
Abstract
Background
For patients starting treatment for depression, current guidelines recommend titrating the antidepressant dosage to the maximum of the licenced range if tolerated. When patients do not achieve remission within several weeks, recommendations include adding or switching to another antidepressant. However, the relative merits of these guideline strategies remain unestablished.
Methods
This multi-centre, open-label, assessor-blinded, pragmatic trial involved two steps. Step 1 used open-cluster randomisation, allocating clinics into those titrating sertraline up to 50 mg/day or 100 mg/day by week 3. Step 2 used central randomisation to allocate patients who did not remit after 3 weeks of treatment to continue sertraline, to add mirtazapine or to switch to mirtazapine. The primary outcome was depression severity measured with the Patient Health Questionnaire-9 (PHQ-9) (scores between 0 and 27; higher scores, greater depression) at week 9. We applied mixed-model repeated-measures analysis adjusted for key baseline covariates.
Results
Between December 2010 and March 2015, we recruited 2011 participants with hitherto untreated major depression at 48 clinics in Japan. In step 1, 970 participants were allocated to the 50 mg/day and 1041 to the 100 mg/day arms; 1927 (95.8%) provided primary outcomes. There was no statistically significant difference in the adjusted PHQ-9 score at week 9 between the 50 mg/day arm and the 100 mg/day arm (0.25 point, 95% confidence interval (CI), − 0.58 to 1.07, P = 0.55). Other outcomes proved similar in the two groups.
In step 2, 1646 participants not remitted by week 3 were randomised to continue sertraline (n = 551), to add mirtazapine (n = 537) or to switch to mirtazapine (n = 558): 1613 (98.0%) provided primary outcomes. At week 9, adding mirtazapine achieved a reduction in PHQ-9 scores of 0.99 point (0.43 to 1.55, P = 0.0012); switching achieved a reduction of 1.01 points (0.46 to 1.56, P = 0.0012), both relative to continuing sertraline. Combination increased the percentage of remission by 12.4% (6.1 to 19.0%) and switching by 8.4% (2.5 to 14.8%). There were no differences in adverse effects.
Conclusions
In patients with new onset depression, we found no advantage of titrating sertraline to 100 mg vs 50 mg. Patients unremitted by week 3 gained a small benefit in reduction of depressive symptoms at week 9 by switching sertraline to mirtazapine or by adding mirtazapine.
Trial registration
ClinicalTrials.gov, NCT01109693. Registered on 23 April 2010.
Similar content being viewed by others


Background
Every year, an estimated five million people in high-income countries alone start new antidepressants to treat their depression [1,2,3]. In the USA, the annual prevalence of prescribed antidepressant use exceeds 10% of the population, almost double that of 10 years before [2]. Antidepressant use is similarly high and increasing in European countries, ranging between 4 and 9% [1], with a 1-year incidence of new antidepressant prescription of approximately 1% [3]. Clinicians need specific, detailed and appropriate guidelines to guide their antidepressant pharmacotherapy.
To initiate antidepressant treatment, modern guidelines recommend new generation antidepressants and in particular selective serotonin reuptake inhibitors (SSRIs) [4]. A network meta-analysis of 12 new generation antidepressants suggested that the SSRI sertraline, because of its favourable balance of benefits, acceptability and cost, may be the best choice when starting treatment for major depression [5]. Sertraline has been the most widely prescribed antidepressant in the USA [6] and elsewhere [7].
Once they choose a first-line antidepressant, practitioners must optimise its dosage, considering the wide approved dose range for most drugs. Many guidelines only list such ranges and do not specify where within this range the initial treatment should aim [4, 8, 9]. The American Psychiatric Association guideline is more specific and recommends that the initial doses be incrementally raised and maximised, side effects permitting ([10], p. 43). However, systematic reviews of randomised controlled trials (RCTs) have provided conflicting results regarding the benefits and harms of lower vs higher doses of various antidepressants within their therapeutic ranges. One review synthesised results from 33 RCTs comparing two or more doses of the same antidepressants and found that a dose level of 100–200 mg imipramine equivalents (or 20–40 mg fluoxetine equivalents) showed the highest response rate, while lower doses showed a reduction in efficacy and higher doses were not accompanied by increased efficacy. Adverse events increased monotonically with dose [11]. However, this study included both old and new generation antidepressants. The dose-response curves may be different for different classes of antidepressants. More recent reviews focussing on a single agent [12] or several agents [13] suggested that there was no dose-response relationship within the approved dose ranges for SSRIs.
Clinicians face another challenge because, after several weeks of the first-line treatment, only 50% of patients respond (i.e. achieve depression severity less than half that at pre-treatment), and only 30% remit (i.e. return to a euthymic state). Patients’ failure to respond or remit requires consideration of alternative treatments. Guideline recommendations for second-line treatment include dose escalation, switching to a different antidepressant or adding a different drug [4, 10]. The last strategy may be divided into ‘augmentation’ when a non-antidepressant drug is added to an antidepressant and ‘combination’ when two antidepressants are used together [4]. Systematic reviews of RCTs agree that dose escalation confers no benefit beyond continuing the initial drug dose [14,15,16,17]; consistent with these results, the German National Guideline clearly states that dose escalation for SSRI does not work [8]. Two previous systematic reviews have found some support for the switching strategy [18], especially to an antidepressant from a different class [19]. However, a recent more rigorous meta-analysis found no high-level evidence that switching the antidepressant is effective when compared to simply continuing the initial antidepressant [20]. There are also reviews that support various augmentation strategies [21] and combination strategies [22]. However, most of the RCTs addressing patients who fail to respond to initial pharmacotherapy for depression compared new strategies against continuing preceding treatment; far less evidence exists comparing alternative second-line strategies against one another. For example, only two reports have directly compared switching vs combination strategies among patients who failed first-line treatment: one study compared combination vs switching vs continuing the prior treatment [23], while another compared dose escalation vs combination vs continuing the prior treatment [24]. Unfortunately, neither study was sufficiently powered (only 32–38 patients in each arm in the first study and 98–99 patients in the second study) to reach meaningful conclusions. The STAR*D trial randomised 1439 patients with major depression who had not remitted on citalopram to various switching strategies or augmentation/combination strategies. However, they used an equipoise-stratified design which gave patients choices in their treatment regimen: only 105 patients consented to randomisation to any of the drug switching or augmentation strategies, and consequently the analyses had to be conducted separately among the switching strategies or among the augmentation/combination strategies [25, 26]. A recently published study compared switching to bupropion against combining with bupropion or augmenting with an antipsychotic aripiprazole among 1522 patients with antidepressant-refractory major depression and found that aripiprazole augmentation significantly increased response over bupropion combination or switching [27]. Most patients in this trial were, however, chronically depressed and highly refractory (the mean duration of index episode was more than 85 months and the mean number of previous medication courses was 2.4). The trial does not, therefore, address the initial treatment of a new depressive episode.
Even less is known about when to institute second-line treatment. The American Psychiatric Association practice guideline recommends 4–8 weeks [10], the American College of Physicians 6–8 weeks [28] and the National Institute for Clinical Excellence (NICE) guideline 3–4 weeks at one place but 6–8 weeks at another [4].
Thus, in trying to optimise antidepressant pharmacotherapy for patients with major depression, clinicians face conflicting guidelines informed by low quality evidence. We therefore conducted a pragmatic RCT — an RCT that mimics the practice environment and thus maximises applicability — to examine the following two questions concerning the first- and second-line treatments for a hitherto untreated episode of major depression:
-
1.
What is the relative effectiveness and safety of initially titrating to the lowest vs the highest value of the therapeutic dose range of an antidepressant? On the basis of the results of prior evidence [5] and existing practices [6, 7], we chose sertraline as the drug to be tested. Since the available evidence suggests that there may be differences between different classes of antidepressants with respect to dose-response relationship [15], the first hypothesis pertains to the choice of the initial target dose of an SSRI, and of sertraline in particular, within its therapeutic range.
-
2.
When patients do not remit on first-line treatment, what is the relative effectiveness and safety of continuing initial treatment, combining with mirtazapine or switching to mirtazapine? We set 3 weeks as this decision point as the earliest time point in guideline recommendations [4]. Our choice of mirtazapine is consistent with a systematic review reporting that combination of a reuptake inhibitor antidepressant such as an SSRI and a blocker of presynaptic alpha-2 autoreceptor (mirtazapine, mianserin, trazodone) was superior to other combinations [22]. In addition, mirtazapine poses a very low risk of drug interaction. Finally, in the recently updated network meta-analysis of 21 antidepressants, mirtazapine was the second most potent antidepressant after amitriptyline [29]; its less favourable acceptability profile renders it less suitable as first-line treatment, but when first-line treatment fails, mirtazapine represents a potentially appropriate second choice.
Methods
Study design and participants
The Strategic Use of New generation antidepressants for Depression (SUN☺D) is a multi-centre, open-label, assessor-blinded, pragmatic RCT that involved two randomisation steps to examine the first- and second-line treatment strategies for untreated unipolar major depressive episodes.
The study was conducted in the departments of psychiatry at 48 clinics and hospitals across Japan between December 2010 and September 2015. We recruited adult men and women between 25 and 75 years who had a primary diagnosis of a non-psychotic unipolar major depressive episode according to the Diagnostic and Statistical Manual of Mental Disorders, 4th Edition (DSM-IV) [30] within the past month as ascertained by the study psychiatrists administering the semi-structured interview using the major depression section of the Primary Care Evaluation of Mental Disorders (PRIME-MD) [31]. All recruited persons were also not allowed to be taking any antidepressant, antipsychotic or mood stabiliser. Other exclusion criteria included history of schizophrenia, schizoaffective disorder, bipolar disorder; current dementia, borderline personality disorder, eating disorder or substance dependence; and imminent high risk of suicide, as judged by the study psychiatrist. No severity threshold was required so long as the participant satisfied the diagnostic criteria for major depressive disorder in the past month. The protocol provides additional details of the eligibility criteria [32]. When the study psychiatrist decided that sertraline was indicated, the patient began taking 25 mg/day of sertraline; only those who tolerated sertraline for 3–16 days were eligible.
An institutional review board at each participating site approved the study. An independent data monitoring committee oversaw the trial. All participants provided written informed consent. The study protocol, its amendment and the statistical analysis plan have been published elsewhere [32, 33].
Randomisation and masking
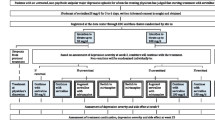
Figure 1 presents the study flow. In step 1, using the minimisation method adjusting for expected recruitment, study sites were randomised to titrate sertraline up to the minimum or the maximum of the licenced dosage in Japan (50 mg/day or 100 mg/day) by week 3. The unit of randomisation was therefore by site. The cluster randomisation design was chosen to avoid any confusion and possible protocol violation that might have arisen had we asked study physicians, who often had their own preference for either the minimum or the maximum target doses, to use different titrating strategies for different patients. Further, we were concerned that requiring patients to undergo two individual randomisations within 3 weeks might decrease the feasibility of this large pragmatic trial.

Trial profile. ® randomised, EDC electronic data capture system
In step 2, participants who had not reached remission, defined as scoring 4 or less on the Patient Health Questionnaire (PHQ-9) [34] at week 3, were randomised 1:1:1 through the web-based central computer system to continue with sertraline, to combine sertraline with mirtazapine or to switch to mirtazapine. The step 2 randomisation used the minimisation method adjusting for site; whether 50% or greater reduction on PHQ-9 had been achieved; and whether patients reported moderate or greater impairment due to side effects.
Physicians and patients were aware of their treatments. Outcome assessors were blinded throughout. The statisticians conducting the analyses and the writing committee were masked to treatment allocation until they signed the agreed-upon interpretations of the results.
Procedures
In step 1, participants in the minimum dosage arm received 50 mg/day of sertraline at week 1 to be continued through week 3, and those in the maximum dosage arm received 50–75 mg/day at week 1 and 100 mg/day at week 2 to be continued through week 3. When these target dosages were not tolerated, lower dosages were accepted.
In step 2, in the continuing sertraline arm, sertraline was administered as 50 or 100 mg/day according to the initial randomisation. In the combination arm, sertraline was continued and mirtazapine was added between 7.5 and 45 mg/day at the discretion of the study psychiatrist. In the mirtazapine switch arm, mirtazapine between 7.5 mg and 45 mg/day was administered; sertraline was tapered and discontinued by week 7.
Co-administration of benzodiazepines, but not of another antidepressant, antipsychotic or mood stabiliser was allowed up to week 9. Neither was any depression-specific psychotherapy such as cognitive behavioural therapy (CBT) or interpersonal psychotherapy (IPT) allowed. Those who withdrew consent to the study treatments by week 9 but still participated in follow-up evaluations received treatment as negotiated with their physician. Between weeks 9 and 25, there were no restrictions on treatments, and the continuation therapy was at the physician’s discretion.
Outcomes
For both the step 1 and step 2 randomisations, the primary outcome was depression severity as measured by masked assessors conducting semi-structured interviews using the PHQ-9 at week 9. The PHQ-9 consists of the nine diagnostic criteria items of major depression from the DSM-IV [35]. Each item is rated between 0 = ’Not at all’ through 3 = ’Nearly every day’, making the total score range 0–27, with higher ratings indicating increased depression severity. The instrument has demonstrated excellent reliability, validity and responsiveness [34].
Secondary outcomes included the Frequency, Intensity and Burden of Side Effects Rating (FIBSER) and the Beck Depression Inventory 2nd edition (BDI-II). The FIBSER collects information regarding the frequency, intensity and burden of side effects, each on a 7-point scale with higher ratings indicating greater severity, and provides a total score between 3 and 21 [25]. The BDI-II is a self-report measure of depression severity addressing 21 symptoms of depression, each on a scale between 0 and 3, with the total score ranging between 0 and 63; higher scores indicate more severe depression [36]. In addition, the study psychiatrist recorded any incident of suicidality according to the Columbia Classification Algorithm of Suicide Assessment (C-CASA) [37] or of manic/hypomanic/mixed episodes at weeks 9 and 25, and reported any serious adverse events throughout the study. Response was defined as 50% or greater reduction from week 1 on PHQ-9, and remission as scoring 4 or less on PHQ-9 [34].
The PHQ-9 and the FIBSER were administered by telephone assessors [38], trained at and operating from the central office and blind to the treatment assignment and the timing of the assessment, at weeks 1, 3, 9 and 25. We had previously established the high inter-rater reliability of our telephone assessors [39]. The BDI-II was filled in by the participants at weeks 0, 1 and then every 2 weeks up to week 9, and every 4 weeks up to week 25.
Sample size
For step 1, we calculated that we would need 66 participants at each of 30 sites in order to ensure adequate power (beta level of 0.20) at a two-sided alpha level of 0.05 to detect a mean difference of 1 point on PHQ-9 with an estimated standard deviation (SD) of 5 or a standardised mean difference of 0.20 between the two arms, for the null hypothesis of no difference and an intracluster correlation coefficient of 0.05 [40]. We reasoned that an effect size of 0.20, corresponding with a small effect according to Cohen [41], would represent an important difference between active treatments. The required total sample size for step 1 was 1980.
For step 2, we calculated that 522 participants per group would ensure adequate power (beta level of 0.20) at a two-sided alpha level of 0.05 to detect a standardised mean difference of 0.20 among any two of the three treatment groups. Assuming a dropout rate of 10% and a remission rate of 10% at week 3, we needed 1934 participants entering step 1.
Statistical analysis
In order to examine the optimum initial target dose of sertraline, we analysed results from all those who were cluster-randomised at week 1 to titrate sertraline up to 50 mg/day or 100 mg/day and, had they followed the protocol, would have stayed on that sertraline dose up to week 9 — that is, those who had remitted and continued on sertraline; those who had not remitted and were randomised at week 3 to continue sertraline; and those who declined the second randomisation. We used a weighted mixed-model repeated-measures analysis to compare model-adjusted least squares means: the primary outcome was the PHQ-9 at week 9 as pre-specified in the study protocol. The model included random effects for subjects and sites and fixed effects of treatment, visit (as categorical) and treatment-by-visit interaction, adjusted for age, sex, education, marital status, number of previous depressive episodes and baseline scores at weeks 0 and 1. At the first blinded interpretation committee meeting, the statistician reported an imbalance in the baseline demographic and clinical variables. The writing committee therefore agreed, at that meeting, without knowledge of the identity of the arms and based on their prior knowledge regarding variables that may be associated with depression severity, on the following variables to be entered in the primary analysis as possible confounders: age, sex, education, marital status, number of previous depressive episodes and baseline PHQ-9 scores at weeks 0 and 1. Each observation was weighted by inverse probability of censoring (IPCW) to account for missing outcomes due to being allocated to the combination or switching arms at step 2 randomisation. The weight for IPCW was calculated by a logistic regression that included age, sex, education, marriage, number of episodes, PHQ-9 scores at weeks 0 and 1 and whether the participant was allocated to continue sertraline or otherwise at week 3 as predictors of missingness. We applied the same modelling approach to the continuous secondary outcomes. We used weighted generalised linear mixed models with the logit link and binomial distributions to account for clustering effects for the dichotomous secondary outcomes. For the time to discontinuation of the allocated or any treatment, we applied Cox regression with the same covariates and calculated the hazard ratio (HR) and its 95% confidence interval (CI). For step 1 analyses, we examined two subgroup hypotheses, pre-specified in the statistical analysis plan [33]: (1) whether the PHQ-9 score at week 1 was less than or greater than 15 and (2) whether the patient had shown improvement from week 0 to week 1 at or above the median of the sample. We also conducted four sensitivity analyses employing mixed models with different assumptions, described in the statistical analysis plan [33].
To compare the second-line strategies, we used the mixed-model repeated-measures analysis comparing model-adjusted least squares means of the PHQ-9 at week 9 among the three treatment arms. This model included the following: fixed effects of PHQ-9 at week 3, treatment, visit (as categorical), treatment-by-visit interaction, minimisation variables for step 2 randomisation. The model included individuals and sites as random effects. We applied the same modelling approach to the continuous secondary outcomes and the logistic regression model for the dichotomous secondary outcomes. In order to facilitate the clinical interpretation of the obtained odds ratio (OR), we calculated the risk difference (RD) and its CI by applying the model-adjusted OR and its CI to the event rate in the sertraline continuation arm. For the time to discontinuation, we applied Cox regression with the same covariates. We applied the Hochberg procedure for adjusting the multiple comparisons involved with three treatment arms in the primary comparison and reported adjusted P values. For step 2 analyses, we examined three pre-specified hypotheses regarding possible effect modifiers [33]: (1) whether patients had achieved a 50% or greater reduction on PHQ-9 from week 1 to week 3, (2) whether FIBSER results indicated ’moderate’ or greater impairment due to side effects or (iii) whether step 1 was high or low dose.
All efficacy and safety analyses followed the intention-to-treat principle, and all patents were analysed in the groups to which they were randomised.
Blinded interpretation of the results
The writing committee reviewed a statistical report in which the treatments were masked, and developed interpretation of the results and associated conclusions under alternative scenarios for all possible permutations of treatments [42]. The treatments were revealed only after the writing committee signed off on the agreed-upon interpretations (See Additional file 1).
Role of the funding source
The funders of the study had no role in study design, data collection, data analysis, data interpretation or in writing of the report. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Changes from the original protocol
No major change was made since the original protocol and statistical analysis plan [32, 33]. Our published protocol [32] lists the several minor changes in the details since the original protocol.
Results
Figure 1 shows the screening, randomisation and follow-up of the study participants. Between December 2010 and March 2015, 56,261 first-visit patients to the participating 48 clinics and hospitals in Japan underwent eligibility assessment, of whom 7895 suffered from untreated unipolar major depressive episodes. Of these, 2011 patients satisfied eligibility criteria and were cluster randomised to titrate sertraline up to the minimum or maximum of the licenced dosage within 3 weeks: 970 in 22 clinics and hospitals to the 50 mg/day arm and 1041 in 26 clinics and hospitals to the 100 mg/day arm. The participants’ characteristics differed between the two arms, especially with regard to sex and PHQ-9 scores at weeks 0 and 1 (Table 1). Of those randomised at week 1, 1927 (95.8%) and 1910 (95.0%) were successfully followed up at week 9 and week 25, respectively.
Of these, 1647 had not remitted by week 3 and were individually randomised to continue sertraline (n = 551), combine sertraline with mirtazapine (n = 538) or switch to mirtazapine (n = 558). Participants’ characteristics proved similar across the three arms (Table 2). Of those randomised at week 3, 1613 (98.0%) and 1597 (97.0%) were successfully followed up at week 9 and week 25, respectively.
With regard to step 1 treatments, in the 50 mg/day arm, 91.7% (889/970) had been prescribed 50 mg/day, 0.1% (1/970) 37.5 mg/day, 1.3% (13/970) 25 mg/day and 0.1% (1/970) 75 mg/day by week 3; in the 100 mg/day arm, 82.0% (854/1041) had reached 100 mg/day, 5.3% (55/1041) 75 mg/day, 6.7% (70/1041) 50 mg/day and 0.9% (9/1041) 25 mg/day. In the 50 mg/day arm 6.8% (66/970) had stopped treatment as had 5.1% (53/1041) in the 100 mg/day arm. For step 2 randomised allocations at week 3, 99.5% (548/551), 96.1% (516/537) and 96.8% (540/558) of the randomised participants started their allocated treatment for the continuation, combination and switch arms, respectively; of the last group, 72.9% (407/558), 83.9% (468/558) and 87.8% (490/558) were successfully tapered off sertraline by weeks 5, 6 and 7, respectively.
In the following analyses, we ascertained the assumptions of normality and the homoscedasticity of the error in both the steps 1 and 2 mixed-effects models. We also ascertained the proportional hazards assumption by visual inspection of the log cumulative hazard curves and by testing the treatment*time interaction in Cox regression models.
Step 1: 50 mg/day vs 100 mg/day as initial target dose of sertraline
According to the adjusted and weighted mixed-model repeated-measures analyses, there was no statistically significant difference (0.25 point, 95% CI − 0.58 to 1.07, P = 0.55) between the 100 mg/day arm and the 50 mg/day arm in the PHQ-9 score at week 9 (Table 3). There were no statistically significant differences in the secondary efficacy outcomes at week 9. Neither were there any statistically significant differences in the global burden of side effects between the 50 mg/day and 100 mg/day arms.
At week 25, the 100 mg/day arm scored lower (− 2.28, 95% CI − 3.91 to − 0.66, P = 0.0059) on self-rated BDI-II than the 50 mg/day arm (Table 4). The two arms did not differ, however, in the other efficacy outcomes of PHQ-9, the proportion of remission or in the global burden of side effects.
The incidence of suicidality, manic switches or serious adverse events was very small, and there were no statistically significant differences between the two arms through 25 weeks (Additional file 2: Tables S1, S2).
Results were similar regardless of baseline depression severity or initial response (Additional file 2: Table S3). All sensitivity analyses differed little from the primary findings (Additional file 2: Table S4).
Step 2: Continue sertraline vs combine with mirtazapine vs switch to mirtazapine at week 3
The adjusted mixed-model repeated-measures analysis revealed that the mirtazapine combination arm and the mirtazapine switch arm were both superior to the sertraline continuation arm; the combination arm by − 0.99 points (95% CI − 1.55 to − 0.43, P = 0.0012) and the switch arm by − 1.01 points (95% CI − 1.56 to − 0.46, P = 0.0012) on PHQ-9 after Hochberg adjustment for multiple comparison. Results were very similar between the combination and switch arms (0.02, 95% CI − 0.54 to 0.57, P = 0.95); see Table 5. Self-rated BDI-II scores and the dichotomised response and remission rates were consistent with the primary analysis. RDs for response and remission, respectively, were 9.1% (95% CI 2.8–15.4%) and 12.4% (6.1–19.0%) for the combination strategy, and 8.2% (1.7–14.3%) and 8.4% (2.5–14.8%) for the switching strategy, over the continuation strategy. They corresponded with numbers needed to treat (NNTs) of 11.0 (6.5–35.7) and 8.1 (5.3–16.4) for the combination and 12.2 (7.0–59) and 11.9 (6.8–40) for the switching strategies.
The proportion of continuation of the allocated treatment was significantly lower in the combination arm (RD 8.5%, 95% CI 2.9–14.9%) and the switching arm (6.4%, 95% CI 1.3–12.5%) than the continuation arm. The overall burden of side effects as measured with FIBSER did not, however, differ among the three strategies (Table 5). Neither were there any material differences regarding incidence of suicidality, manic switches or serious adverse events (Additional file 2: Table S5).
At week 25, the treatment arms did not differ in any of the efficacy (PHQ-9, BDI-II, remission), acceptability (time to discontinuation of the allocated treatment or of any treatment) or harm outcomes (Table 6, Additional file 2: Table S6).
There was no evidence of effect modification in any of the three pre-planned subgroup analyses (Additional file 2: Table S7).
Discussion
The SUN☺D trial involved two randomisations to examine the first- and second-line antidepressant pharmacotherapies for the acute phase treatment of hitherto untreated major depressive disorder.
The step 1 randomisation examined the impact of titrating to the minimum or maximum of the licenced dose of sertraline by week 3. In patients starting treatment for major depressive disorder, there were no important differences in effectiveness or adverse effects between these two starting sertraline doses. The results of the primary outcome at week 9 (0.25 point difference in adjusted PHQ-9 score, 95% CI − 0.58 to 1.07, P = 0.55) excluded an important difference in favour of the higher dose. Neither were there any important differences between the minimum vs the maximum target doses in side effects, burden or treatment acceptability up to either week 9 or week 25.
When patients do not remit after 3 weeks of sertraline treatment, however, adding mirtazapine or switching sertraline to mirtazapine resulted in approximately a one-point benefit in PHQ-9 at week 9, a standardised mean difference of around 0.16. This difference corresponded to RDs in response of 9.1% (95% CI 2.8–15.4%) for the combination and 8.2% (1.7–14.3%) for the switching strategy and, in remission, of 12.4% (6.1–19.0%) for the combination and 8.4% (2.5–14.8%) for the switching strategy. These values are slightly below or above the usually accepted clinically significant threshold of 10% RD, but they may be important to patients when one considers that they are differences between alternative active treatments and that RDs for antidepressants over placebo are approximately 20% only [29, 43]. In addition, these results were consistent with patients’ self-reports by BDI-II. The benefits of combination and switching did not, however, persist after continuation treatment at the treating physician’s discretion at week 25.
Combination or switching strategies resulted in 6–8% fewer patients continuing the allocated treatments up to week 9 than continuing on sertraline. However, given the greater improvement in depressive symptoms with the strategies that included mirtazapine; similar global ratings of side effects among the treatment arms at weeks 9 and 25; and similar time to discontinuation of allocated treatment or any treatment when considered up to 25 weeks; this finding does not raise concern regarding the use of combination and switching strategies.
The most recent comprehensive network meta-analysis of antidepressants found that mirtazapine may have a greater response rate than sertraline (OR = 1.15, 95% CI 0.93–1.43) when used as first-line monotherapy [29]. The current study findings are compatible with this network meta-analysis, and this superior efficacy of mirtazapine may be responsible for some of the benefits of switching or combination strategies over sertraline continuation found in the current study.
Some guidelines recommend dose increase within the approved range after non-response to a lower dose ([4], p. 356, [10], p.53). Two previous studies have tested this strategy for sertraline and found that there was no additional benefit: one study compared 50 mg vs 150 mg of sertraline for 5 weeks after an initial 3 weeks on 50 mg and reported no difference [44]. Another study compared 100 mg vs 200 mg of sertraline for 5 weeks after an initial 6 weeks in which patients received up to 100 mg and reported that the increase to 200 mg resulted in a lower response rate [24]. Although the current study did not specifically address this strategy, our results with regard to the initial target dose are consistent with these previous studies.
There are limitations of our study. First, the step 1 cluster randomisation by clinic was performed at the start of the study. As a result, there was no concealment at the level of randomisation of individual patients, which explains the differences in characteristics of patients enrolled in the two arms. Clinicians at study sites allocated to the lower dose were less inclined to enrol severe patients or male patients than were clinicians at sites allocated to the higher dose. We dealt with this prognostic imbalance by adjusting for key variables in the mixed-model repeated-measures analyses and confirmed robustness of the primary findings against model assumptions through sensitivity analyses. Unknown confounders may still, however, have biased our findings, limiting strength of inference in the step 1 findings.
Secondly, the open-label design may have created some undetected performance bias that may threaten the internal validity of both the step 1 and step 2 comparisons, such as differential administration of co-prescriptions or psychological support. Such bias is, however, likely to be minimal, because the protocol did not allow any depression-specific psychotherapies such as CBT or IPT or concomitant administration of antipsychotics or mood stabilisers up to week 9. Co-prescription of benzodiazepines was permitted, but they were prescribed very similarly in all arms (60–70% of the participants received either anxiolytics or hypnotics). Further, a differential placebo effect associated with adding or switching to mirtazapine vs continuing with sertraline could explain some of the apparent effect of the switching/adding arms in step 2. Blinded assessment of outcomes somewhat ameliorates this concern. Moreover, the open-label design was consonant with the pragmatic nature of the study, in which we made comparisons including such possible practice variability.
Thirdly, the tapering speed of sertraline in the switching arm was relatively slow and allowed some patients to take the combination of sertraline and mirtazapine for several weeks. The efficacy of the switching strategy might therefore be contaminated by that of the combination treatment. However, gradual tapering was appropriate for this pragmatic trial and allowed us to address the impact of interventions as clinicians would implement them in the real world.
Fourthly, one might question the generalisability of the current study findings beyond the specific drugs and dosages employed. As the initial target dose of the antidepressant, we compared the minimum and the maximum of the licenced dosage in Japan, 50 mg/day and 100 mg/day of sertraline. The maximum dosage for sertraline in the USA and in some other countries is 200 mg/day. The study specified dosing for mirtazapine between 7.5 and 45 mg, which is also lower than the dosing sometimes used in other countries. These differences may reflect differences in body weights and other ethnically specific variations in the genetic as well as non-genetic mechanisms affecting the pharmacokinetics and dynamics of psychotropic drugs [45]. Up to now, however, inquiry has failed to identify ethnicity as a convincing modifier of antidepressant effect [46]. Had we compared 50 mg/day vs 200 mg/day, or had we required higher dose ranges for mirtazapine, the results might have differed. However, consistent with our step 1 findings, RCTs using other drugs have in most cases also suggested no additional benefit with higher doses [12, 47, 48]. It is also uncertain whether results would be similar had we chosen to begin with an antidepressant other than sertraline or chosen to combine or switch with a drug other than mirtazapine. Our step 2 findings for adding mirtazapine to sertraline are consistent with the previous systematic review examining various combination strategies [22]. One might consider our step 2 findings for switching to mirtazapine at odds with the recent meta-analysis on this topic that found no additional benefit in switching to various antidepressants [20]. However, the only study in this review specific to mirtazapine reported that using mirtazapine was more effective than continuing the prior medication [49].
A further limitation is that, although it provides evidence that changing therapy after failure to remit at an early point (in our study 3 weeks) may be preferable to waiting a longer period (e.g. the oft-recommended 6 to 8 weeks), the design did not specifically address the optimal timing of the decision to switch or combine. One RCT compared switching to duloxetine immediately after non-response to 4weeks of escitalopram against continuing 4 more weeks on escitalopram and then switching if non-responsive: the two arms did not differ in their primary outcome of time to response, while there was significant increase in a secondary outcome of remission in the early switch arm than in the later switch arm [50]. Another recent RCT examined the value of switching escitalopram to venlafaxine if patients have shown minimal improvement after 2 weeks; remission rates were 8% higher in the switched than in the continuation arm, but this difference did not reach statistical significance, possibly due to a small sample size (n = 192, p = 0.21) [51]. Thus, the optimal time to combine or switch remains uncertain.
Strengths of this pragmatic trial primarily relate to design features that enhance the real-world application of the results. We employed a large number of study sites using broad eligibility criteria, and thus enrolled sufficient patients to achieve high power to establish or refute differences between groups. Because Japan does not have a primary care system, patients with new onset depressive episodes usually consult office practice psychiatrists directly. Thus, in many other countries, primary care doctors would see and begin treatment of most participants entered in SUN☺D. In comparison to many multi-centre trials, we were able to enrol a large proportion (2011 of 7895) of potentially eligible patients. Enrolment of patients not yet treated for their current episode is another strength, eliminating a potential source of variability. For step 1 randomisation, we excluded patients who did not tolerate a low dose of sertraline, faithfully addressing the clinical question in which patients and clinicians are interested: In patients who tolerate an initial low dose of sertraline, should clinicians titrate to the maximum dose? For step 2 randomisation, we selected alternative interventions that are recommended in practice guidelines [4, 10] and are widely used [52].
We limited risk of bias through centralised, blinded, telephone assessments that allowed us to achieve more than 95% follow-up through 25 weeks, unique among large trials of psychiatric interventions. The centralised training of the raters enhanced the reliability and validity of the depression assessments [38]. A pre-published protocol [32], statistical analysis plan [33] and blinded interpretation of the results to minimise the researchers’ interpretation bias further enhance the trustworthiness of this RCT.
Conclusions
The SUN☺D trial suggests that, for the initial antidepressant therapy for a new major depressive episode, titrating sertraline to the maximum over the minimum within the licenced dosage confers no additional benefit but increases cost. The confidence in this conclusion is limited by the failure of the cluster randomisation to achieve prognostic balance, and the possible residual confounding after our adjusted analysis. When patients fail to remit on this initial treatment, early combination or switching using mirtazapine resulted in a small benefit in reducing depression without an increase in adverse effects. Inferences apply to the strategies as implemented, which reflect clinical practice, including gradual tapering of sertraline in the switching arm. Factors bearing on the decision to combine or switch are likely to include costs (combination will be more costly), the current burden of side effects of the first-line treatment, the expected burden of combination or switching and the patient’s readiness to change antidepressants.
The many drugs available allow clinicians considerable options in the selection of an initial antidepressant and of second-line antidepressants to switch to or combine with. Clinicians and patients may consider starting the treatment with a low dose of agents for which evidence suggests satisfactory efficacy and acceptability in the current trial and in the comprehensive network meta-analysis [29]. Should they choose to combine antidepressants as the second-line strategy, the relative merits of potential combinations remain largely untested in RCTs, and it is implausible that RCTs will ultimately evaluate all such alternatives. A previous review suggested that a combination of a reuptake inhibitor antidepressant and an antagonist of presynaptic alpha-2 autoreceptor is more effective than other combinations [22], and the current findings were consistent with this suggestion. Those who place a high value in treatments that have been tested in large trials that both minimise risk of bias and mimic real-world conditions may prefer the specific strategies used in this trial.
References
Abbing-Karahagopian V, Huerta C, Souverein PC, de Abajo F, Leufkens HG, Slattery J, Alvarez Y, Miret M, Gil M, Oliva B, et al. Antidepressant prescribing in five European countries: application of common definitions to assess the prevalence, clinical observations, and methodological implications. Eur J Clin Pharmacol. 2014;70(7):849–57.
Kantor ED, Rehm CD, Haas JS, Chan AT, Giovannucci EL. Trends in prescription drug use among adults in the United States from 1999-2012. JAMA. 2015;314(17):1818–31.
Kjosavik SR, Hunskaar S, Aarsland D, Ruths S. Initial prescription of antipsychotics and antidepressants in general practice and specialist care in Norway. Acta Psychiatr Scand. 2011;123(6):459–65.
NICE. Depression: the treatment and management of depression in adults (partial update of NICE clinical guideline 23). London: National Institute for Clinical Excellence; 2009.
Cipriani A, Furukawa TA, Salanti G, Geddes JR, Higgins JP, Churchill R, Watanabe N, Nakagawa A, Omori IM, McGuire H, et al. Comparative efficacy and acceptability of 12 new-generation antidepressants: a multiple-treatments meta-analysis. Lancet. 2009;373:746–58.
Medicines use and spending in the U.S. — a review of 2016 and outlook for 2021. https://www.iqvia.com/institute/reports/medicines-use-and-spending-in-the-us-a-review-of-2016. Accessed 20 June 2018.
Furukawa TA, Onishi Y, Hinotsu S, Tajika A, Takeshima N, Shinohara K, Ogawa Y, Hayasaka Y, Kawakami K. Prescription patterns following first-line new generation antidepressants for depression in Japan: a naturalistic cohort study based on a large claims database. J Affect Disord. 2013;150:916–22.
S3-Guideline/National Disease Management Guideline Unipolar Depression. Short version. https://www.leitlinien.de/nvl/depression. Accessed 20 June 2018.
Bauer M, Pfennig A, Severus E, Whybrow PC, Angst J, Moller HJ. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of unipolar depressive disorders, part 1: update 2013 on the acute and continuation treatment of unipolar depressive disorders. World J Biol Psychiatry. 2013;14(5):334–85.
American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder (third edition). Am J Psychiatry. 2010;167(Suppl):1–152.
Bollini P, Pampallona S, Tibaldi G, Kupelnick B, Munizza C. Effectiveness of antidepressants. Meta-analysis of dose-effect relationships in randomised clinical trials. Br J Psychiatry. 1999;174:297–303.
Purgato M, Gastaldon C, Papola D, Magni LR, Rossi G, Barbui C. Drug dose as mediator of treatment effect in antidepressant drug trials: the case of fluoxetine. Acta Psychiatr Scand. 2015;131(6):408–16.
Hieronymus F, Nilsson S, Eriksson E. A mega-analysis of fixed-dose trials reveals dose-dependency and a rapid onset of action for the antidepressant effect of three selective serotonin reuptake inhibitors. Transl Psychiatry. 2016;6(6):e834.
Corruble E, Guelfi JD. Does increasing dose improve efficacy in patients with poor antidepressant response: a review. Acta Psychiatr Scand. 2000;101(5):343–8.
Adli M, Baethge C, Heinz A, Langlitz N, Bauer M. Is dose escalation of antidepressants a rational strategy after a medium-dose treatment has failed? A systematic review. Eur Arch Psychiatry Clin Neurosci. 2005;255(6):387–400.
Ruhe HG, Huyser J, Swinkels JA, Schene AH. Dose escalation for insufficient response to standard-dose selective serotonin reuptake inhibitors in major depressive disorder: systematic review. Br J Psychiatry. 2006;189:309–16.
Dold M, Bartova L, Rupprecht R, Kasper S. Dose escalation of antidepressants in unipolar depression: a meta-analysis of double-blind, randomized controlled trials. Psychother Psychosom. 2017;86(5):283–91.
Ruhe HG, Huyser J, Swinkels JA, Schene AH. Switching antidepressants after a first selective serotonin reuptake inhibitor in major depressive disorder: a systematic review. J Clin Psychiatry. 2006;67(12):1836–55.
Papakostas GI, Fava M, Thase ME. Treatment of SSRI-resistant depression: a meta-analysis comparing within- versus across-class switches. Biol Psychiatry. 2008;63(7):699–704.
Bschor T, Kern H, Henssler J, Baethge C. Switching the antidepressant after nonresponse in adults with major depression: a systematic literature search and meta-analysis. J Clin Psychiatry. 2018;79(1). https://doi.org/10.4088/JCP.16r10749.
Zhou X, Ravindran AV, Qin B, Del Giovane C, Li Q, Bauer M, Liu Y, Fang Y, da Silva T, Zhang Y, et al. Comparative efficacy, acceptability, and tolerability of augmentation agents in treatment-resistant depression: systematic review and network meta-analysis. J Clin Psychiatry. 2015;76(4):e487–98.
Henssler J, Bschor T, Baethge C. Combining antidepressants in acute treatment of depression: a meta-analysis of 38 studies including 4511 patients. Can J Psychiatr. 2016;61(1):29–43.
Ferreri M, Lavergne F, Berlin I, Payan C, Puech AJ. Benefits from mianserin augmentation of fluoxetine in patients with major depression non-responders to fluoxetine alone. Acta Psychiatr Scand. 2001;103(1):66–72.
Licht RW, Qvitzau S. Treatment strategies in patients with major depression not responding to first-line sertraline treatment. A randomised study of extended duration of treatment, dose increase or mianserin augmentation. Psychopharmacology. 2002;161(2):143–51.
Rush AJ, Trivedi MH, Wisniewski SR, Stewart JW, Nierenberg AA, Thase ME, Ritz L, Biggs MM, Warden D, Luther JF, et al. Bupropion-SR, sertraline, or venlafaxine-XR after failure of SSRIs for depression. N Engl J Med. 2006;354(12):1231–42.
Trivedi MH, Fava M, Wisniewski SR, Thase ME, Quitkin F, Warden D, Ritz L, Nierenberg AA, Lebowitz BD, Biggs MM, et al. Medication augmentation after the failure of SSRIs for depression. N Engl J Med. 2006;354(12):1243–52.
Mohamed S, Johnson GR, Chen P, Hicks PB, Davis LL, Yoon J, Gleason TC, Vertrees JE, Weingart K, Tal I, et al. Effect of antidepressant switching vs augmentation on remission among patients with major depressive disorder unresponsive to antidepressant treatment: the VAST-D randomized clinical trial. JAMA. 2017;318(2):132–45.
Qaseem A, Snow V, Denberg TD, Forciea MA, Owens DK. Using second-generation antidepressants to treat depressive disorders: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2008;149(10):725–33.
Cipriani A, Furukawa TA, Salanti G, Chaimani A, Atkinson LZ, Ogawa Y, Leucht S, Ruhe HG, Turner EH, Higgins JPT, et al. Comparative efficacy and acceptability of 21 antidepressant drugs for the acute treatment of adults with major depressive disorder: a systematic review and network meta-analysis. Lancet. 2018;391(10128):1357–66.
American Psychiatric Association. Diagnostic and statistical manual of mental disorders, 4th ed. Washington, DC: American Psychiatric Association; 1994.
Spitzer RL, Williams JB, Kroenke K, Linzer M, deGruy FV 3rd, Hahn SR, Brody D, Johnson JG. Utility of a new procedure for diagnosing mental disorders in primary care. The PRIME-MD 1000 study. JAMA. 1994;272(22):1749–56.
Furukawa TA, Akechi T, Shimodera S, Yamada M, Miki K, Watanabe N, Inagaki M, Yonemoto N. Strategic use of new generation antidepressants for depression: SUN☺D study protocol. Trials. 2011;12(1):116.
Yonemoto N, Tanaka S, Furukawa TA, Kato T, Mantani A, Ogawa Y, Tajika A, Takeshima N, Hayasaka Y, Shinohara A, et al. Strategic use of new generation antidepressants for depression: SUN☺D protocol update and statistical analysis plan. Trials. 2015;16:459.
Kroenke K, Spitzer RL, Williams JB. The PHQ-9: validity of a brief depression severity measure. J Gen Intern Med. 2001;16(9):606–13.
Spitzer RL, Kroenke K, Williams JB. Validation and utility of a self-report version of PRIME-MD: the PHQ primary care study. Primary Care Evaluation of Mental Disorders. Patient Health Questionnaire. JAMA. 1999;282(18):1737–44.
Beck AT, Steer RA, Brown GK. BDI-II: BeckDepression Inventory, second edition, manual. San Antonio: The Psychological Corporation; 1996.
Posner K, Oquendo MA, Gould M, Stanley B, Davies M. Columbia Classification Algorithm of Suicide Assessment (C-CASA): classification of suicidal events in the FDA's pediatric suicidal risk analysis of antidepressants. Am J Psychiatry. 2007;164(7):1035–43.
Pinto-Meza A, Serrano-Blanco A, Penarrubia MT, Blanco E, Haro JM. Assessing depression in primary care with the PHQ-9: can it be carried out over the telephone? J Gen Intern Med. 2005;20(8):738–42.
Shimodera S, Kato T, Sato H, Miki K, Shinagawa Y, Kondo M, Fujita H, Morokuma I, Ikeda Y, Akechi T, et al. The first 100 patients in the SUN☺D trial (strategic use of new generation antidepressants for depression): examination of feasibility and adherence during the pilot phase. Trials. 2012;13(1):80.
Wells KB, Sherbourne C, Schoenbaum M, Duan N, Meredith L, Unutzer J, Miranda J, Carney MF, Rubenstein LV. Impact of disseminating quality improvement programs for depression in managed primary care: a randomized controlled trial. JAMA. 2000;283(2):212–20.
Cohen J. Statistical power analysis in the behavioral sciences. Hillsdale: Erlbaum; 1988.
Jarvinen TL, Sihvonen R, Bhandari M, Sprague S, Malmivaara A, Paavola M, Schunemann HJ, Guyatt GH. Blinded interpretation of study results can feasibly and effectively diminish interpretation bias. J Clin Epidemiol. 2014;67(7):769–72.
Turner EH, Matthews AM, Linardatos E, Tell RA, Rosenthal R. Selective publication of antidepressant trials and its influence on apparent efficacy. N Engl J Med. 2008;358:252–60.
Schweizer E, Rynn M, Mandos LA, Demartinis N, Garcia-Espana F, Rickels K. The antidepressant effect of sertraline is not enhanced by dose titration: results from an outpatient clinical trial. Int Clin Psychopharmacol. 2001;16(3):137–43.
Chaudhry I, Neelam K, Duddu V, Husain N. Ethnicity and psychopharmacology. J Psychopharmacol. 2008;22(6):673–80.
Kessler RC, van Loo HM, Wardenaar KJ, Bossarte RM, Brenner LA, Ebert DD, de Jonge P, Nierenberg AA, Rosellini AJ, Sampson NA, et al. Using patient self-reports to study heterogeneity of treatment effects in major depressive disorder. Epidemiol Psychiatr Sci. 2017;26(1):22–36.
Furukawa TA, McGuire H, Barbui C. Meta-analysis of effects and side effects of low dosage tricyclic antidepressants in depression: systematic review. BMJ. 2002;325(7371):991–5.
Meeker AS, Herink MC, Haxby DG, Hartung DM. The safety and efficacy of vortioxetine for acute treatment of major depressive disorder: a systematic review and meta-analysis. Syst Rev. 2015;4:21.
Zhu H, Jinlong Y, HOngbo Z. A study of switchig to mirtazapine for treatment-resistant depression. Shanghai Arch Psychiatry. 2003;15:355–7.
Romera I, Perez V, Menchon JM, Schacht A, Papen R, Neuhauser D, Abbar M, Svanborg P, Gilaberte I. Early switch strategy in patients with major depressive disorder: a double-blind, randomized study. J Clin Psychopharmacol. 2012;32(4):479–86.
Tadic A, Wachtlin D, Berger M, Braus DF, van Calker D, Dahmen N, Dreimuller N, Engel A, Gorbulev S, Helmreich I, et al. Randomized controlled study of early medication change for non-improvers to antidepressant therapy in major depression—the EMC trial. Eur Neuropsychopharmacol. 2016;26(4):705–16.
Kornbluh R, Papakostas GI, Petersen T, Neault NB, Nierenberg AA, Rosenbaum JF, Fava M. A survey of prescribing preferences in the treatment of refractory depression: recent trends. Psychopharmacol Bull. 2001;35(3):150–6.
Acknowledgements
Group information: A complete list of participating centres and investigators in the Strategic Use of New generation antidepressants for Depression (SUN☺D trial) is provided in Additional file 3.
Funding
The study was funded by the Ministry of Health, Labor and Welfare, Japan (H-22-Seishin-Ippan-008) from April 2010 through March 2012 to TAF (http://www.mhlw.go.jp/english/) and thereafter by the Japan Foundation for Neuroscience and Mental Health (JFNMH) to TAF (http://www.jfnm.or.jp/). The JFNMH received donations from Asahi Kasei, Eli Lilly, GlaxoSmithKline (GSK), Janssen, Merck Sharp & Dohme (MSD), Meiji, Mochida, Otsuka, Pfizer, Shionogi, Taisho and Mitsubishi-Tanabe. The funders of the study had no role in study design, data collection, data analysis, data interpretation or in writing of the report.
Availability of data and materials
Sharing of identifiable, individual-level data is prohibited by Japanese law. Fully anonymised, unidentifiable data with age, sex and site deleted are available from the University Hospital Medical Information Network (UMIN-ICDR http://www.umin.ac.jp/icdr/index.html), an academic network information centre for biomedical sciences funded by the Japanese Ministry of Education, Culture, Science, Sports and Technology (MEXT) at https://upload.umin.ac.jp/cgi-open-bin/icdr_e/ctr_view.cgi?recptno=R000034028.
Author information
Authors and Affiliations
Consortia
Contributions
TAF is the principal investigator and had overall responsibility for the management of the study. TAF, NY, ST and QZ had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analyses. TAF conceived the study. YOg, NTa, YH, AT and KSh performed data curation; NY, ST, QZ and TAF performed the formal analysis. TAF and MY acquired the funding. TK, TAF, AM, KK, HK, SH, HS, KSu, BC, KI, YI, YS, MK, YOk, HF, MS, SY, NTs, TI, NF, TA, MY, SS, NW, MI, KM, YOg, NTa, YH, AT, KSh and GHG performed the investigation. TAF, TA, MY, SS, NW, MI, KM and NY developed the methodology.MK, YOk, HF, MS, SY, NTs, TI, NF, YOg, NTa, YH, AT and KSh administered the project. TAF and GHG supervised the project. TAF and GHG wrote the original draft. TK, AM, KK, HK, SH, HS, KSu, BC, KI, YI, YS, MK, YOk, HF, MS, SY, NTs, TI, NF, TA, MY, SS, NW, MI, KM, YOg, NTa, YH, AT, KSh, NY, ST and QZ reviewed and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study protocol was approved by the ethics committees/institutional review boards of Kyoto University Graduate School of Medicine (C0446), Nagoya City University Hospital (45–10-0004), Kochi Medical School (22–96), Kumamoto University (No. 1341), Yatsushiro Kosei Hospital (dated October 28, 2011), Yuge Hospital (No. 86), Kurume University Medical School (No. 11151), St. Lucia Hospital (dated January 11, 2012), Hiroshima University Hospital (Rin 297), University of Tokyo Hospital (P2011062-11X), Toho University School of Medicine (No. 23053), Hokkaido University Hospital (JI011–0292) and Sapporo Hanazono Hospital (dated June 4, 2014). All participants provided written informed consent.
Competing interests
TK has received lecture fees from Eli Lilly and Mitsubishi-Tanabe and has contracted research with GSK, MSD and Mitsubishi-Tanabe. He has received royalties from Kyowa Yakuhin. TAF has received lecture fees from Eli Lilly, Janssen, Meiji, MSD, Otsuka, Pfizer and Mitsubishi-Tanabe and consultancy fees from Takeda Science Foundation. He has received research support from Mochida and Mitsubishi-Tanabe. AM has received lecture fees from Mochida, Eli Lilly and Meiji. SH has received lecture fees from MSD, Pfizer, Meiji, Tanabe-Mitsubishi and Otsuka. HS has received lecture fees from Daiichi-Sankyo and Mochida. KSu has received lecture and/or consultant fees from Eli Lilly, Mochida, Eisai, Meiji and Daiichi-Sankyo. BC has received lecture fees from Eli Lilly, Meiji and Mitsubishi-Tanabe. KI has received lecture fees from Dainippon-Sumitomo, Eli Lilly, Otsuka, Takeda, Kyowa Yakuhin, Mitsubishi-Tanabe, Meiji and Shionogi. YI has received lecture fees from Meiji, Eli Lilly, Janssen and Otsuka. YS has received lecture fees from Janssen, Kyowa Yakuhin, Meiji, MSD, Otsuka and Mitsubishi-Tanabe. MK has received lecture fees from Yoshitomi and a research grant from Novartis. YOk has received lecture fees from Otsuka, Dainippon-Sumitomo, Astellas, Pfizer, Eli Lilly, Janssen, Meiji, Mochida, Yoshitomi, Eisai and GSK. HF has received lecture fees from Meiji, Mochida, MSD and Otsuka. SY has received lecture fees from Daiichi-Sankyo and Otsuka. NTs has received lecture fees from Astellas, Shionogi, Novartis, Fujifilm RI Pharma, Meiji, Mochida, Janssen, Eli Lilly and Dainippon-Sumitomo. TI has received lecture fees from GSK, Mochida, Asahi Kasei, Shionogi, Otsuka, Dainippon-Sumitomo, Eli Lilly, Eisai, Mitsubishi-Tanabe, Pfizer, AbbVie, MSD, Yoshitomi, Takeda and Meiji. He has received grants from Shionogi, Astellas, Otsuka, Dainippon-Sumitomo, Eli Lilly, Eisai, Mitsubishi-Tanabe, Pfizer, AbbVie, MSD, Yoshitomi, Takeda and Meiji. He is on advisor boards for GSK, Mochida, Otsuka, Eli Lilly, Mitsubishi-Tanabe, Pfizer and Takeda. NF has received lecture fees from Pfizer, Eisai, Mochida, Ono, Shionogi, MSD and Otsuka. TA has received lecture fees and/or research funds from Daiichi-Sankyo, Eisai, Hisamitsu, Eli Lilly, MSD, Meiji, Mochida, Otsuka, Pfizer, Novartis and Terumo. MY has received lecture fees from Meiji, MSD and Asahi Kasei and has contracted research with Nippon Chemiphar. SS has received lecture fees from Otsuka, MSD, Meiji, Eli Lilly, Mochida and Shionogi. MI has received a grant from Novartis Pharma. He has received lecture fees from Pfizer, Mochida, Shionogi, Sumitomo Dainippon Pharma, Daiichi-Sankyo, Meiji and Takeda. KM has received lecture fees from Eisai, GSK, Meiji, MSD, Otsuka, Pfizer, Eli Lilly, Mochida, Yoshitomi, Dainippon-Sumitomo, Takeda and Shionogi. YOg has received a lecture fee from Eli Lilly. NTa has received lecture fees from Otsuka and Meiji. YH has received a lecture fee from Yoshitomi. AT has received lecture fees from Eli Lilly and Mitsubishi-Tanabe. ST has received lecture fees from AstraZeneca, Taiho and Ono. He has received consultation fees from DeNA Life Science and CanBus. He has received outsourcing fees from Satt and Asahi Kasei Pharma. His wife has been engaged in a research project of Bayer. All these fees have been outside the submitted work. KK, HK, MS, NW, KSh, NY, QZ and GHG declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Blinded data analyses statement of interpretation. (PDF 336 kb)
Additional file 2:
Figure S1. Schedule of the assessments. Table S1. Incidence of suicidality, manic switches or any serious adverse events up to week 9 for step 1 randomisation. Table S2. Incidence of suicidality, manic switches or any serious adverse events up to week 25 for step 1 randomisation. Table S3. Two pre-specified subgroup analyses for step 1 randomisation. Table S4. Four pre-specified sensitivity analyses for step 1 randomisation. Table S5. Incidence of suicidality, manic switches or any serious adverse events up to week 9 for step 2 randomisation. Table S6. Incidence of suicidality, manic switches or any serious adverse events up to week 25 for step 2 randomisation. Table S7. Three pre-specified subgroup analyses for step 2 randomisation. (DOCX 32 kb)
Additional file 3:
SUN☺D Investigators and committee members. (DOCX 17 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Kato, T., Furukawa, T.A., Mantani, A. et al. Optimising first- and second-line treatment strategies for untreated major depressive disorder — the SUN☺D study: a pragmatic, multi-centre, assessor-blinded randomised controlled trial. BMC Med 16, 103 (2018). https://doi.org/10.1186/s12916-018-1096-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12916-018-1096-5