used as instrumental variables, serving as a genetic proxy for age at menarche.
Whereas self-reported age at menarche may be associated with several different confounders (even if precisely measured), the genetic instrument is assumed to be independent of such confounding. Both the widespread genetic influence on age at menarche [52] and the high accuracy and reliability of self-reported age at menarche [54] jointly increase the strength of the genetic instrument employed here, which serves to improve study power and minimise weak instrument bias [55]. The strength of the genetic instrument makes MR especially valuable for advancing menarche research. Provided that some important assumptions of MR hold true, we can estimate the causal effects of age at menarche on adolescent mental health.
A previous study found preliminary evidence that the relationship between age at menarche and depression in early adolescence may be causal, using MR in ALSPAC (N = 2404) [56]. Specifically, they found that early age at menarche resulted in more depressive symptoms at age 14 (independent of BMI), but not later in adolescence. However, this study had low power due to a modest sample size for MR. Here, we aim to replicate the 14-year analyses in adolescents from a larger birth cohort, the Norwegian Mother, Father, and Child Cohort Study (MoBa) [57]. This replication will allow for a confirmatory and higher-powered test of the hypothesis that earlier age at menarche is causally related to adolescent depression.
Beyond replicating its key finding, we will also extend the previous approach [56] in several key ways. First, we will test whether the effects of earlier age at menarche extend to other domains of mental health (anxiety disorders, conduct disorder (CD), oppositional defiant disorder (ODD), and ADHD), independent of associations with depression. Second, we will use multivariable methods to examine different confounders or mechanisms, by simultaneously including genetic instruments for childhood body size, adult BMI, or estradiol in the MR model together with age at menarche. Third, in line with recommendations to triangulate evidence across approaches for robust causal inference [58], we will combine MR with negative control analyses using symptoms prior to puberty as a negative control outcome. This triangulation is particularly important in the context of replication studies, given that the same sources of bias could lead to results being replicated in another study using the same methodology [59, 60].
A previous hypothesis-free MR phenome-wide association study identified potential causal effects of age at menarche on adult mental health [61], but these were not followed up with replication in any independent cohorts. Here, we take a confirmatory approach, testing causal hypotheses about the role of age at menarche in the aetiology of developing mental health disorders. This is important in part because a causal effect of age at menarche may help explain the sharp rise in depression rates among females from early adolescence [22]. This research might further help with identifying female adolescents at increased risk, facilitating early identification and prevention of mental health problems in adolescence and beyond.
To test our hypotheses, we make use of the Registered Report format, demonstrating its applicability to epidemiological analyses of cohort data when a new wave of data collection ensures that the exposure and outcome data have not been observed prior to the analytic choices being made. This format, combined with several sensitivity tests, will strengthen our statistical inferences by preserving false-positive rates at the specified level [62] and ultimately increase confidence in the causal conclusions that are drawn.
We addressed the following research questions: (1) To what extent is age at menarche associated with adolescent depression? (2) Does age at menarche associate with symptoms or diagnoses in other domains of mental health, independent of depression? (3) What is the evidence for a causal link between age at menarche and depression? and (4) Is there evidence of causal links between age at menarche and other domains of mental health? The specific hypotheses for each research question are listed in Table 1.
Methods
Design
Sample
The Norwegian Mother, Father, and Child Cohort Study (MoBa) is a population-based pregnancy cohort study conducted by the Norwegian Institute of Public Health [57]. Pregnant women and their partners were recruited from all over Norway at approximately 17 weeks gestation between 1999 and 2008. The women consented to participation in 41% of the pregnancies. The cohort includes approximately 114,500 children, 95,200 mothers, and 75,200 fathers. The current study is based on version 12 of the quality-assured data files released for research in January 2019. We also used data from the Medical Birth Registry of Norway (MBRN).
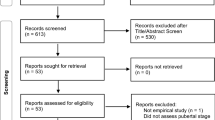
In MoBa, phenotype data have been collected by questionnaires from early pregnancy to middle childhood, provided primarily by mothers (around weeks 17, 22, and 30 of pregnancy and at child ages 0.5, 1.5, 3, 5, and 8 years). This project also made use of an ongoing wave of data collection in adolescence (questionnaires returned at ~ 14.4 years; hereafter age 14). The 14-year data were not available to us during the preparation of the stage 1 element of the Registered Report (see Fig. 1).

Overview of the Registered Report process
Inclusion criteria and sample size
We included all MoBa females (as registered at birth in MBRN) with any available phenotype data at age 14. There were 13,398 females with 14-year data, of which 9832 were genotyped.
Measures
Exposures
Self-reported age at menarche (in years) from the 14-year questionnaire was included as the main exposure. We ran the observational analyses with both a continuous and a categorical (early/average/late) variable based on reported age at menarche (see Additional file 1 for further details). Values were imputed for those who had not yet reached menarche at age 14, using information about the stage of pubertal development, as well as all the covariates and outcomes (see Additional file 1). We also included the self-reported stage of breast development at age 14 as an additional exposure for sensitivity analyses.
Mental health problems
Depressive symptoms were assessed through the Short Mood and Feelings Questionnaire (SMFQ; 13 items) [63]. We also computed a dichotomised version of the SMFQ (see Additional file 1). Anxiety symptoms were assessed through a short form of the Screen for Child Anxiety-Related Disorders (SCARED; 5 items) [64]. Behaviour problems (CD, ODD, and ADHD) were assessed with the Rating Scale for Disruptive Behaviour Disorders (RS-DBD; 34 items) [65]. All symptom outcomes were log or square root transformed due to non-normal distributions. The measures were treated as continuous, and scores were standardised to have a mean of 0 and a standard deviation of 1. Information about the psychometric properties of the scales is provided in Additional file 2: Table S1. An overview of all variables included in the study and their processing is in Additional file 3: Table S2.
Psychiatric diagnoses
We linked to the “control and payment of health refunds” database (KUHR) and the Norwegian Patient Registry (NPR) to obtain psychiatric diagnoses from medical records (see Additional file 1 for diagnostic codes and further details). Individuals were classified as a “case” in the case–control analysis if they had received a relevant diagnosis in either primary (covered by KUHR) or secondary health care (covered by NPR) during adolescence (between ages 10 and 17).
Covariates
We included BMI at ages 8 and 14, age at questionnaire return, maternal and paternal age, parental education and income, financial problems, parental cohabitation, parity, and maternal prenatal and postnatal depression as covariates (see Additional file 3: Table S2).
Genotyping and quality control
In MoBa, blood samples were obtained from children (umbilical cord) at birth [66]. The genotyping and quality control have been described in detail elsewhere [67].
Genetic instruments for Mendelian randomisation
A recent genome-wide association study (GWAS) meta-analysis of 42 studies involving 329,345 post-pubertal women of European ancestry found 389 independent signals associated with self-reported age at menarche, reaching the conventional threshold for genome-wide significance (p < 5 × 10−8) in the discovery sample [52]. These variants were largely replicated in a sample of 39,543 post-pubertal women from the Icelandic deCODE study, explaining 7.4% of the variance in age at menarche. First, we subset these genome-wide significant variants to single nucleotide polymorphisms (SNPs) only by removing insertions and deletions. Then, we extracted these SNPs (as available) from the genetic data in MoBa, which did not contribute to the GWAS meta-analysis. Having subset to genome-wide significant SNPs available in the MoBa cohort, we then clumped them for independence (linkage disequilibrium R2 = 0.001, clumping window = 10,000 kb). For the one-sample MR, we used this set of SNPs to construct a weighted genetic risk score based on published GWAS effect estimates. The score was computed as the weighted sum of the age-at-menarche-increasing alleles across the selected SNPs. Specifically, we multiplied the number of effect alleles (0, 1, or 2; or if imputed, probabilities of effect alleles) at each SNP by their weight (GWAS SNP-trait association), then summed and divided by the total number of SNPs used. Genotyping batch and the first 20 principal components were regressed out of the genetic instruments, the latter to control for confounding by population stratification.
We also prepared the age at menarche summary statistics—along with summary statistics for estradiol [68], adult BMI [69], recalled childhood body size [69], major depression [70], and the 14-year symptom outcomes in MoBa—for use in two-sample MR analyses (see Additional file 4 for details [69,70,71,72,73,74]). We also employed Steiger filtering [75] to create another genetic instrument for age at menarche, excluding SNPs that were more predictive of depression at age 14 than age at menarche. This primarily served to prevent reverse causation and to remove potential pleiotropic pathways other than the causal pathway of interest.
Statistical analysis
Observational analyses
First, we ran linear regression analyses to estimate the observational associations between age at menarche and continuous symptom outcomes, accounting for the effects of covariates. In addition, we ran logistic regression analyses to estimate observational associations with the diagnostic outcomes from registry data, accounting for the effects of covariates. All models were run with and without the covariates described above to obtain adjusted and unadjusted estimates, and inferences were based on the adjusted estimates.
Mendelian randomisation analyses
To avoid problems related to confounding and reverse causation common to traditional observational methods, MR uses j genetic variants G1, G2, …, Gj as a proxy for the exposure X to estimate the association between the exposure X and the outcome Y (see Fig. 2 for an illustrative diagram) [50]. The obtained estimate is assumed to be independent of potential confounders U. This assumption builds on Mendel’s first and second law of inheritance [53]. The two laws are (1) the segregation of alleles at the same locus is independent (equal segregation) and (2) the alleles of different genes are inherited independently of each other during gamete formation (independent assortment).

Directed acyclic graph illustrating the Mendelian randomisation design. Gj is the jth genetic variant, with direct effect γj on exposure X, and direct effect αj on outcome Y; θ is the estimated causal effect of the exposure on the outcome; ϕj is the relationship between confounders U and Gj; dotted lines represent possible violations of the MR assumptions
MR assumptions
The three main assumptions of MR are (1) that the instrument Gj is associated with the exposure X, called the relevance assumption; (2) that there are no unmeasured confounders of the gene-outcome association U, called the independence assumption; and (3) that the genetic variants Gj affect the outcome Y only through the exposure X, called exclusion restriction. While assumption 1 can be verified empirically, assumptions 2 and 3 are empirically unverifiable (but potentially falsifiable). Owing to how instrumental variable analyses are estimated, violations of these assumptions may lead to strong biases in the estimates; therefore, such estimates should be interpreted with care and in conjunction with other evidence [76]. Several sensitivity analyses that have been developed to address potential bias from violations of the MR assumptions were employed here [51].
One-sample MR
In the one-sample MR analyses of continuous symptom variables, we used two-stage least squares (2SLS) regression. In the 2SLS approach, self-reported age at menarche X was first regressed on the genetic variants Gj, obtaining the predicted values. In the second stage, the regression of the outcome Y on the exposure X is estimated as usual, replacing self-reported age at menarche with the predicted values from the first stage, hereafter referred to as “genetically predicted age at menarche”. For binary outcomes, a logistic model was used in the second stage. We applied a post-estimation correction of the standard errors (the HC1 option in the sandwich R package [77]) for both continuous and binary outcomes. In addition to one-sample MR, we conducted two-sample MR analyses based on GWAS of the symptom outcomes in MoBa. Combining one- and two-sample MR is beneficial even when the same outcome sample is used, since any bias from weak instruments would skew the one-sample estimate towards the (confounded) observational estimate and the two-sample estimate towards the null [78]. In addition, conducting two-sample MR maximises the availability of sensitivity analyses to test the MR assumptions.
Two-sample MR
In the two-sample MR analyses, only the genotype-outcome (G-Y) association was estimated in MoBa. For these analyses (using the TwoSampleMR package [71]), we extracted estimates for the genotype-exposure (G-X) association from summary-level data from the age-at-menarche GWAS [52] and produced a set of SNP-specific Wald estimates by calculating the ratio between the G-X and the G-Y associations. We tested the heterogeneity between the Wald ratios using SNP-specific Q statistics. Then, these estimates were combined using the inverse variance weighted (IVW) meta-analysis approach to obtain an estimate of the causal effect.
Multivariable MR and MR sensitivity analyses
We conducted a battery of sensitivity analyses across the one- and two-sample MR (described in Additional file 5 [79,80,81,82,83,84,85]) to assess the robustness of results and the impact of horizontal pleiotropy—where the genetic variants affect the outcome through other pathways than the exposure of interest. A likely source of horizontal pleiotropy is via BMI, which is therefore included as an additional exposure in multivariable Mendelian randomisation (MVMR) analyses [83]. The employed two-sample MR sensitivity analyses (MR-Egger [79], MR pleiotropy residual sum and outlier (MR-PRESSO) [80], weighted median [81], and contamination mixture methods [82]) make different assumptions about horizontal pleiotropy, and we consider effects that are consistent across these approaches to be more likely causal, in line with a triangulation approach.
Equivalence testing
We additionally used equivalence testing to assess whether estimated effects could be considered practically equivalent to 0. The region of practical equivalence to 0 was set, for each analysis, by pre-defining the smallest effect size of interest (SESOI). Equivalence tests were carried out with a 5% alpha. Details of the full procedure for setting the SESOI and carrying out the equivalence testing are described in Additional file 6 [20, 24, 29, 31, 38, 42, 49, 56, 86,87,88,89,90].
Negative control analyses
Since an individual’s age at menarche cannot directly influence their mental health prior to puberty, childhood symptoms can serve as a negative control outcome in our study. Such analyses can be used to detect unmeasured confounding in the context of MR, given that the negative control outcome is associated with confounders in a similar way to the outcome of interest [87]. Here, we estimate the causal effect of age at menarche on symptoms of depression, anxiety CD, ODD, and ADHD measured before puberty (at 8 years). We formally compare the estimate from the main analysis with this negative control by testing whether the 14-year estimate is consistent with an effect more extreme than the lower 95% CI of the 8-year estimate.
Missingness/handling of missing data
Within the 14-year sample, we used multiple imputation (MI) to account for missing data in all variables (see Additional file 3 for the amount of missing data per variable). Importantly, some individuals reported having not yet had their first menstrual period in the 14-year questionnaire. Imputed values for age at menarche for these individuals were not allowed to be lower than 15 years. Further details on the MI and handling of outliers are presented in Additional file 1. In addition to MI, we used inverse probability weighting to address potential bias from selective attrition out of the study over time (see Additional file 1).
Power calculations
For the stage 1 submission, power analyses were conducted in R by simulation for all null hypothesis significance tests (NHSTs) and equivalence tests used to investigate each hypothesis (see summary in Table 2, and further details in Additional file 7).
Software and analysis code
We conducted all statistical analyses in R version 4.1.2. Data preparation and analysis code is publicly available via GitHub: https://github.com/psychgen/aam-psych-adolesc-rr.
Overview of hypothesis tests and inference criteria
Pre-specified statistical tests and inference criteria for each hypothesis are summarised in Table 2, including the interpretation of all potential patterns of results. Further details about the main and sensitivity analyses, equivalence bounds, and inference criteria are in Additional file 6. Note that for directional hypotheses, we pre-specified one-sided null hypothesis significance tests and equivalence tests; therefore, some reported p-values are one-tailed.
Results
The pre-registered analyses were conducted according to the stage 1 protocol [91], and all deviations are detailed and justified in Table 3.
Descriptive statistics
The average age at menarche after multiple imputation (since 7.25% had not reached menarche at age 14) was 12.69 years (SD = 1.18). We conducted a sensitivity analysis setting an equal number of age at menarche values to missing, testing the imputation accuracy (see Additional file 8). The genetic instrument for age at menarche (based on 235 independent SNPs present in MoBa) explained 6.9% of the variation in age at menarche (R2 = 0.069, F = 996.4) and was associated with BMI at 8 (β = − 0.08, 95% CI [− 0.10, − 0.05], p < 0.01) and 14 years (β = − 0.10, 95% CI [− 0.12, − 0.08], p < 0.01). There were no notable associations with other covariates (Additional file 9: Table S3). The mean number of depressive symptoms at age 14 was 9.20 (SD = 6.56).
Depressive symptoms and diagnoses
Main analyses of symptom outcomes
An earlier age at menarche was observationally associated with more depressive symptoms at age 14 (β = − 0.11, 95% CI [− 0.12, − 0.09], pone-tailed < 0.01) after adjusting for covariates and pre-pubertal symptoms (see Fig. 3). The one-sample MR analysis indicated a small, causal relationship (β = − 0.07, 95% CI [− 0.13, 0.00], pone-tailed = 0.03), corresponding to an increase of 0.06 standard deviations in depression symptoms score per earlier year of menarche (after re-scaling the estimate). The adjusted observational estimate was consistent with an effect as extreme as our smallest effect size of interest, but the causal estimate was just within the region of practical equivalence to 0. In our negative control one-sample MR analyses, pubertal timing was not associated with depressive symptoms prior to puberty (β = − 0.03, 95% CI [− 0.12, 0.05], pone-tailed = 0.22), and the 14-year estimate was consistent with an effect more extreme than this.

Observational and causal links between age at menarche and depression. A Standardised betas of age at menarche predicting adolescent depressive symptoms, based on linear regressions unadjusted and adjusted for covariates and symptoms at age 8, one-sample Mendelian randomisation (MR), and negative control MR with 8-year depressive symptoms as the outcome. B Standardised odds ratios of age at menarche predicting depression diagnoses during adolescence, based on logistic regressions unadjusted and adjusted for covariates, and one-sample MR. In both panels, the orange dashed line represents the smallest effect size of interest for the observational analysis; the blue dashed line represents the smallest effect size of interest for the MR. NB: 95% confidence intervals are presented to show the precision of the estimates, but all statistical tests for depression outcomes were pre-specified to be one-tailed, meaning that the visual interpretation of the CIs in relation to the point null and smallest effect sizes of interest differs from the test result (described in text) in places
Main analyses of diagnostic outcomes
For depression diagnoses during adolescence (see Fig. 3B), we also found evidence of a small association in the observational analysis (OR = 0.78, 95% CI [0.72, 0.84], pone-tailed < 0.01) and one-sample MR (OR = 0.74, 95% CI [0.54, 1.01], pone-tailed = 0.03). The MR estimate was equivalent to a 29% increase in the likelihood of receiving a depression diagnosis with each year of earlier menarche. The number of depression cases in childhood (i.e., 7) was too small to include this as a negative control outcome. Only the MR estimate, which was less precise than the observational estimates, was consistent with values at least as extreme as our SESOI.
Sensitivity analyses
The two-sample MR sensitivity analyses with depressive symptoms as the outcome yielded mixed results (see Fig. 4). The IVW (β = − 0.05, 95% CI [− 0.11, 0.01], pone-tailed = 0.04), contamination mixture (β = − 0.18, 95% CI [− 0.28, − 0.04], pone-tailed < 0.01), and weighted median methods (β = − 0.02, 95% CI [− 0.11, 0.06], pone-tailed = 0.31) gave estimates in a consistent direction with the one-sample MR. The MR-Egger estimate was positive (β = 0.08, 90% CI [− 0.07, 0.23], pone-tailed = 0.14). Moreover, after Steiger filtering, the IVW estimate was attenuated (β = − 0.02, 95% CI [− 0.08, 0.04], pone-tailed = 0.22), whereas the contamination mixture estimate was unchanged (β = − 0.17, 95% CI [− 0.25, − 0.04], pone-tailed = 0.01). The MR-Egger intercept provided little evidence of directional pleiotropy, with an intercept value of − 0.006 (95% CI [− 0.01, 0.00], p = 0.05). The MR PRESSO global test did not detect any outliers.

Two-sample MR sensitivity analyses of age at menarche and depressive symptoms. MR sensitivity analyses showing broadly consistent directions of effect (except for MR-Egger), with an earlier age at menarche related to elevated adolescent depressive symptoms. MR, Mendelian randomisation; SNP, single nucleotide polymorphism
MVMR analyses accounting for overlap with childhood body size and adult BMI were limited by weak instruments (conditional F-statistic range 7.2–10.3). The estimate for age at menarche predicting depressive symptoms at age 14 was in a consistent direction with the IVW estimate (which was β = − 0.05) but partly attenuated when accounting for childhood body size (β = − 0.03, 95% CI [− 0.07, 0.02], pone-tailed = 0.11) and substantially attenuated when accounting for adult BMI (β = − 0.01, 95% CI [− 0.05, 0.04], pone-tailed = 0.40). Modified Q-statistics indicated pleiotropy when including childhood body size (Q1866 = 2044.3, p < 0.01) and adult BMI (Q1812 = 1924.0, p = 0.03). When including genetically predicted estradiol as a second exposure, the estimate was only somewhat attenuated (β = − 0.04, 95% CI [− 0.09, − 0.00], pone-tailed = 0.02). Again, there was evidence of pleiotropy (Q1804 = 1951.0, p < 0.01). The MVMR sensitivity analyses provided broadly similar results (Additional file 10: Table S4).
Furthermore, we incorporated the stage of breast development as an additional exposure in the observational models. A more advanced breast stage was associated with more depression diagnoses but including it in the same model left estimates for age at menarche largely unchanged (see Additional file 8). We also conducted the analyses based on a categorised age at menarche exposure and dichotomised SFMQ—as in Sequeira et al. [56]—which showed consistent results (see Additional file 8). Finally, we also incorporated IP weights to account for selective attrition, which attenuated much of the differences in baseline covariates between participants (n = 13,398) and non-participants (n = 41,832) at age 14 (Additional file 11: Fig. S9). The weighted results for symptoms and diagnoses of depression were similar to the unweighted results, although with somewhat reduced precision due to the inclusion of the weights (Additional file 12: Fig. S10).
Symptoms and diagnoses in other domains
Main analyses of symptom outcomes
For anxiety symptoms at age 14 (see Fig. 5), the observational analysis adjusted for covariates, concurrent depressive symptoms and pre-pubertal anxiety symptoms showed little evidence of an association (β = − 0.02, 95% CI [− 0.04, − 0.01], p < 0.01). Also, the one-sample MR provided no evidence of a causal relationship (β = 0.02, 95% CI [− 0.05, 0.09], p = 0.64). Similarly, there was little evidence for a relationship with ADHD traits in the fully adjusted observational analysis (β = − 0.02, 95% CI [− 0.04, − 0.01], p = 0.01) or the MR (β = 0.02, 95% CI [− 0.06, 0.09], p = 0.67). There was a small adjusted observational relationship with CD symptoms (β = − 0.06, 95% CI [− 0.08, − 0.05], p < 0.01), with which the MR estimate was consistent, but confidence intervals included 0 (β = − 0.06, 95% CI [− 0.13, 0.01], p = 0.08). The results for ODD symptoms showed no evidence of an observational (β = 0.01, 95% CI [− 0.01, 0.03], p = 0.27) or causal relationship (β = 0.01, 95% CI [− 0.06, 0.08], p = 0.80). Only the unadjusted (but not adjusted) observational and causal estimates for CD symptoms were consistent with effects outside the range of practical equivalence to 0. Negative control analyses for these outcomes were consistent with these results.

Observational and causal links between age at menarche and other domains. A Standardised betas for age at menarche predicting symptoms in other domains of mental health in adolescence, based on linear regressions unadjusted and adjusted for covariates, concurrent depressive symptoms (age 14) and pre-pubertal symptoms (age 8), one-sample Mendelian randomisation (MR), and negative control MR with 8-year symptoms as outcomes. B Standardised odds ratios of age at menarche predicting diagnoses in other domains of mental health during adolescence (ages 10–17), based on logistic regressions unadjusted and adjusted for covariates, adolescent (ages 10–17) depression diagnoses and childhood diagnoses for each domain (ages 0–8), and one-sample MR. In both panels, the orange dashed line represents the smallest effect size of interest for the observational analysis; the blue dashed line represents the smallest effect size of interest for the MR; 95% confidence intervals are presented. ANX, anxiety; CD, conduct disorder; ODD, oppositional defiant disorder; ADHD, attention-deficit hyperactivity disorder; DBD, disruptive behaviour disorder
Main analyses of diagnostic outcomes
For anxiety diagnoses during adolescence (see Fig. 5), the observational analysis adjusting for covariates, adolescent depression, and childhood anxiety diagnoses showed a small observational association (OR = 0.86, 95% CI [0.80, 0.91], p < 0.01), and the MR estimate was consistent, but confidence intervals included the null (OR = 0.82, 95% CI [0.63, 1.07], p = 0.14). Similarly, age at menarche showed no evidence of an observational (OR = 0.90, 95% CI [0.80, 1.01], p = 0.07) or causal relationship with ADHD diagnoses (OR = 0.69, 95% CI [0.47, 1.02], p = 0.06), although the MR point estimate was more extreme than the effect found for depression diagnoses. For DBD diagnoses, there was no evidence of an observational (OR = 0.90, 95% CI [0.76, 1.07], p = 0.24) or causal relationship (OR = 1.04, 95% CI [0.52, 2.07], p = 0.92). Across the observational analyses, all estimates fell within our defined range of practical equivalence to 0, while MR findings were generally too imprecise to draw clear conclusions. There were too few diagnoses in childhood to conduct negative control analyses (see Table 3).
Sensitivity analyses
The two-sample MR sensitivity analyses for symptoms in other domains showed little evidence of any associations apart from CD, where all estimates were consistent with earlier age at menarche leading to more CD symptoms (Additional file 13: Fig. S11). MVMR analyses accounting for overlap with major depression provided little evidence of causal relationships between age at menarche and 14-year symptoms of anxiety (β = − 0.01, 95% CI [− 0.05, 0.03], p = 0.63), CD (β = 0.00, 95% CI [− 0.04, 0.04], p = 0.94), ODD (β = 0.00, 95% CI [− 0.04, 0.04], p = 0.87), and ADHD (β = 0.01, 95% CI [− 0.03, 0.05], p = 0.80). MVMR sensitivity analyses were sometimes imprecise but consistent with the pattern of null findings (Additional file 10: Table S4). The MR-Egger test showed little evidence of directional pleiotropy for any of the outcomes. The MR PRESSO global test also did not detect any outliers.
Exploratory analyses
We conducted unregistered follow-up analyses of the causal effect of age at menarche on diagnostic outcomes since the MR results were imprecise. To explore whether the estimates for anxiety and ADHD were attenuated when accounting for comorbid depression and corresponding childhood diagnoses, we ran one-sample MR models with these included as covariates. This somewhat attenuated the estimate for anxiety (OR = 0.86, 95% CI [0.66, 1.13]) but not for ADHD (OR = 0.64, 95% CI [0.43, 0.97]). We also sought to explore the impact of the timing of diagnosis further, since this may have an impact on estimates. To this end, we divided the outcomes into new diagnoses in (1) preadolescence (ages 9–11), (2) early adolescence (ages 12–14), and (3) mid-late adolescence (ages 15–17). For depression, there was only evidence of a causal relationship in early (OR = 0.50, 95% CI [0.26, 0.95]) and not mid-late adolescence (OR = 0.95, 95% CI [0.64, 1.41]). The pattern for ADHD diagnoses was the same, with only a relationship in early adolescence (OR = 0.47, 95% CI [0.23, 0.95]). There were no relationships with anxiety diagnoses in either time window (see Additional file 8).
Discussion
In this Registered Report, we assessed the causal link between age at menarche and adolescent mental health in a large, population-based cohort. In observational analyses, we found evidence of an association between earlier age at menarche and elevated depressive symptoms at age 14, which was robust to the inclusion of measured covariates, pre-pubertal symptoms, and across all sensitivity analyses. In contrast, age at menarche was not associated with symptoms in the other domains apart from CD, once depressive symptoms were accounted for. One-sample MR analyses mirrored the observational results—with evidence of a small, causal effect of earlier age at menarche on elevated depressive symptoms—but not the other outcome domains. Negative control MR analyses using pre-pubertal symptoms as outcomes corroborated this pattern of findings. The results for diagnostic outcomes were generally less precise, indicating a causal effect of earlier age at menarche on more depression diagnoses during adolescence, but not diagnoses in other domains. Taken together, the main analyses supported our hypothesis of a causal effect of earlier age at menarche on diagnoses of adolescent depression and suggest that this effect is specific to depression, rather than influencing adolescent mental health in general.
Our adjusted observational estimates, negative control analyses, and one-sample MR analyses were all consistent with small causal effects of age at menarche on symptoms and diagnoses of depression. This finding effectively replicates a previous finding in adolescents [56] and aligns with previous findings in adults [61, 70, 92]. For each year earlier menarche, the odds of being diagnosed with depression during adolescence (ages 10–17) increased by approximately 29%. The robustness of this effect—which received broadly consistent empirical support across different outcomes and methodologies—is striking. Nonetheless, some considerations are important to its interpretation. The effects of age at menarche on symptoms and diagnoses of depression were very small, and the extent to which they should be considered clinically meaningful is a matter of debate. On the symptom outcome, individuals with a year earlier menarche would score less than half a point higher, on average, on the SMFQ scale. For diagnoses, the absolute risk of adolescent depression diagnoses changes from 5.2 to 6.6% with 1 year earlier menarche. Our analyses defined a region of practical equivalence to 0 based on pre-defined smallest effect sizes of interest, and while some estimates were consistent with effects outside this region, the majority of plausible values fell within. It should be noted that our smallest effect sizes of interest were set based on existing empirical evidence, and not based on clinical change thresholds—which may be preferable [86]. Overall, the effect sizes obtained here are consistent with the small associations obtained for a range of mental health and behavioural outcomes in other studies [20, 36] and demonstrate a reduction in effect sizes when accounting for confounding.
While the evidence from our main analyses of depression outcomes was relatively consistent, potential complexities in the interpretation of these relationships prompted us to perform extensive sensitivity analyses. The two-sample MR sensitivity analyses, which make different assumptions about the role of horizontal pleiotropy, provided mixed results regarding the causal relationship of age at menarche with depressive symptoms. The results based on Steiger filtering suggested that reverse causation, or other pleiotropic pathways, may be involved in the relationship between age at menarche and adolescent depressive symptoms. It should be noted that these analyses were relatively imprecise because they required that we perform GWAS of the symptom outcomes in MoBa with a sample size (9832) well below what is typically required for genomic discovery. We also conducted multivariable MR analyses, which were designed to estimate the direct effect of age at menarche while accounting for BMI. The results indicated that the association between age at menarche and depressive symptoms was confounded by BMI. The estimate was in a consistent direction but partly attenuated when including childhood body size and markedly attenuated when including adult BMI. While childhood body size is a likely confounder, post-pubertal BMI could also be on the causal pathway to depression; therefore, the impact of adjusting for adult BMI should not be taken primarily as evidence of confounding. These analyses were limited by weak instruments, so may have been biassed towards the null by the violation of the relevance assumption. Nonetheless, these and previous findings [56, 61] are in line with the role of BMI as a confounder of the link between pubertal development and adolescent depression. Future research utilising stronger genetic instruments would be required to quantify the extent of confounding by BMI, and its potential mediating role.
The pattern of results in our study suggests that age at menarche may contribute to symptom differentiation in adolescence, affecting risk for depression diagnoses specifically, rather than acting across the range of mental health outcomes included here. There were some possible exceptions, including conduct symptoms (where the adjusted observational estimate suggested a small effect, and the one-sample MR result was consistent in magnitude and direction but non-significant) and ADHD (where observational results suggested no association, but the MR point estimate—while non-significant—was more extreme than that for depression). However, we saw very little signal for anxiety, disruptive behaviour disorders, and ADHD when accounting for co-occurring depression and other confounders. Previous observational studies have shown associations with a wide range of conditions, including anxiety and behavioural conditions [20, 21]. Notably, our unadjusted estimates for each condition—both symptoms and diagnoses—were also indicative of effects. As a result of the general attenuation of these effects after control for confounders (both explicitly in the adjusted observational estimate and implicitly in MR), we can infer that some of the previously identified associations may not be causal or that they may not remain after accounting for the effect of co-occurring depression. We recommend that future studies account for the comorbidity between mental health conditions when assessing pubertal timing effects, given the potential for condition-specific mechanisms.
Interestingly, sex differences in depression emerge and then peak during adolescence, before declining across adulthood [93]. To explain this phenomenon, future research could benefit from taking a lifespan approach to reproductive development and women’s mental health. Given some evidence of converging aetiology of depression, age at menarche and menopause [70, 94], future studies could aim to explore their joint genetic architecture and potential shared biological underpinnings [95]. A hypothesis that remains to be tested is that the associations between depression and earlier female reproductive events (including age at menarche) may be caused by the duration of sex hormone exposure. If so, we would expect that the risk of depression would increase whenever menarche occurs—and that in the longer term, those with a later menarche would eventually “catch up” with others experiencing it earlier. Indeed, evidence of this “catching up” has recently been found in ALSPAC [96]. In line with this pattern of results, our exploratory analyses indicated an ~ 80% increase in the odds of depression diagnoses and a ~ 90% increase in the odds of ADHD diagnoses per year of earlier menarche in early adolescence (ages 12–14), but not in the years prior to or after this period. This may suggest that menarche causes a transient increase in the prevalence of depression (and possibly ADHD, although this finding should be considered hypothesis-generating rather than conclusive). Future waves of data collection in MoBa could be used to corroborate this further.
We also conducted MVMR analyses utilising genetic variants associated with estradiol as an additional exposure [68], showing limited attenuation of the relationship between earlier age at menarche and elevated depressive symptoms when including estradiol. When including the stage of breast development as an additional predictor in observational analyses, results for depressive symptoms and diagnoses remained unchanged. This conflicts with previous analyses in ALSPAC which suggested that breast stage is driving the association with depressive symptoms rather than age at menarche [42]. To shed some light on this complex pattern of findings, future studies could directly investigate the role of estradiol and other pubertal sex hormones in mediating the effects of pubertal development, rather than relying on proxy measures.
Future studies could also test the causal relationship between pubertal timing and mental health using other genetically informed methods. For instance, co-twin control studies have found that pubertal timing effects on adolescent mental health were largely due to shared genetic influences [97, 98]. MR can also be conducted within families, accounting for population phenomena that may bias genetic associations, such as population stratification, dynastic effects, and assortative mating [99]. Although our sample of female adolescents was too small for conducting well-powered within-family MR, consortium-based analyses could offer a solution.
Limitations
This study features a relatively large sample of genotyped adolescents and a strong genetic instrument for age at menarche. Furthermore, the Registered Report format and extensive sensitivity analyses strengthen the support for our conclusions. However, there are some important limitations to our study. Although our sample size was larger than previous studies, the precision of MR analyses was low for less prevalent conditions, especially DBD. Another limiting factor to the precision of estimates was the censoring of diagnoses in the registry data, which we proposed to handle with multiple imputation in the stage 1 protocol. However, this turned out not to be feasible, resulting in a deviation from the registered protocol (see Table 3 for a further description and justification). The accuracy of the imputation of missing age at menarche values was also lower than anticipated, but this was a minor issue since values were known to be 15 or higher.
As well as some issues with low precision, limitations in the MR components of our study are linked to the assumptions upon which the method rests. An advantage of our one-sample MR design is that the relevance and independence assumptions could be tested directly. Here, the genetic instrument was strongly associated with age at menarche—but not measured covariates, besides BMI and to some extent maternal age—providing some support for the validity of these assumptions. Furthermore, the two-sample MR sensitivity analyses provided insufficient evidence of directional horizontal pleiotropy, a particularly likely violation of the assumptions in the context of mental health outcomes [100]. However, we reiterate that the final two MR assumptions cannot be verified empirically and that violations may give biassed estimates. It is worth highlighting that the MVMR analyses including BMI may violate these assumptions. First, weak instruments may have biassed the estimates from these analyses towards the null. On a related note, despite broadly consistent results from the MVMR sensitivity analyses, BMI SNPs are highly pleiotropic and may have introduced bias in the MVMR. Overall, the potential of bias means that triangulation is of key importance, and results that are consistent across different methods and outcomes should be given more weight than isolated findings.
Since different methods may lead to different sources of bias, triangulation of multiple analytic approaches has been suggested as a way forward in aetiological epidemiology [58]. However, a key distinction may be between deciding which methods will be combined—and how—before conducting the analyses, or after the fact. “Prospective triangulation”, or pre-specifying a triangulation strategy and specific inference criteria such as in our Registered Report, may further increase the confidence we can have in the results.
There are also limitations to the generalisability of this study. First, MoBa is not fully representative of the general population due to non-random participation at the recruitment stage. Those less represented are the youngest women, those living alone, smokers, and women with previous stillbirths or more than two previous births [101]. However, previous research has suggested that non-random initial participation may have a limited impact on exposure-outcome associations [101, 102]. Furthermore, selective attrition could be expected to have an important impact on our results, due to the substantial drop-out at age 14 in MoBa. Yet, our IPW analyses showed consistent results when accounting for selective attrition. Finally, this study is based on a predominantly white European cohort. Future research in cohorts from non-European countries would advance the field further.
Conclusions
Our findings—based on extensive analyses and hypotheses registered prior to the availability of data [91]—provided support for the hypothesis that an earlier age at menarche causally increases the risk of adolescent depression. After accounting for depression and other confounders, we found no clear evidence of this effect being present for anxiety, disruptive behaviour disorders, or ADHD. A range of sensitivity analyses corroborated our results but suggested that the causal relationship with depressive symptoms may be partly confounded by BMI and/or influenced by low-level genetic pleiotropy. In sum, although the associations of age at menarche with symptoms and diagnoses of depression are likely partly confounded, our results supported small causal relationships. Since the effects were specific rather than shared across all the mental health domains included here, the timing of menarche may contribute to the developmental differentiation of depression from other mental health conditions in adolescence.
Availability of data and materials
The MoBa data are not publicly available as the consent given by the participants does not open for storage of data on an individual level in repositories or journals. Researchers who want access to data sets for replication should submit an application to datatilgang(at)fhi.no. Access to datasets requires approval from the Regional Committee for Medical and Health Research Ethics in Norway and an agreement with MoBa.
Data preparation and analysis code for all elements of the project are publicly available on GitHub at https://github.com/psychgen/aam-psych-adolesc-rr.
Abbreviations
- 2SLS:
-
Two-stage least squares
- ADHD:
-
Attention-deficit hyperactivity disorder
- ALSPAC:
-
Avon Longitudinal Study of Parents and Children
- BMI:
-
Body mass index
- CD:
-
Conduct disorder
- CI:
-
Confidence interval
- GWAS:
-
Genome-wide association study
- IPW:
-
Inverse probability weighting
- IVW:
-
Inverse variance weighted
- KUHR:
-
Control and payment of health refunds
- MI:
-
Multiple imputation
- MoBa:
-
Norwegian Mother, Father, and Child Cohort Study
- MR:
-
Mendelian randomisation
- MR-Egger:
-
Mendelian randomisation-Egger
- MR-PRESSO:
-
MR pleiotropy residual sum and outlier
- MVMR:
-
Multivariable Mendelian randomisation
- NHST:
-
Null hypothesis significance testing
- NPR:
-
Norwegian Patient Registry
- ODD:
-
Oppositional defiant disorder
- RCT:
-
Randomised controlled trial
- RS-DBD:
-
Rating Scale for Disruptive Behaviour Disorders
- SCARED:
-
Screen for Child Anxiety-Related Disorders
- SESOI:
-
Smallest effect size of interest
- SMFQ:
-
Short Mood and Feelings Questionnaire
- SNPs:
-
Single nucleotide polymorphisms
References
Benoit A, Lacourse E, Claes M. Pubertal timing and depressive symptoms in late adolescence: the moderating role of individual, peer, and parental factors. Dev Psychopathol. 2013;25(2):455–71.
Conley CS, Rudolph KD, Bryant FB. Explaining the longitudinal association between puberty and depression: sex differences in the mediating effects of peer stress. Dev Psychopathol. 2012;24(2):691–701.
Copeland W, Shanahan L, Miller S, Costello EJ, Angold A, Maughan B. Outcomes of early pubertal timing in young women: a prospective population-based study. Am J Psychiatry. 2010;167(10):1218–25.
Ge X, Conger RD, Elder GH Jr. Pubertal transition, stressful life events, and the emergence of gender differences in adolescent depressive symptoms. Dev Psychol. 2001;37(3):404–17.
Ge X, Kim IJ, Brody GH, Conger RD, Simons RL, Gibbons FX, et al. It’s about timing and change: pubertal transition effects on symptoms of major depression among African American youths. Dev Psychol. 2003;39(3):430–9.
Ge X, Brody GH, Conger RD, Simons RL. Pubertal maturation and African American children’s internalizing and externalizing symptoms. J Youth Adolesc. 2006;35(4):528–37.
Graber JA, Seeley JR, Brooks-Gunn J, Lewinsohn PM. Is pubertal timing associated with psychopathology in young adulthood? J Am Acad Child Adolesc Psychiatry. 2004;43(6):718–26.
Graber JA, Brooks-Gunn J, Warren MP. Pubertal effects on adjustment in girls: moving from demonstrating effects to identifying pathways. J Youth Adolesc. 2006;35(3):391–401.
Hamlat EJ, Stange JP, Abramson LY, Alloy LB. Early pubertal timing as a vulnerability to depression symptoms: differential effects of race and sex. J Abnorm Child Psychol. 2014;42(4):527–38.
Keenan K, Culbert KM, Grimm KJ, Hipwell AE, Stepp SD. Timing and tempo: exploring the complex association between pubertal development and depression in African American and European American girls. J Abnorm Psychol. 2014;123(4):725–36.
Mendle J, Harden KP, Brooks-Gunn J, Graber JA. Development’s tortoise and hare: pubertal timing, pubertal tempo, and depressive symptoms in boys and girls. Dev Psychol. 2010;46(5):1341–53.
Nadeem E, Graham S. Early puberty, peer victimization, and internalizing symptoms in ethnic minority adolescents. J Early Adolesc. 2005;25(2):197–222.
Rudolph KD, Troop-Gordon W. Personal-accentuation and contextual-amplification models of pubertal timing: predicting youth depression. Dev Psychopathol. 2010;22(2):433–51.
Blumenthal H, Leen-Feldner EW, Babson KA, Gahr JL, Trainor CD, Frala JL. Elevated social anxiety among early maturing girls. Dev Psychol. 2011;47(4):1133–40.
Deardorff J, Hayward C, Wilson KA, Bryson S, Hammer LD, Agras S. Puberty and gender interact to predict social anxiety symptoms in early adolescence. J Adolesc Health. 2007;41(1):102–4.
Bakker MP, Ormel J, Lindenberg S, Verhulst FC, Oldehinkel AJ. Generation of interpersonal stressful events: the role of poor social skills and early physical maturation in young adolescents—the TRAILS study. J Early Adolesc. 2011;31(5):633–55.
Haynie DL. Contexts of risk? Explaining the link between girls’ pubertal development and their delinquency involvement. Soc Forces. 2003;82(1):355–97.
Lynne SD, Graber JA, Nichols TR, Brooks-Gunn J, Botvin GJ. Links between pubertal timing, peer influences, and externalizing behaviors among urban students followed through middle school. J Adolesc Health. 2007;40(2):181.e7–181.e13.
Mrug S, Elliott M, Gilliland MJ, Grunbaum JA, Tortolero SR, Cuccaro P, et al. Positive parenting and early puberty in girls: protective effects against aggressive behavior. Arch Pediatr Adolesc Med. 2008;162(8):781–6.
Ullsperger JM, Nikolas MA. A meta-analytic review of the association between pubertal timing and psychopathology in adolescence: are there sex differences in risk? Psychol Bull. 2017;143(9):903–38.
Hamlat EJ, Snyder HR, Young JF, Hankin BL. Pubertal timing as a transdiagnostic risk for psychopathology in youth. Clin Psychol Sci. 2019;7(3):411–29.
Hankin BL, Abramson LY, Moffitt TE, Silva PA, McGee R, Angell KE. Development of depression from preadolescence to young adulthood: emerging gender differences in a 10-year longitudinal study. J Abnorm Psychol. 1998;107(1):128–40.
Graber JA. Pubertal timing and the development of psychopathology in adolescence and beyond. Horm Behav. 2013;64(2):262–9.
Black SR, Klein DN. Early menarcheal age and risk for later depressive symptomatology: the role of childhood depressive symptoms. J Youth Adolesc. 2012;41(9):1142–50.
Ge X, Conger RD, Elder GH Jr. Coming of age too early: pubertal influences on girls’ vulnerability to psychological distress. Child Dev. 1996;67(6):3386–400.
Joinson C, Heron J, Lewis G, Croudace T, Araya R. Timing of menarche and depressive symptoms in adolescent girls from a UK cohort. Br J Psychiatry. 2011;198(1):17–23.
Lam TH, Stewart SM, Leung GM, Lee PW, Wong JP, Ho LM, et al. Depressive symptoms among Hong Kong adolescents: relation to atypical sexual feelings and behaviors, gender dissatisfaction, pubertal timing, and family and peer relationships. Arch Sex Behav. 2004;33(5):487–96.
Kaltiala-Heino R, Kosunen E, Rimpelä M. Pubertal timing, sexual behaviour and self-reported depression in middle adolescence. J Adolesc. 2003;26(5):531–45.
Kaltiala-Heino R, Marttunen M, Rantanen P, Rimpelä M. Early puberty is associated with mental health problems in middle adolescence. Soc Sci Med. 2003;57(6):1055–64.
Rierdan J, Koff E. Depressive symptomatology among very early maturing girls. J Youth Adolesc. 1991;20(4):415–25.
Stice E, Presnell K, Bearman SK. Relation of early menarche to depression, eating disorders, substance abuse, and comorbid psychopathology among adolescent girls. Dev Psychol. 2001;37(5):608–19.
Hayward C, Gotlib IH, Schraedley PK, Litt IF. Ethnic differences in the association between pubertal status and symptoms of depression in adolescent girls. J Adolesc Health. 1999;25(2):143–9.
Carter R, Caldwell CH, Matusko N, Antonucci T, Jackson JS. Ethnicity, perceived pubertal timing, externalizing behaviors, and depressive symptoms among black adolescent girls. J Youth Adolesc. 2011;40(10):1394–406.
Martino S, Lester D. Menarche and eating disorders. Psychol Rep. 2013;113(1):315–7.
McGuire TC, McCormick KC, Koch MK, Mendle J. Pubertal maturation and trajectories of depression during early adolescence. Front Psychol. 2019;10:1362.
Smith-Woolley E, Rimfeld K, Plomin R. Weak associations between pubertal development and psychiatric and behavioral problems. Transl Psychiatry. 2017;7(4):e1098.
Toffol E, Koponen P, Luoto R, Partonen T. Pubertal timing, menstrual irregularity, and mental health: results of a population-based study. Arch Womens Ment Health. 2014;17(2):127–35.
Joinson C, Heron J, Araya R, Lewis G. Early menarche and depressive symptoms from adolescence to young adulthood in a UK cohort. J Am Acad Child Adolesc Psychiatry. 2013;52(6):591–8.
Angold A, Costello EJ, Erkanli A, Worthman CM. Pubertal changes in hormone levels and depression in girls. Psychol Med. 1999;29(5):1043–53.
Balzer BW, Duke SA, Hawke CI, Steinbeck KS. The effects of estradiol on mood and behavior in human female adolescents: a systematic review. Eur J Pediatr. 2015;174(3):289–98.
Skovlund CW, Mørch LS, Kessing LV, Lidegaard Ø. Association of hormonal contraception with depression. JAMA Psychiat. 2016;73(11):1154–62.
Joinson C, Heron J, Araya R, Paus T, Croudace T, Rubin C, et al. Association between pubertal development and depressive symptoms in girls from a UK cohort. Psychol Med. 2012;42(12):2579–89.
Horvath G, Knopik VS, Marceau K. Polygenic influences on pubertal timing and tempo and depressive symptoms in boys and girls. J Res Adolesc. 2020;30(1):78–94.
Hooper L, Ness AR, Smith GD. Antioxidant strategy for cardiovascular disease. The Lancet. 2001;357(9269):1705–6.
Bell JA, Carslake D, Wade KH, Richmond RC, Langdon RJ, Vincent EE, et al. Influence of puberty timing on adiposity and cardiometabolic traits: a Mendelian randomisation study. PLoS Med. 2018;15(8):e1002641.
Hartwig FP, Bowden J, de Mola CL, Tovo-Rodrigues L, Smith GD, Horta BL. Body mass index and psychiatric disorders: a Mendelian randomization study. Sci Rep. 2016;6(1):1–11.
Wray NR, Ripke S, Mattheisen M, Trzaskowski M, Byrne EM, Abdellaoui A, et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat Genet. 2018;50(5):668–81.
Tyrrell J, Mulugeta A, Wood AR, Zhou A, Beaumont RN, Tuke MA, et al. Using genetics to understand the causal influence of higher BMI on depression. Int J Epidemiol. 2019;48(3):834–48.
Lien L, Haavet OR, Dalgard F. Do mental health and behavioural problems of early menarche persist into late adolescence? A three year follow-up study among adolescent girls in Oslo. Norway Soc Sci Med. 2010;71(3):529–33.
Davey Smith G, Ebrahim S. ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. 2003;32(1):1–22.
Sanderson E, Glymour MM, Holmes MV, Kang H, Morrison J, Munafò MR, et al. Mendelian randomization. Nat Rev Methods Primer. 2022;2(1):1–21.
Day FR, Thompson DJ, Helgason H, Chasman DI, Finucane H, Sulem P, et al. Genomic analyses identify hundreds of variants associated with age at menarche and support a role for puberty timing in cancer risk. Nat Genet. 2017;49(6):834–41.
Davey Smith G, Holmes MV, Davies NM, Ebrahim S. Mendel’s laws, Mendelian randomization and causal inference in observational data: substantive and nomenclatural issues. Eur J Epidemiol. 2020;35(2):99–111.
Lundblad MW, Jacobsen BK. The reproducibility of self-reported age at menarche: the Tromsø Study. BMC Womens Health. 2017;17(1):1–7.
Lawlor DA, Harbord RM, Sterne JA, Timpson N, Davey SG. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med. 2008;27(8):1133–63.
Sequeira ME, Lewis SJ, Bonilla C, Smith GD, Joinson C. Association of timing of menarche with depressive symptoms and depression in adolescence: Mendelian randomisation study. Br J Psychiatry. 2017;210(1):39–46.
Magnus P, Birke C, Vejrup K, Haugan A, Alsaker E, Daltveit AK, et al. Cohort profile update: the Norwegian Mother and Child Cohort Study (MoBa). Int J Epidemiol. 2016;45(2):382–8.
Lawlor DA, Tilling K, Davey SG. Triangulation in aetiological epidemiology. Int J Epidemiol. 2016;45(6):1866–86.
Munafò MR, Davey SG. Robust research needs many lines of evidence. Nature. 2018;553(7686):399–402.
Munafò MR, Higgins JP, Smith GD. Triangulating evidence through the inclusion of genetically informed designs. Cold Spring Harb Perspect Med. 2021;11(8):a040659.
Magnus MC, Guyatt AL, Lawn RB, Wyss AB, Trajanoska K, Küpers LK, et al. Identifying potential causal effects of age at menarche: a Mendelian randomization phenome-wide association study. BMC Med. 2020;18(1):1–17.
Munafò MR, Nosek BA, Bishop DV, Button KS, Chambers CD, Du Sert NP, et al. A manifesto for reproducible science. Nat Hum Behav. 2017;1(1):1–9.
Angold A, Costello EJ, Messer SC, Pickles A. Development of a short questionnaire for use in epidemiological studies of depression in children and adolescents. Int J Methods Psychiatr Res. 1995;5(4):237–49.
Birmaher B, Khetarpal S, Brent D, Cully M, Balach L, Kaufman J, et al. The Screen for Child Anxiety Related Emotional Disorders (SCARED): scale construction and psychometric characteristics. J Am Acad Child Adolesc Psychiatry. 1997;36(4):545–53.
Silva RR, Alpert M, Pouget E, Silva V, Trosper S, Reyes K, et al. A rating scale for disruptive behavior disorders, based on the DSM-IV item pool. Psychiatr Q. 2005;76(4):327–39.
Paltiel L, Anita H, Skjerden T, Harbak K, Bækken S, Kristin SN, et al. The biobank of the Norwegian Mother and Child Cohort Study–present status. Nor Epidemiol. 2014;24(1–2):29–35.
Corfield EC, Frei O, Shadrin AA, Rahman Z, Lin A, Athanasiu L, et al. The Norwegian Mother, Father, and Child cohort study (MoBa) genotyping data resource: MoBaPsychGen pipeline v. 1. 2022. Preprint at https://www.biorxiv.org/content/10.1101/2022.06.23.496289v3.
Schmitz D, Ek WE, Berggren E, Höglund J, Karlsson T, Johansson Å. Genome-wide association study of estradiol levels, and the causal effect of estradiol on bone mineral density. J Clin Endocrinol Metab. 2021;106(11):e4471–86.
Richardson TG, Sanderson E, Elsworth B, Tilling K, Davey SG. Use of genetic variation to separate the effects of early and later life adiposity on disease risk: Mendelian randomisation study. BMJ. 2020;369:m1203.
Howard DM, Adams MJ, Clarke TK, Hafferty JD, Gibson J, Shirali M, et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat Neurosci. 2019;22(3):343–52.
Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife. 2018;7:e34408.
Schmitz D, Ek WE, Berggren E, Höglund J, Karlsson T, Johansson Å. Genome-wide association study of estradiol levels and the causal effect of estradiol on bone mineral density. J Clin Endocrinol Metab. 2021;106(11):e4471–86.
Felix JF, Bradfield JP, Monnereau C, van der Valk RJP, Stergiakouli E, Chesi A, et al. Genome-wide association analysis identifies three new susceptibility loci for childhood body mass index. Hum Mol Genet. 2016;25(2):389–403.
Mbatchou J, Barnard L, Backman J, Marcketta A, Kosmicki JA, Ziyatdinov A, et al. Computationally efficient whole-genome regression for quantitative and binary traits. Nat Genet. 2021;53(7):1097–103.
Hemani G, Tilling K, Davey SG. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLOS Genet. 2017;13(11):e1007081.
Hernan MA, Robins J. Causal inference: what if. Boca Raton: Chapman & Hill/CRC; 2020.
Zeileis A, Köll S, Graham N. Various versatile variances: an object-oriented implementation of clustered covariances in R. J Stat Softw. 2020;95(1):1–36.
Inoue A, Solon G. Two-sample instrumental variables estimators. Rev Econ Stat. 2010;92(3):557–61.
Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23(R1):R89–98.
Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50(5):693–8.
Burgess S, Bowden J, Fall T, Ingelsson E, Thompson SG. Sensitivity analyses for robust causal inference from Mendelian randomization analyses with multiple genetic variants. Epidemiol Camb Mass. 2017;28(1):30–42.
Burgess S, Foley CN, Allara E, Staley JR, Howson JM. A robust and efficient method for Mendelian randomization with hundreds of genetic variants. Nat Commun. 2020;11(1):1–11.
Burgess S, Thompson SG. Multivariable Mendelian randomization: the use of pleiotropic genetic variants to estimate causal effects. Am J Epidemiol. 2015;181(4):251–60.
Sanderson E, Spiller W, Bowden J. Testing and correcting for weak and pleiotropic instruments in two-sample multivariable Mendelian randomization. Stat Med. 2021;40:5434–52.
Grant AJ, Burgess S. Pleiotropy robust methods for multivariable Mendelian randomization. Stat Med. 2021;40(26):5813–30.
Lakens D, Scheel AM, Isager PM. Equivalence testing for psychological research: a tutorial. Adv Methods Pract Psychol Sci. 2018;1(2):259–69.
Sanderson E, Macdonald-Wallis C, Davey SG. Negative control exposure studies in the presence of measurement error: implications for attempted effect estimate calibration. Int J Epidemiol. 2018;47(2):587–96.
Simonsohn U. Small telescopes: detectability and the evaluation of replication results. Psychol Sci. 2015;26(5):559–69.
Brion MJA, Shakhbazov K, Visscher PM. Calculating statistical power in Mendelian randomization studies. Int J Epidemiol. 2013;42(5):1497–501.
Elwert F, Winship C. Endogenous selection bias: the problem of conditioning on a collider variable. Annu Rev Sociol. 2014;40(1):31–53.
Askelund AD, Wootton RE, Torvik FA, Lawn RB, Ask H, Corfield EC, et al. Assessing causal links between age at menarche and adolescent mental health: a Mendelian randomisation study [Registered Report Stage 1 Protocol]. figshare; 2022. Available from: https://springernature.figshare.com/articles/dataset/Assessing_causal_links_between_age_at_menarche_and_adolescent_mental_health_a_Mendelian_randomisation_study_Registered_Report_Stage_1_Protocol_/20101841/2
Hirtz R, Hars C, Naaresh R, Laabs BH, Antel J, Grasemann C, et al. Causal effect of age at menarche on the risk for depression: results from a two-sample multivariable Mendelian randomization study. Front Genet. 2022;13:918584.
Salk RH, Hyde JS, Abramson LY. Gender differences in depression in representative national samples: meta-analyses of diagnoses and symptoms. Psychol Bull. 2017;143(8):783–822.
Ong KK, Elks CE, Li S, Zhao JH, Luan J, Andersen LB, et al. Genetic variation in LIN28B is associated with the timing of puberty. Nat Genet. 2009;41(6):729–33.
Wei YB, Liu JJ, Villaescusa JC, Åberg E, Brené S, Wegener G, et al. Elevation of Il6 is associated with disturbed let-7 biogenesis in a genetic model of depression. Transl Psychiatry. 2016;6(8):e869–e869.
Prince C, Joinson C, Kwong AS, Fraser A, Heron J. The relationship between timing of onset of menarche and depressive symptoms from adolescence to adulthood. Epidemiol Psychiatr Sci. 2023;32:e60.
Padrutt ER, Harper J, Schaefer JD, Nelson KM, McGue M, Iacono WG, et al. Pubertal timing and adolescent outcomes: investigating explanations for associations with a genetically informed design. J Child Psychol Psychiatry. 2023;64(8):1232–41.
Harden KP, Mendle J. Gene-environment interplay in the association between pubertal timing and delinquency in adolescent girls. J Abnorm Psychol. 2012;121(1):73–87.
Brumpton B, Sanderson E, Heilbron K, Hartwig FP, Harrison S, Vie GÅ, et al. Avoiding dynastic, assortative mating, and population stratification biases in Mendelian randomization through within-family analyses. Nat Commun. 2020;11(1):3519.
Wootton RE, Jones HJ, Sallis HM. Mendelian randomisation for psychiatry: how does it work, and what can it tell us? Mol Psychiatry. 2022;27(1):53–7.
Nilsen RM, Vollset SE, Gjessing HK, Skjaerven R, Melve KK, Schreuder P, et al. Self-selection and bias in a large prospective pregnancy cohort in Norway. Paediatr Perinat Epidemiol. 2009;23(6):597–608.
Nohr EA, Liew Z. How to investigate and adjust for selection bias in cohort studies. Acta Obstet Gynecol Scand. 2018;97(4):407–16.
Acknowledgements
We thank the Norwegian Institute of Public Health (NIPH) for generating high-quality genomic data. This research is part of the HARVEST collaboration, supported by the Research Council of Norway (#229624). We also thank deCODE Genetics and the NORMENT Centre for providing genotype data, funded by the Research Council of Norway (#223273), South-Eastern Norway Health Authority, and KG Jebsen Stiftelsen. We further thank the Center for Diabetes Research, University of Bergen for providing genotype data and performing quality control and imputation of the data funded by the ERC AdG project SELECTionPREDISPOSED, Stiftelsen Kristian Gerhard Jebsen, Trond Mohn Foundation, the Research Council of Norway, the Novo Nordisk Foundation, the University of Bergen, and the Western Norway Health Authorities (Helse Vest). The Norwegian Mother, Father and Child Cohort Study is supported by the Norwegian Ministry of Health and Care Services and the Ministry of Education and Research. We are grateful to all the participating families in Norway who take part in this ongoing cohort study.
This work was performed on the TSD (Tjeneste for Sensitive Data) facilities, owned by the University of Oslo, operated and developed by the TSD service group at the University of Oslo, IT Department (USIT). The computations were performed on resources provided by Sigma2—the National Infrastructure for High Performance Computing and Data Storage in Norway. Data from NPR has been used in this publication. The interpretation and reporting of these data are the sole responsibility of the authors, and no endorsement by NPR is intended nor should be inferred.
Funding
Open access funding provided by Norwegian Institute of Public Health (FHI) The Research Council of Norway supports F.A.T., C.S., E.C., H.A., T.R.-K., N.M.D., and O.A.A. (#300668; #274611; #273659, #273659, #324620, #274611, #295989, #229129; #213837; #248778; #223273; #249711). The South-Eastern Regional Health Authority supports A.D.A., R.E.W., O.A.A., A.H., L.J.H., and E.C. (#2020023, #2020024, 2017–112, #2020022, #2018058, #2021045). N.M.D. and G.D.S. work in a unit that receives support from the University of Bristol and the UK Medical Research Council (MC_UU_00011/1). O.A.A. is also supported by Stiftelsen Kristian Gerhard Jebsen and H2020 grant CoMorMent (#847776). This work was partly supported by the Research Council of Norway through its Centres of Excellence funding scheme (#262700). The funders have/had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Author contributions are presented according to the CRediT (Contributor Roles Taxonomy). A.D.A: conceptualisation, methodology, formal analysis, software, visualisation, writing—original draft, writing—review and editing, and project administration. R.E.W: conceptualisation, methodology, software, and writing—review and editing. F.A.T: writing—review and editing. R.B.L: writing—review and editing. H.A: writing—review and editing and funding acquisition. E.C: data curation, software, and writing—review and editing. M.C.M: conceptualisation, methodology, and writing—review and editing. T.R.-K: writing—review and editing and funding acquisition. P.M: investigation and writing—review and editing. O.A.A: investigation and writing—review and editing. C.S.: writing—review and editing. G.D.S: writing—review and editing. N.M.D: methodology and writing—review and editing. A.H: conceptualisation, methodology, writing—review and editing, supervision, and funding acquisition. L.J.H: conceptualisation, methodology, formal analysis, software, data curation, writing—original draft, writing—review and editing, supervision, and funding acquisition. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The establishment of MoBa and initial data collection was based on a licence from the Norwegian Data Protection Agency and approval from the Regional Committees for Medical and Health Research Ethics. The MoBa cohort is now based on regulations related to the Norwegian Health Registry Act. The current study was approved by the Regional Committees for Medical and Health Research Ethics (REK numbers 2016/1702). By consenting to MoBa, participants have also agreed to linkage to KUHR, NPR, and MBRN. MBRN is a national health registry containing information about all births in Norway.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Supplementary methods with information about the: a) categorised age at menarche, b) dichotomised depressive symptoms, c) multiple imputation, d) diagnostic codes, e) inverse probability weighting, f) psychometric properties of scales, g) definition/removal of outliers.
Additional file 2: Table S1.
With psychometric properties of 8y symptom scales (ordinal Cronbach’s alphas).
Additional file 3: Table S2.
With overview of variables: details about items, missingness, and processing.
Additional file 4.
Description of genetic instruments for two-sample MR analyses.
Additional file 5.
Description of MR sensitivity analyses, including: a) one-sample MR sensitivity analyses, b) two-sample MR sensitivity analyses, c) multivariable MR analyses.
Additional file 6.
Outline of analyses for each hypothesis, including: a) main analyses, b) negative control analyses, c) the smallest effect size of interest, d) sensitivity analyses, e) inference criteria.
Additional file 7.
Description of power analyses, including: a) projected prevalence, b) data generation, c) power calculation, d) Figs. S1-S8 showing results of power analysis for all hypotheses.
Additional file 8.
Supplementary results, including: a) imputation of age at menarche, b) breast stage as an additional exposure, c) categorised age at menarche, d) exploratory analyses.
Additional file 9: Table S3.
With associations of genetic instrument for age at menarche with the covariates.
Additional file 10: Table S4.
With results of multivariable Mendelian randomisation sensitivity analyses.
Additional file 11: Fig. S9.
With differences between participants and non-participants with IP weighting.
Additional file 12: Fig. S10.
Showing the impact of inverse probability weighting on results for depression.
Additional file 13: Fig. S11.
With 2-sample MR sensitivity analyses for other mental health domains.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Askelund, A.D., Wootton, R.E., Torvik, F.A. et al. Assessing causal links between age at menarche and adolescent mental health: a Mendelian randomisation study. BMC Med 22, 155 (2024). https://doi.org/10.1186/s12916-024-03361-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12916-024-03361-8