
Abstract
The majority of breast cancers present with estrogen receptor (ER)-positive and human epidermal growth factor receptor (HER2)-negative features and might benefit from endocrine therapy. Although endocrine therapy has notably evolved during the last decades, the invariable appearance of endocrine resistance, either primary or secondary, remains an important issue in this type of tumor. The improvement of our understanding of the cancer genome has identified some promising targets that might be responsible or linked to endocrine resistance, including alterations affecting main signaling pathways like PI3K/Akt/mTOR and CCND1/CDK4-6 as well as the identification of new ESR1 somatic mutations, leading to an array of new targeted therapies that might circumvent or prevent endocrine resistance. In this review, we have summarized the main targeted therapies that are currently being tested in ER+ breast cancer, the rationale behind them, and the new agents and combinational treatments to come.
Similar content being viewed by others


Introduction
Endocrine therapy represents a major treatment in all settings of the disease for breast cancers expressing estrogen receptor (ER)-α, which accounts for around 70 % of tumors [1, 2]. During the last two decades, third-generation aromatase inhibitors (AIs), such as anastrozole, letrozole, and exemestane, have become the standard endocrine treatment in postmenopausal women both in advanced and early disease, contributing to an improvement in median survival from 28 to 45 months between the late 1980s and late 1990s [3]. Despite the efficacy of these compounds, response rates for first-line metastatic patients have been described as up to 40 %, with all initial responders eventually developing resistance over time [4]. After progression on an AI, it might still be indicated to pursue with another endocrine agent like fulvestrant, unless there is significant visceral burden and rapid tempo of disease [5]. Other possibilities include treatment with a selective estrogen receptor modulator like tamoxifen or even hormone additive therapies, such as the use of progestins (medroxyprogesterone acetate) [6] and estrogen (ethinyl estradiol) [7, 8].
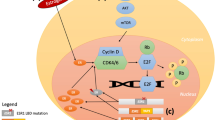
Due to its clinical significance, extensive research has been made in order to determine the potential mechanisms of endocrine resistance. Initial studies had identified the loss of ER expression as responsible for primary resistance, as well as polymorphisms of CYP2D6 and CYP19A1 as being responsible for the lack of benefit from tamoxifen and aromatase inhibitors, respectively [9–12], although further studies have not been able to confirm these findings [12, 13]. For both primary as well as secondary resistance, one of the main responsible mechanisms is thought to be the interaction between ER and growth factor receptor signaling via either the phosphatidylinositol-3-kinase (PI3K)/protein kinase B (Akt)/mammalian target of rapamycin (mTOR) pathway, or the mitogen-activated protein kinase (MAPK) pathway which promotes ER phosphorylation (therefore activation) via a non-classical genomic pathway [14] (Fig. 1). More recently, high-throughput technologies studies in ER-positive metastatic breast cancer samples have identified a large number of molecular aberrations in potential driver genes such as PIK3CA mutations, FGFR1 and CCND1 amplifications (11 %), and ESR1 mutations (4 %) [12, 15–19], some of them previously linked to endocrine resistance. This, in addition to the recent interest in the cell cycle regulation pathway cyclin D1/cyclin-dependent kinases [20], has resulted in the appearance of several therapies targeting these pathways in order to circumvent or delay the development of endocrine resistance.

Cross-talk between ER signaling and growth factor signaling pathways described as linked to resistance to endocrine therapy. Classical ER signaling needs to bind to estrogens and HSP90 chaperone protein before binding to transcription start site of target genes such as cyclin D. This transcription activity is partly mediated by histone deacetylation by HDAC6. CyclinD activates E2F transcription via Rb phosphorylation and promotes G1-S transition into the cell cycle for cell proliferation. Suppression of classical ER signaling by endocrine therapy might promote activation of the tyrosine kinase receptor signaling pathways PI3K/Akt/mTOR and RAS-RAF-MAPK via its effectors S6K1 and 4EBP1 to promote ligand-independent activation of ER. Numbers shown in this figure correspond to the function sites of the target agents described in the manuscript. ①mTOR inhibitor: inhibition of mTORC1 down-regulated S6K1 and 4EBP1. In mTOR inhibitor resistance, feedback signaling seems to be activated indicated by white arrow. ②, ③PI3K inhibitors and Akt inhibitors. ④CDK4/6 inhibitors. ⑤FGFR inhibitors. ⑥HDAC6 inhibitors. ⑦Specific inhibitory agents for mutant ER (ex. HSP90 inhibitors). This figure was exclusively drawn for this article
In this review, we summarize the rationale and the key clinical data obtained to date with targeting therapies for ER+/human epidermal growth factor receptor (HER)2- advanced breast cancer. This review is complementary to the one reported in the same journal by Migliaccio et al. [21], since it will discuss mostly new targeted therapies and mechanisms of resistance.
mTOR inhibitors
The PI3K (phosphatidylinositol 3-kinase), Akt/PKB (protein kinase B), and mTOR (mammalian target of rapamycin) pathway is an intracellular pathway which mediates gene activation, cell cycle, survival, metabolism motility, and genomic instability [22]. The pathway also contributes to cancer-promoting aspects of the tumor environment, such as angiogenesis [23].
The PI3K pathway is the most frequently altered pathway in breast cancer: the PIK3CA gene (encoding the catalytic isoform p110α) is the second most frequently mutated oncogene, and PTEN (encoding the phosphatase and tensin homolog) is among the most mutated tumor suppressor genes [24, 25]. In addition, many other molecular alterations within different components of the pathway, including PIK3CA amplifications, AKT1 mutations, and PTEN loss, have been observed in ER+ breast cancer [16, 25]. Moreover, the PI3K/Akt/mTOR pathway has been described as potentially intervening in secondary endocrine resistance in ER+ breast cancer [16, 26, 27]. In preclinical models, long-term estrogen-deprived breast cancer cells show an up-regulation of the PI3K pathway leading to a ligand-independent activation of ER by its phosphorylation through the mTOR complex 1 (mTORC1)/S6K1 axis [26, 28]. A series of first-generation mTOR inhibitors have been developed including everolimus (Afinitor, Novartis) [29] and temsirolimus (Torisel, Wyeth) [30] as rapamycin derivatives that inhibit mTOR through allosteric binding to mTORC1. In preclinical models, the use of everolimus in combination with aromatase inhibitors (AIs) results in synergistic inhibition of proliferation and induction of apoptosis [31]. In a randomized, phase II study comparing neoadjuvant everolimus plus letrozole with letrozole alone in patients with newly diagnosed ER-positive breast cancer, the response rate for the combination was higher than that for letrozole alone [32]. Several phase II and III studies including an mTOR inhibitor have been completed in patients with advanced hormone receptor (HR)+ breast cancer, and so far three major randomized trials have reported consistent data in efficacy [33–35] (Table 1). The phase III trial BOLERO (Breast cancer trials of oral everolimus)-2 enrolled 724 patients who were randomized to receive everolimus combined with exemestane (a steroidal AI) versus exemestane plus placebo in postmenopausal patients with HR+ advanced breast cancer previously treated with a non-steroidal AI (letrozole or anastrozole). At the time of the pre-planned analysis, median progression-free survival (PFS) was significantly better for the everolimus plus exemestane arm compared to the control arm (6.9 versus 2.8 months, HR 0.43, 95 % CI 0.35 to 0.54, P < 0.001 according to local assessment) [34]. The results of this study led to the approval by the FDA and EMA of everolimus in combination with exemestane in postmenopausal patients with advanced HR+ breast cancer previously exposed to letrozole or anastrozole. Final study results with median 18-month follow-up show that median PFS remained significantly longer with everolimus plus exemestane versus placebo plus exemestane in the overall population [investigator review: 7.8 versus 3.2 months, respectively; HR = 0.45 (95 % CI 0.38 to 0.54); P < 0.0001; central review: 11.0 versus 4.1 months, respectively; HR = 0.38 (95 % CI 0.31 to 0.48); P < 0.0001] [36]. Updated results have not found a significant benefit for overall survival (OS) with the combination arm, although a trend was observed, with a median OS of 31 months versus 27 months for everolimus versus the placebo arm, respectively; (HR = 0.89; 95 % CI 0.73 to 1.10; P = 0.14) [37]. Similarly, the French phase II study TAMRAD (tamoxifen plus everolimus) randomized endocrine therapy alone (in this case tamoxifen) versus tamoxifen plus everolimus in patients again with metastatic ER+ breast cancer previously treated with endocrine therapy [33]. In this trial including a total of 111 patients, clinical benefit rate (CBR) at 6 months (the primary endpoint) was clearly superior for the combination arm as compared with tamoxifen alone (61 % versus 42 % for combined therapy versus tamoxifen alone, respectively (exploratory P = 0.045). Time to progression (TTP) was also favorable in the combination arm (8.6 versus 4.5 months; HR 0.54, 95 % CI 0.36 to 0.81, P = 0.0021). There was also a benefit in OS for the mTOR-inhibitor arm (not reached versus 32.9 months, HR 0.45, 95 % CI 0.24 to 0.81, P = 0.007) [33]. Interestingly, the HORIZON trial, a phase III study in postmenopausal women with HR+ breast cancer that randomized 1,112 patients to receive the mTOR inhibitor temsirolimus in combination with letrozole versus letrozole plus placebo as first-line endocrine treatment, was closed prematurely following an intermediate analysis due to futility [35]. The analysis showed no difference in PFS, the primary endpoint, between the two arms (median PFS of 9 months; HR 0.90, 95 % CI 0.76 to 1.07, P = 0.25). There are several ongoing major randomized trials with everolimus in HR-positive advanced breast cancer including BOLERO-4, which will evaluate the benefit from the combination of everolimus and letrozole as first-line treatment (NCT01698918) and might be able to determine if the lack of benefit observed with temsirolimus in the HORIZON study was related to patient population, as preclinical studies have observed that the PI3K/Akt/mTOR pathway is mostly activated after previous endocrine therapy exposure. Finally, the BOLERO-6 trial is an ongoing three-arm phase II randomized study comparing everolimus plus exemestane, exemestane alone, and capecitabine (NCT01783444) in postmenopausal patients with HR+ breast cancer already exposed to endocrine therapy.
Many efforts have been performed in order to identify potential biomarkers of benefit from mTOR inhibition in patients with breast cancer. Immunohistochemistry (IHC) studies conducted on 55 formalin-fixed paraffin-embedded primary samples from the TAMRAD trial suggested that everolimus is more effective for tumors presenting with high levels of p4EBP1 (a downstream effector of the mTOR pathway), suggesting that baseline mTOR activation might be associated with sensitivity to mTOR inhibition [38]. In parallel, next-generation sequencing studies performed in 309 samples from the BOLERO-2 trial found that the presence of more than one molecular alteration (from four key pathways including FGFR1/2 amplification, PIK3CA mutation, PTEN loss, or CCDN1 amplification) was associated with a lack of benefit from everolimus treatment (HR = 0.78; 95 % CI 0.39-1.54) [17]. These findings suggest that primary resistance to mTOR inhibition might depend on the coexistence of mutations or amplifications in other pathways; therefore, combination therapy with other target agents should be considered for this population. Interestingly, the presence of a PIK3CA mutation was not predictive of benefit from everolimus treatment.
PI3K inhibitor/Akt inhibitor
As mentioned before, PI3K pathway alterations occur in about 70 % of breast cancers and include mutations and/or amplifications of the genes encoding the PI3K catalytic subunits, p110α (PIK3CA) and p110β (PIK3CB), the PI3K regulatory subunit p85α (PIK3R1), and the PI3K effectors AKT1, AKT2, and PDK1. The loss of lipid phosphatases such as PTEN can also activate the pathway [17, 39–42]. Preclinically, activation of RTK signaling has been seen to induce transcription of growth-related genes and cause decreases in ER levels and activity, leading to an inferior response to endocrine therapy [43]. Cotargeting this pathway with ER and PI3K inhibitors therefore appears to be a promising therapeutic opportunity for patients with ER+ breast cancer.
The development of PI3K inhibitors is rapidly evolving with newer and more potent compounds entering clinical trials including pan-PI3K inhibitors targeting all isoforms of PI3K, as well as the isoform-specific inhibitors, like inhibitors of the PI3K catalytic subunit p110α, which offer the potential of achieving greater selective target blockade while minimizing off-target effects due to inhibition of other isoforms. Some of the pan-PI3K inhibitors include XL147 [44] and GDC-0941 [45], although the most advanced in clinical research in HR-positive breast cancer is the pan-PI3K inhibitor BKM120 (buparlisib) [46] (Table 1). So far, single-agent clinical trials with pan-PI3K inhibitors have shown modest effect [44, 45, 47]. BKM120 has been evaluated for safety, tolerability, and preliminary activity in combination with letrozole in ER+/HER2- metastatic breast cancer patients refractory to endocrine therapy [48]. The CBR, its primary objective, was of 31 out of 51 patients. Buparlisib's maximum-tolerated dose (MTD) was 100 mg/d. Common drug-related adverse events included ≤ grade 2 hyperglycemia, nausea, fatigue, transaminitis, and mood disorders. Buparlisib is currently being tested in two phase III clinical trials in combination with fulvestrant for patients previously treated with an AI (BELLE-2, NCT01610284) and after resistance of mTOR inhibitor (BELLE-3, NCT01633060). Of note, another phase II/III trial evaluating the benefit of paclitaxel in combination with BKM120 or placebo (BELLE-4, NCT01572727) in first-line advanced HER2-negative breast cancer was recently terminated after an interim analysis due to futility. Another phase II trial of GDC-0941 in combination with fulvestrant (NCT01437566), both in HR+ postmenopausal breast cancer patients, was updated with a result of no PFS significance in the combination group (HR = 0.74; 95 % CI 0.51-1.05), otherwise effective in the ER and PR positive subgroup (HR = 0.44; 95 % CI 0.28-0.69). The combination group showed no correlation in the subgroup with PIK3CA mutation, but the patients with PIK3CA mutation showed an accurately higher objective response rate (15.8 % versus 3.1 %). Other clinical trials, including the phase II study of XL147 in combination with letrozole (NCT01082068), are currently ongoing.
Preliminary reports about BYL719, a PI3K-α inhibitor, have shown promising activity in patients with heavily pretreated PIK3CA mutant breast cancer in a phase I study. Out of the 17 patients treated, 8 (47 %) presented a tumor shrinkage of >20 % [49]. BYL719 is currently being tested in several phase I clinical trials in different types of combinations including with letrozole in postmenopausal patients harboring advanced breast cancer (NCT01791478), with either letrozole or exemestane for the same population (NCT01870505), or in endocrine-sensitive premenopausal HR+ cancer with combined endocrine therapy of tamoxifen and goserelin (NCT02058381). Whether selective PIK3CA isoform inhibitors may be superior to pan-PI3K inhibition in safety and efficacy, and which patient populations may benefit the most from their use, are questions yet to be addressed.
In addition, the presence of a negative feedback loop in the PI3K/Akt/mTOR pathway has been demonstrated, in which activation of mTORC1/S6K1 inhibits growth factor signaling to PI3K, exerting negative feedback to restrict insulin and IGF-1 signaling. Loss of this negative feedback mechanism has been shown to occur in cells and tumors exposed to mTOR inhibitors, preferentially those that inhibit mTORC1, which leads to mTORC2 assembly and an increase in phosphorylation of Akt Ser473 [50]. mTOR inhibition also leads to an escape signaling to RAS/RAF/MEK (MAPK signaling) [50, 51] and to an up-regulation of platelet-derived growth factor receptor (PDGFR) signaling [51, 52]. Thus, inhibition upstream to mTOR in the PI3K-Akt pathway might be expected to enhance mTOR inhibition and to exert an anti-tumor effect [17, 39–46, 48, 49, 53].
In order to compensate this Akt activation by this feedback loop caused by mTORC1 inactivation, several different approaches are currently being studied. The first one includes the dual blockade of PI3K and mTOR by the combination of a PI3K inhibitor and an mTOR inhibitor as is currently being tested in a phase II trial of BYL719 in combination with everolimus and exemestane (NCT02077933). Several dual PI3K/mTOR inhibitors are also currently being investigated in phase II studies in different types of tumors including HR+ advanced breast cancer. A phase II randomized trial testing GDC-0941 in combination with fulvestrant (NCT01437566) in HR+ postmenopausal breast cancer patients did not report a significant benefit on PFS (HR = 0.74; 95 % CI 0.51-1.05) [54]. PIK3CA mutations were not predictive for the efficacy of GDC-0941. Another phase II trial is ongoing with XL765 in combination with letrozole (NCT01082068). Another approach is the use of mTORC1/mTORC2 complex inhibitors like the four-arm phase II study with AZD2014 in two different schedules (continuous or intermittent) in association with fulvestrant versus fulvestrant + everolimus versus fulvestrant alone as the control arm (NCT02216786).
Of note, several Akt inhibitors are currently being tested in clinical trials to determine their potential benefit, some of them including patients with advanced breast cancer (Table 1), although the trials are still at early stages.
CDK inhibitor
The cyclin D1 and cyclin-dependent kinase 4 and 6 (CDK4/6) complex pathway is involved in cell cycle regulation and several downstream signals. During cell cycle progression, the cyclin D1-CDK4/6 complex mediates the phosphorylation and inactivation of the retinoblastoma protein (pRb), allowing for cells to progress from the G1 phase to the S phase [55]. In ER-positive breast cancer, the presence of cyclin D1 amplification has been observed, which causes cell cycle deregulation and results in over-proliferation of cancer cells [56]. Therefore, inhibition of the cyclin D1-CDK4/6 complex and the role it might play in restoring cell cycle control in breast cancer is a critical area of study. Results from early in vitro and in vivo studies have demonstrated that treatment with PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal ER-positive human breast cancer cell lines in vitro [57]. Three different oral small-molecule CDK4/6 inhibitors are currently being investigated: palbociclib (Ibrance, Pfizer), abemaciclib (LY2835219, Lilly), and LEE011 (Novartis) (Table 2).
The phase II clinical trial PALOMA-1/TRIO-18 (NCT00721409), testing the efficacy of letrozole with or without palbociclib, was conducted as first-line treatment in HR+ postmenopausal breast cancer patients. Final results have showed a median PFS of 10.2 months (95 % CI 5.7-12.6) for patients in the letrozole alone group, compared with 20.2 months (95 % CI 13.8-27.5) for those given palbociclib plus letrozole (HR = 0.488, 95 % CI 0.319 to 0.748; one-sided P = 0.0004) [58]. Notably, the benefit of palbociclib was not outweighed by excess toxic effects, with neutropenia (without an increase in febrile neutropenia) being the most common grade 3-4 adverse event. Several other adverse events were seen in more than 20 % of patients, with increases noted in the palbociclib group, but most were mild or manageable. These results have led to approval of palbociclib in early 2015 by the Food and Drug Administration (FDA) for the treatment of postmenopausal women with ER-positive, HER2-negative advanced breast cancer as initial endocrine-based therapy for their metastatic disease. Palbociclib is also currently being tested in different phase III clinical trials in patients with HR+ postmenopausal advanced breast cancer with different combinations including palbociclib plus letrozole versus letrozole monotherapy in first-line therapy (PALOMA-2, NCT01740427), palbociclib plus fulvestrant versus fulvestrant monotherapy (PALOMA-3, NCT01942135), and palbociclib plus exemestane versus capecitabine (PEARL, NCT02028507), these latter two studies in patients with resistance to AI. Another CDK4/6 inhibitor, LEE011, is currently being investigated in a phase III clinical trial in association with fulvestrant in first-line HR-positive advanced breast cancer (MONALEESA-2: NCT01958021) for postmenopausal patients, and in association with nsAI/TAM plus goserelin for premenopausal breast cancer (MONALEESA-7: NCT02278120). Similarly, abemaciclib is currently being tested in a phase III clinical trial in combination with non-steroidal aromatase inhibitors (letrozole or anastrazole) in postmenopausal women with breast cancer (MONARCH 3: NCT02246621). Results from a previous phase I trial demonstrated that more than 75 % of patients with HR+ breast cancer experienced either partial response or stable disease after a second-line treatment with abemaciclib [59].
Preclinical studies had shown that increased expression of cyclin D1 and pRb were associated with response in vitro, as was decreased expression of p16 (a natural CDK4/6 inhibitor) [57]. Unfortunately, in the phase II PALOMA-1/TRIO-18, patient selection on the basis of cyclin D1 amplification or p16 loss was not associated with an improved outcome from palbociclib treatment [58].
A combinatorial drug screen preclinical study has recently identified that CDK 4/6 inhibition sensitizes cells with acquired and intrinsic resistance to PI3K inhibition on multiple PIK3CA mutant cancers with decreased sensitivity to PI3K inhibitors. In fact, the combination of CDK 4/6 and PI3K inhibitors exhibited synergistic activity against PIK3CA mutant breast cancer cell lines. The reason behind this is the fact that cancers resistant to PI3K inhibitors present with persistence to cyclin D1 pathway activation as determined by the presence of Rb phosphorylation. In vivo, the combination of PI3K and CDK 4/6 inhibitors leads to tumor regression in PIK3CA mutant xenografts, overcoming intrinsic and adaptive resistance to PI3K inhibition [60].
Based on these findings, several phase I/II studies are currently ongoing with the combination of LEE011 with fulvestrant and BYL719 or BKM120 (NCT02088684), as well as LEE011, BYL719, and letrozole (NCT01872260) in postmenopausal advanced HR+ breast cancer.
FGFR inhibitor
Fibroblast growth factor receptors (FGFRs) are a family of transmembrane tyrosine kinase receptors belonging to the fibroblast growth factor (FGF) pathway, that upon activation promote cell proliferation, migration, angiogenesis, and survival in cancer cells by the activation of the Ras-dependent MAPK signaling pathway and PI3K/Akt/mTOR. FGFR1 amplification has been identified in around 10 % of HR+ breast cancers, and it has been associated with a worse prognosis, higher Ki67 expression, and resistance to endocrine therapy [61, 62]. Several other less frequent alterations in the FGF pathway have been identified including FGFR2 amplifications, FGFR3 translocations, and amplifications of different ligands like FGF3 and FGF4 that might potentially activate the pathway [41]. Several FGFR inhibitors are currently being investigated in HR+ advanced breast cancer in order to reverse resistance to endocrine therapy (Table 2). Dovitinib (TKI258) is a first-generation oral tyrosine kinase inhibitor (TKI) which inhibits FGFR1-3, VEGFR, and PDGFR. Preclinical data showed that dovitinib inhibits proliferation in FGFR1- and FGFR2- amplified, but not in FGFR-normal breast cancer cell lines [63]. Treatment with dovitinib as monotherapy was evaluated in a phase II clinical trial in women presenting with advanced HR+ breast cancer [63]. Patients were stratified based on the presence of FGFR1 amplification and/or FGF pathway activation determined by qPCR assay. Overall, unconfirmed response or stable disease for more than 6 months was observed in 5 (25 %) and 1 (3 %) patient(s) with FGFR1-amplified and FGFR1-nonamplified breast cancers, respectively. Interestingly, the response rate was 21 % in patients with activated FGF-pathway breast cancer based on qPCR, compared with a 12 % increase in target lesions in patients who did not present with FGF pathway amplification [63]. Dovitinib is currently being investigated in a randomized, placebo-controlled phase II study in combination with fulvestrant (NCT01528345). Another agent, AZD4547, specifically inhibits FGFR1 to 3 and is currently being investigated in ongoing phase I/II trials in patients with advanced HR+ breast cancer after exposure to non-steroidal aromatase inhibitors (NCT01791985), initially in combination with exemestane and posteriorly, after the results of the BOLERO-2 study, with fulvestrant. Both studies include patients with and without alterations in the FGF pathway to determine if there is a role for FGFR inhibition in endocrine-resistant breast cancer and if potential benefit is limited to the presence of a determinate molecular aberration.
HDAC inhibitor
Numerous epigenetic mechanisms have increasingly being revealed and relate to regulation of gene expression without changing DNA sequence. One of these mechanisms is modification of histone structure by acetylation which contributes to the dilatation of nucleosomal structure and the gathering of transcript factors followed by induction of transcription. The key enzymes, the histone deacetylases (HDACs), remove acetylation to stop the transcription, playing an important role in regulating gene expression [64, 65]. As alterations in HDACs are found in many human cancers [66–68], histone deacetylase inhibitors (HDACi) have aroused interest as a potential treatment for cancer. The first of these new HDACi, vorinostat (suberoylanilide hydroxamic acid), has received FDA approval as monotherapy for treating patients with cutaneous T-cell lymphoma. Moreover, HDAC inhibition has proven to be synergistic or additive with different anticancer agents, including radiation therapy [66], chemotherapy, and new targeted agents [66, 68–70]. In the case of breast cancer, the epigenetic silencing of ER target genes is crucial to ER-independent growth and has been described as a mechanism of endocrine resistance [71]. Based on that, different HDAC inhibitors are being investigated in combination with endocrine therapy in tumors resistant to endocrine therapy (Table 2). Vorinostat has been assessed in combination with tamoxifen in a non-randomized phase II study in patients previously treated with endocrine therapy [72]. The overall response rate was 19 %, and the clinical benefit rate (defined as stable disease > 24 weeks) was 40 %. Similarly, the results from the randomized double-blind phase II study of exemestane with or without entinostat, a benzamide HDAC inhibitor, enrolled 130 patients with resistance to non-steroidal AI. The PFS was 4.3 versus 2.3 months (HR = 0.73, 95 % CI: 0.50-1.07, P = 0.055), and the OS was 28.1 versus 19.8 months (HR = 0.59, 95 % CI: 0.36-0.97) for the combination versus the exemestane alone arm, respectively [73]. There is currently ongoing a phase III trial with the same treatment design for the same population (NCT02115282), as well as a randomized phase II study of fulvestrant with or without entinostat (NCT02115594), a phase II trial of vorinostat in combination with AI treatment (NCT00616967), and a phase I trial of abexinostat (S78454/PCI-24781), an oral pan-HDAC inhibitor in combination with tamoxifen. The most important dose-limiting toxicity of these compounds is thrombocytopenia, which is constantly observed and might limit drug combinations [74].
Targeting ESR1 mutation
Several reports have recently described the appearance of somatic ESR1 mutations as a potential mechanism of secondary endocrine resistance in HR+ breast cancer. Robinson et al. [75, 76] identified ESR1 mutations in 6 of 11 (55 %) HR+ advanced breast tumors. Further, Toy et al. [66] identified somatic ESR1 mutations in 9 of 36 (25 %) and in 5 of 44 (11%) ER+ metastatic breast cancers obtained from participants in the BOLERO-2 clinical trial whose disease had progressed during treatment with aromatase inhibitors [34]. A more recent report from Jeselsohn et al. [77] found that, overall, the frequency of these mutations was 12% (9/76; 95 % CI, 6 % to 21 %) in metastatic tumors, although it increased up to 20 % (5/25; 95 % CI, 7 % to 41 %) in a subgroup of patients who received an average of 7 lines of treatment. Interestingly, sequencing of ER-positive primary tumors did not identify ESR1 mutations, including some primary tumors obtained before therapy from a subset of cases with known ESR1 mutation at metastases [25, 76, 77]. Only Toy et al. identified ESR1 mutations in only 3 % of 183 pretreatment tumor biopsies from BOLERO-2 trial participants [76]. Moreover, none of these groups identified any ESR1 mutations when sequencing ER-negative breast tumors [75–77]. All these results suggest that ESR1 mutations are rare in newly diagnosed, untreated breast cancers but appear to be frequently acquired during progression to hormone resistance, especially in the context of estrogen deprivation therapy. To support this theory, these mutations seem also to affect the ligand-binding domain (LBD), encoding p.Tyr537Ser and p.Asp538Gly, which strongly promote classical ER signaling of target genes in the absence of ligand, resulting in the synthesis of receptors with ligand-independent activity and could promote resistance to AI treatment. Both Toy et al. [76] and Robinson et al. [75] showed that mutant ERα protein can still bind antiestrogens such as tamoxifen and fulvestrant, although higher doses of these drugs were required to inhibit this mutant ERα. This raises the possibility that altered dosing or the development of more potent and/or selective ER antagonists might inhibit residual ER activity and thus overcome resistance in the presence of a mutated ERα.
Yu et al. [78] recently reported that targeting heat shock protein (HSP) 90, which is the chaperone protein of ER, may be useful to treat Y537S ESR1-mutated tumors. The authors showed that mutant ESR1 tumors are highly dependent on HSP90, and preclinical studies with the HSP90 inhibitor STA9090 demonstrated cytotoxicity alone and in combination with raloxifene and fulvestrant to ex vivo cultured circulating breast tumor cells [78]. Interestingly, they also described that the allele frequency of ESR1 mutation correlated with the sensitivity to HSP90 inhibition. These findings suggest that ESR1-mutation targeted therapy will be possibly oriented by genomic portraits from each patient and that there is a need for more potent or specific antagonists of the mutant forms to block ER signaling as next-generation selective ER modulators (SERMs) and selective ER down-regulators (SERDs).
Conclusion
The mechanism of resistance to endocrine therapy in patients with ER-positive breast cancer remains a major issue. Previous studies had already identified a cross-talk between the ER pathway and growth factors pathways, mostly PI3K/Akt/mTOR and RAS/RAF/MAPK, as a main potential mechanism responsible for endocrine resistance. Moreover, the use of high-throughput technologies has identified several molecular aberrations present in breast tumors including PIK3CA mutations, AKT, FGFR1, and CCDN1 amplifications, as well as PTEN loss that contribute to the activation of these pathways and therefore might propitiate endocrine resistance via non-classical activation of ER. These findings have been made in parallel to the development of targeted therapies against these driver genes, leading to the approval of two new targeted therapies: everolimus and palbociclib against mTOR and CDK4/6, respectively, in combination with hormonotherapy to circumvent endocrine resistance. More recently, the discovery of somatic ESR1 mutations in tumors previously treated with endocrine therapy has directed attention to a new mechanism of resistance to endocrine deprivation. This, in addition to the results of currently ongoing clinical trials including combinations of different targeted therapies and a more comprehensive knowledge of the main molecular aberrations, will revolutionize the future management of ER-positive breast cancer.
Many challenges still remain though, as we try to identify the subsets of patients most likely to benefit from these novel targeted agents. A strategy for biological markers-driven selection of target agents for each patient and an integrated form for detecting reproducible key molecular alterations which cause endocrine resistance are mandatory for future precision medicine in this subset of breast cancer.
Abbreviations
- AI:
-
Aromatase inhibitor
- Akt:
-
Protein kinase B
- CBR:
-
Clinical benefit rate
- CDK:
-
Cyclin-dependent kinases
- CI:
-
Confidential interval
- ER:
-
Estrogen receptor
- FGFR:
-
Fibroblast growth factor receptor
- GPCR:
-
G-protein-coupled receptor
- HDAC:
-
Histone deacetylases
- HER:
-
Human epidermal growth factor receptor
- HR:
-
Hazard ratio
- HR:
-
Hormone receptor
- HSP:
-
Heat shock protein
- IGFR:
-
Insulin-like growth factor receptor
- IRS:
-
Insulin receptor substrate
- MAPK:
-
Mitogen-activated protein kinases
- mTOR:
-
mammalian target of rapamycin
- mTORC:
-
mTOR complex 1
- nsAI:
-
non-steroidal AI
- OS:
-
Overall survival
- PFS:
-
Progression-free survival
- PGDFR:
-
Platelet-derived growth factor receptor
- PgR:
-
Progesterone receptor
- PI3K:
-
Phosphatidylinositol-3-kinase
- qPCR:
-
quantitative polymerase chain reaction
- S6K1:
-
Ribosomal protein S6 kinase beta-1
- SERD:
-
Selective ER down-regulator
- SERM:
-
Selective estrogen receptor modulator
- TTP:
-
Time to progression
- VEGFR:
-
Vascular endothelial growth factor receptor
References
Theriault RL, Carlson RW, Allred C, Anderson BO, Burstein HJ, Edge SB, et al. Breast cancer, version 3.2013: featured updates to the NCCN guidelines. J Natl Compr Canc Netw. 2013;11:753–60. quiz 761.
Cardoso F, Costa A, Norton L, Senkus E, Aapro M, Andre F, et al. ESO-ESMO 2nd international consensus guidelines for advanced breast cancer (ABC2)dagger. Ann Oncol. 2014;25:1871–88.
Andre F, Slimane K, Bachelot T, Dunant A, Namer M, Barrelier A, et al. Breast cancer with synchronous metastases: trends in survival during a 14-year period. J Clin Oncol. 2004;22:3302–8.
Buzdar AU. Phase III, study of letrozole versus tamoxifen as first-line therapy of advanced breast cancer in postmenopausal women: analysis of survival and update of efficacy from the International Letrozole Breast Cancer Group. J Clin Oncol. 2004;22:3199–200. author reply 3200–3191.
Andre F, Neven P, Marinsek N, Zhang J, Baladi JF, Degun R, et al. Disease management patterns for postmenopausal women in Europe with hormone-receptor-positive, human epidermal growth factor receptor-2 negative advanced breast cancer. Curr Med Res Opin. 2014;30:1007–16.
Muss HB, Case LD, Atkins JN, Bearden 3rd JD, Cooper MR, Cruz JM, et al. Tamoxifen versus high-dose oral medroxyprogesterone acetate as initial endocrine therapy for patients with metastatic breast cancer: a Piedmont Oncology Association study. J Clin Oncol. 1994;12:1630–8.
Ellis MJ, Gao F, Dehdashti F, Jeffe DB, Marcom PK, Carey LA, et al. Lower-dose vs high-dose oral estradiol therapy of hormone receptor-positive, aromatase inhibitor-resistant advanced breast cancer: a phase 2 randomized study. JAMA. 2009;302:774–80.
Iwase H, Yamamoto Y, Yamamoto-Ibusuki M, Murakami KI, Okumura Y, Tomita S, et al. Ethinylestradiol is beneficial for postmenopausal patients with heavily pre-treated metastatic breast cancer after prior aromatase inhibitor treatment: a prospective study. Br J Cancer. 2013;109:1537–42.
Abraham JE, Maranian MJ, Driver KE, Platte R, Kalmyrzaev B, Baynes C, et al. CYP2D6 gene variants: association with breast cancer specific survival in a cohort of breast cancer patients from the United Kingdom treated with adjuvant tamoxifen. Breast Cancer Res. 2010;12:R64.
Wang L, Ellsworth KA, Moon I, Pelleymounter LL, Eckloff BW, Martin YN, et al. Functional genetic polymorphisms in the aromatase gene CYP19 vary the response of breast cancer patients to neoadjuvant therapy with aromatase inhibitors. Cancer Res. 2010;70:319–28.
Jin Y, Desta Z, Stearns V, Ward B, Ho H, Lee KH, et al. CYP2D6 genotype, antidepressant use, and tamoxifen metabolism during adjuvant breast cancer treatment. J Natl Cancer Inst. 2005;97:30–9.
Ferraldeschi R, Arnedos M, Hadfield KD, A'Hern R, Drury S, Wardley A, et al. Polymorphisms of CYP19A1 and response to aromatase inhibitors in metastatic breast cancer patients. Breast Cancer Res Treat. 2012;133:1191–8.
Binkhorst L, Mathijssen RH, Jager A, van Gelder T. Individualization of tamoxifen therapy: much more than just CYP2D6 genotyping. Canc Treat Rev. 2015;41:289–99.
Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med. 2011;62:233–47.
Gewinner C, Wang ZC, Richardson A, Teruya-Feldstein J, Etemadmoghadam D, Bowtell D, et al. Evidence that inositol polyphosphate 4-phosphatase type II is a tumor suppressor that inhibits PI3K signaling. Cancer Cell. 2009;16:115–25.
Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400–4.
Hortobagyi GN, Piccart-Gebhart MJ, HS R. Correlation of molecular alterations with efficacy of everolimus in hormone receptor–-positive, HER2-negative advanced breast cancer: Results from BOLERO-2. J Clin Oncol 31, 2013 (suppl; abstr LBA509).
Andre F, Job B, Dessen P, Tordai A, Michiels S, Liedtke C, et al. Molecular characterization of breast cancer with high-resolution oligonucleotide comparative genomic hybridization array. Clin Cancer Res. 2009;15:441–51.
Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–52.
Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer. 2011;11:558–72.
Migliaccio I, Malorni L, Hart CD, Guarducci C, Di Leo A. Endocrine therapy considerations in postmenopausal patients with hormone receptor positive, human epidermal growth factor receptor type 2 negative advanced breast cancers. BMC Med. 2015;13:280.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Hirsch E, Ciraolo E, Franco I, Ghigo A, Martini M. PI3K in cancer-stroma interactions: bad in seed and ugly in soil. Oncogene. 2014;33:3083–90.
Samuels Y, Ericson K. Oncogenic PI3K and its role in cancer. Curr Opin Oncol. 2006;18:77–82.
Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70.
Miller TW, Hennessy BT, Gonzalez-Angulo AM, Fox EM, Mills GB, Chen H, et al. Hyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor-positive human breast cancer. J Clin Invest. 2010;120:2406–13.
Simoncini T, Hafezi-Moghadam A, Brazil DP, Ley K, Chin WW, Liao JK. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature. 2000;407:538–41.
DeGraffenried LA, Fulcher L, Friedrichs WE, Grunwald V, Ray RB, Hidalgo M. Reduced PTEN expression in breast cancer cells confers susceptibility to inhibitors of the PI3 kinase/Akt pathway. Ann Oncol. 2004;15:1510–6.
Tabernero J, Rojo F, Calvo E, Burris H, Judson I, Hazell K, et al. Dose- and schedule-dependent inhibition of the mammalian target of rapamycin pathway with everolimus: a phase I tumor pharmacodynamic study in patients with advanced solid tumors. J Clin Oncol. 2008;26:1603–10.
Chan S, Scheulen ME, Johnston S, Mross K, Cardoso F, Dittrich C, et al. Phase II study of temsirolimus (CCI-779), a novel inhibitor of mTOR, in heavily pretreated patients with locally advanced or metastatic breast cancer. J Clin Oncol. 2005;23:5314–22.
Boulay A, Rudloff J, Ye J, Zumstein-Mecker S, O'Reilly T, Evans DB, et al. Dual inhibition of mTOR and estrogen receptor signaling in vitro induces cell death in models of breast cancer. Clin Cancer Res. 2005;11:5319–28.
Baselga J, Semiglazov V, van Dam P, Manikhas A, Bellet M, Mayordomo J, et al. Phase II randomized study of neoadjuvant everolimus plus letrozole compared with placebo plus letrozole in patients with estrogen receptor-positive breast cancer. J Clin Oncol. 2009;27:2630–7.
Bachelot T, Bourgier C, Cropet C, Ray-Coquard I, Ferrero JM, Freyer G, et al. Randomized phase II trial of everolimus in combination with tamoxifen in patients with hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer with prior exposure to aromatase inhibitors: a GINECO study. J Clin Oncol. 2012;30:2718–24.
Baselga J, Campone M, Piccart M, Burris 3rd HA, Rugo HS, Sahmoud T, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366:520–9.
Wolff AC, Lazar AA, Bondarenko I, Garin AM, Brincat S, Chow L, et al. Randomized phase III placebo-controlled trial of letrozole plus oral temsirolimus as first-line endocrine therapy in postmenopausal women with locally advanced or metastatic breast cancer. J Clin Oncol. 2013;31:195–202.
Yardley DA, Noguchi S, Pritchard KI, Burris 3rd HA, Baselga J, Gnant M, et al. Everolimus plus exemestane in postmenopausal patients with HR(+) breast cancer: BOLERO-2 final progression-free survival analysis. Adv Ther. 2013;30:870–84.
Piccart M, Hortobagyi GN, Campone M, Pritchard KI, Lebrun F, Ito Y, et al. Everolimus plus exemestane for hormone-receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: cancer: overall survival results from BOLERO-2. Ann Oncol. 2014;25:2357–62.
Treilleux IAM, Cropet C, Ferrero J, Lacourtoisie S, Spaeth D. Predictive markers of everolimus efficacy in hormone receptor positive (HR+) metastatic breast cancer (MBC): final results of the TAMRAD Trial Translational Study. J Clin Oncol. 2013;31:510.
Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, Neve RM, Kuo WL, Davies M, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008;68:6084–91.
Ellis MJ, Lin L, Crowder R, Tao Y, Hoog J, Snider J, et al. Phosphatidyl-inositol-3-kinase alpha catalytic subunit mutation and response to neoadjuvant endocrine therapy for estrogen receptor positive breast cancer. Breast Cancer Res Treat. 2010;119:379–90.
Campbell IG, Russell SE, Choong DY, Montgomery KG, Ciavarella ML, Hooi CS, et al. Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Res. 2004;64:7678–81.
Perez-Tenorio G, Alkhori L, Olsson B, Waltersson MA, Nordenskjold B, Rutqvist LE, et al. PIK3CA mutations and PTEN loss correlate with similar prognostic factors and are not mutually exclusive in breast cancer. Clin Cancer Res. 2007;13:3577–84.
Fu X, Osborne CK, Schiff R. Biology and therapeutic potential of PI3K signaling in ER+/HER2-negative breast cancer. Breast. 2013;22:S12–8.
Shapiro GI, Rodon J, Bedell C, Kwak EL, Baselga J, Brana I, et al. Phase I safety, pharmacokinetic, and pharmacodynamic study of SAR245408 (XL147), an oral pan-class I PI3K inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2014;20:233–45.
Sarker D, Ang JE, Baird R, Kristeleit R, Shah K, Moreno V, et al. First-in-human phase I study of pictilisib (GDC-0941), a potent pan-class I phosphatidylinositol-3-kinase (PI3K) inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2015;21:77–86.
Maira SM, Pecchi S, Huang A, Burger M, Knapp M, Sterker D, et al. Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Mol Canc Therapeut. 2012;11:317–28.
Rodon J, Brana I, Siu LL, De Jonge MJ, Homji N, Mills D, et al. Phase I dose-escalation and -expansion study of buparlisib (BKM120), an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. Investig New Drugs. 2014;32:670–81.
Mayer IA, Abramson VG, Isakoff SJ, Forero A, Balko JM, Kuba MG, et al. Stand up to cancer phase Ib study of pan-phosphoinositide-3-kinase inhibitor buparlisib with letrozole in estrogen receptor-positive/human epidermal growth factor receptor 2-negative metastatic breast cancer. J Clin Oncol. 2014;32:1202–9.
Juric D, Rodon J, Gonzalez-Angulo A, Burris HA, Bendel J, Berlin J, et al. BYL719, a next generation PI3K alpha specific inhibitor: Preliminary safety, PK, and efficacy results from the first-in-human study. Cancer Res. 2012;72:1.
Shah OJ, Wang Z, Hunter T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr Biol. 2004;14:1650–6.
Gupta S, Ramjaun AR, Haiko P, Wang Y, Warne PH, Nicke B, et al. Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell. 2007;129:957–68.
Zhang H, Bajraszewski N, Wu E, Wang H, Moseman AP, Dabora SL, et al. PDGFRs are critical for PI3K/Akt activation and negatively regulated by mTOR. J Clin Invest. 2007;117:730–8.
Edelman G, Bedell C, Shapiro G, Pandya SS, Kwak EL, Scheffold C, et al. A phase I dose-escalation study of XL147 (SAR245408), a PI3K inhibitor administered orally to patients (pts) with advanced malignancies. J Clin Oncol. 28:15s, 2010 (suppl; abstr 3004).
Krop I, Johnston S, Mayer A, Dickler M, Ganju V, Forero-Torres A. FERGI phase II study of PI3K inhibitor Pictilisibpictilisib (GDC-0941) plus fulvestrant vs Fulvestrant plus placebo in patients with ER+, aromatase inhibitor (AI)-resistant advanced or metastatic breast cancer - Part I results. Cancer Research. 05/2015; 75(9 Supplement).
Roberts PJ, Bisi JE, Strum JC, Combest AJ, Darr DB, Usary JE, et al. Multiple roles of cyclin-dependent kinase 4/6 inhibitors in cancer therapy. J Natl Cancer Inst. 2012;104:476–87.
Lange CA, Yee D. Killing the second messenger: targeting loss of cell cycle control in endocrine-resistant breast cancer. (Endocr Relat. Cancer. 2011;18:C19–24.
Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11:R77.
Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. 2015;16:25–35.
Patnaik A, Rosen LS, Tolaney SM, Tolcher AW, Goldman JW, Leena Gandhi L, et al. Clinical activity of LY2835219, a novel cell cycle inhibitor selective for CDK4 and CDK6, in patients with metastatic breast cancer. Cancer Res 2014;74:Abstract nr CT232.
Vora SR, Juric D, Kim N, Mino-Kenudson M, Huynh T, Costa C, et al. CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell. 2014;26:136–49.
Turner N, Pearson A, Sharpe R, Lambros M, Geyer F, Lopez-Garcia MA, et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 2010;70:2085–94.
Andre F, Bachelot T, Commo F, Campone M, Arnedos M, Dieras V, et al. Comparative genomic hybridisation array and DNA sequencing to direct treatment of metastatic breast cancer: a multicentre, prospective trial (SAFIR01/UNICANCER). Lancet Oncol. 2014;15:267–74.
Andre F, Bachelot T, Campone M, Dalenc F, Perez-Garcia JM, Hurvitz SA, et al. Targeting FGFR with dovitinib (TKI258): preclinical and clinical data in breast cancer. Clin Cancer Res. 2013;19:3693–702.
Sengupta N, Seto E. Regulation of histone deacetylase activities. J Cell Biochem. 2004;93:57–67.
Fischle W, Wang Y, Allis CD. Binary switches and modification cassettes in histone biology and beyond. Nature. 2003;425:475–9.
Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–84.
Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer. 2001;1:194–202.
Dokmanovic M, Marks PA. Prospects: histone deacetylase inhibitors. J Cell Biochem. 2005;96:293–304.
Piekarz RL, Sackett DL, Bates SE. Histone deacetylase inhibitors and demethylating agents: clinical development of histone deacetylase inhibitors for cancer therapy. Cancer J (Sudbury, Mass). 2007;13:30–9.
Yu C, Rahmani M, Conrad D, Subler M, Dent P, Grant S. The proteasome inhibitor bortezomib interacts synergistically with histone deacetylase inhibitors to induce apoptosis in Bcr/Abl+ cells sensitive and resistant to STI571. Blood. 2003;102:3765–74.
Margueron R, Duong V, Castet A, Cavailles V. Histone deacetylase inhibition and estrogen signalling in human breast cancer cells. Biochem Pharmacol. 2004;68:1239–46.
Munster PN, Thurn KT, Thomas S, Raha P, Lacevic M, Miller A, et al. A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. Br J Cancer. 2011;104:1828–35.
Yardley DA, Ismail-Khan RR, Melichar B, Lichinitser M, Munster PN, Klein PM, et al. Randomized phase II, double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive breast cancer progressing on treatment with a nonsteroidal aromatase inhibitor. J Clin Oncol. 2013;31:2128–35.
Ali A, Bluteau O, Messaoudi K, Palazzo A, Boukour S, Lordier L, et al. Thrombocytopenia induced by the histone deacetylase inhibitor abexinostat involves p53-dependent and -independent mechanisms. Cell Death Dis. 2013;4:e738.
Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, Cao X, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet. 2013;45:1446–51.
Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet. 2013;45:1439–45.
Jeselsohn R, Yelensky R, Buchwalter G, Frampton G, Meric-Bernstam F, Gonzalez-Angulo AM, et al. Emergence of constitutively active estrogen receptor-alpha mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin Cancer Res. 2014;20:1757–67.
Yu M, Bardia A, Aceto N, Bersani F, Madden MW, Donaldson MC, et al. Cancer therapy. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science. 2014;345:216–20.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
MY-I and MA declare that they have no competing interests. FA has a Research contract and is on the Advisory Board for Novartis and Astra Zeneca.
Authors’ contributions
MY-I and MA wrote the manuscript. FA discussed the initial outline and read the final document. All authors approved the final version of the manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Yamamoto-Ibusuki, M., Arnedos, M. & André, F. Targeted therapies for ER+/HER2- metastatic breast cancer. BMC Med 13, 137 (2015). https://doi.org/10.1186/s12916-015-0369-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12916-015-0369-5