
Abstract
Background
Explaining the emergence of the hallmarks of bilaterians is a central focus of evolutionary developmental biology—evodevo—and evolutionary genomics. For this purpose, we must both expand and also refine our knowledge of non-bilaterian genomes, especially by studying early branching animals, in particular those in the metazoan phylum Porifera.
Results
We present a comprehensive analysis of the first whole genome of a glass sponge, Oopsacas minuta, a member of the Hexactinellida. Studying this class of sponge is evolutionary relevant because it differs from the three other Porifera classes in terms of development, tissue organization, ecology, and physiology. Although O. minuta does not exhibit drastic body simplifications, its genome is among the smallest of animal genomes sequenced so far, and surprisingly lacks several metazoan core genes (including Wnt and several key transcription factors). Our study also provides the complete genome of a symbiotic Archaea dominating the associated microbial community: a new Thaumarchaeota species.
Conclusions
The genome of the glass sponge O. minuta differs from all other available sponge genomes by its compactness and smaller number of encoded proteins. The unexpected loss of numerous genes previously considered ancestral and pivotal for metazoan morphogenetic processes most likely reflects the peculiar syncytial tissue organization in this group. Our work further documents the importance of convergence during animal evolution, with multiple convergent evolution of septate-like junctions, electrical-signaling and multiciliated cells in metazoans.
Similar content being viewed by others


Background
Understanding the early steps of animal evolution is one of the major challenges of evolutionary biology. One way to achieve this goal is to compare genomic data across non-bilaterian animals (Ctenophora, Placozoa, Porifera, Cnidaria) [1,2,3].
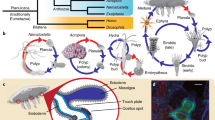
As one of the best candidates for sister group to all other animals [4,5,6,7,8,9,10] (Fig. 1a), sponges (Porifera) are of particular interest. Although the biology of Porifera is still poorly known [11], their ancient origin (> 600 million years (Myrs)) [12] has given rise to a phylum with over 9500 described species [13] distributed among four classes, with diverse ecological, embryological, cellular, and morphological features [14,15,16,17] (Fig. 1a, b).

General features of the hexactinellid sponge Oopsacas minuta. a Phylogenetic position of O. minuta: this species belongs to the phylum Porifera (sponges), one of the best candidates as sister group to all other metazoans. Free animal silhouettes were downloaded from PhyloPic (http://phylopic.org/). Among the four lineages of Porifera (> 9500 species), O. minuta belongs to Hexactinellida (glass sponges). b Hexactinellida (in red) have particular features (e.g., syncytial tissues) compared to the three other sponge classes, Demospongiae (in orange), Homoscleromorpha (in blue), and Calcarea (in green). c O. minuta adults inhabit Mediterranean canyons and shallow caves (photo credit Dorian Guillemain), where individuals are 1–7 cm long and have a clear basal–apical polarity (ap apical pole, bp basal pole) (scale bar = 1 cm)
The first sponge genome sequenced, from the demosponge Amphimedon queenslandica, revealed a larger size and gene content than expected [18,19,20]. However, transcriptomic data from other species indicated that A. queenslandica was not representative of the diversity of sponges [17, 21,22,23,24,25,26]. Several sponge genomes now sequenced [27,28,29,30,31] illustrate the disparity of sponge genomes in terms of size, features of non-coding regions, and gene repertoire [3, 28]. However, the lack of a complete genome from the Hexactinellida (termed “glass sponges” for their often heavily siliceous skeleton) (Fig. 1b) has remained a major gap in our knowledge of Porifera, despite the insights provided by snapshots of their gene contents [3, 23, 26, 32, 33]. A comprehensive description of the genomic features of a glass sponge will show exactly how much they differ from sponges in other classes and will identify key genomic changes associated with the diversification of sponges into four markedly different classes.
Here, we provide the first high-quality genome of a glass sponge. The species Oopsacas minuta (Topsent 1927) (Fig. 1c) was chosen for its unambiguous taxonomic identification, its accessibility at shallow depths in Mediterranean caves, and because of important aspects of its embryology and biology that have been studied previously [34,35,36]. Given the range of unusual characteristics of Hexactinellida including their syncytial organization of tissues, their ability to propagate electrical signals, and their adaptation to the deep-sea habitat, we focused our bioinformatic analyses on genes involved in development, epithelia and multiciliogenesis, silicate biogenesis, and signal transduction. We also gathered data on the dominant microbiome associated with O. minuta.
Results
Initial metagenomic data processing
The data obtained from the DNA (deoxyribonucleic acid) mixture extracted from non-clonal specimens collected from their natural environment were initially processed as sequences from a low-complexity metagenome consisting of the sponge nuclear genome, its mitochondrial genome, and of an undescribed population of microorganisms including non-resident (food) and resident (symbionts sensu lato [37]) species.
The combination of sequence data (from two complementary platforms Pacific Biosciences (PacBio) and Illumina) yielded an initial dataset of 1759 scaffolds larger than 1 kb (kilo base) that were first classified as Eukaryota, Bacteria, Archaea, or virus sequences. Further steps in refining the assembly resulted in three distinctive sets of super-scaffolds associated with large coverage values: the genome of a Thaumarchaeota (coverage = 1206), the O. minuta nuclear genome (coverage = 186), and the mitochondrial genome (coverage = 455). A number of other scaffolds with smaller coverage were attributed to the residual microbiome of the sponge (Additional file 1: Table S1). The fully assembled mitochondrial genome was published previously [38].
The dominance of a Thaumarchaeota symbiont
Data on hexactinellid microbiomes are scarce, and histological observations suggest that bacterial symbionts are rare [36, 39, 40].
Of the 1759 scaffolds, 107 scaffolds were assigned to Bacteria (more than 11 phyla) dominated by γ-proteobacteria (Fig. 2a; Additional file 1: Table S2). In addition, one of the two viral contigs was affiliated with the Circoviridae, a family of small single-stranded DNA viruses. Surprisingly for an animal that filters sea water where viruses are abundant [41,42,43], viruses have rarely been observed in sponges [44, 45] and few sponge viromes are available [46,47,48,49]. Nevertheless, Circoviridae-related sequences have been reported previously in two demosponges [49]. Known circoviruses tend to be associated with vertebrate hosts and are considered rare in marine invertebrates [50], highlighting the novelty of our finding [51].

Characterization of the microbiome associated with O. minuta. a Relative coverage and sequence size of non-eukaryotic contigs according to their taxa (Table S1, Additional file 1). The microbiome of O. minuta is dominated by a new Thaumarchaeota species that we propose to name Candidatus Cenarchaeum massiliensis and various proteobacteria. b Microsynteny analysis of genomic segments bearing an amo gene cluster in marine Thaumarchaeota showing that the genome Candidatus Cenarchaeum massiliensis has features highly conserved with other marine Thaumarchaeota (arrows of the same color indicate orthologous genes and black arrows represent genes without orthology relationship in these regions from OrthoMCL analysis). Other data concerning Ca. C. massiliensis are available in Tables S3 and S4 and Figs. S1, S2, and S3 of Additional file 1. c Localization of Ca. C. massiliensis in the tissues of O. minuta by Card-FISH using a specific probe (blue = DAPI staining; green = labeled probe) shows the presence of this symbiont in the whole trabecular syncytium including the dermal layer and the flagellated chambers (see also Fig. S4, Additional file 1)
Besides a more diverse microbiome than previously reported [34,35,36], the most visible feature emerging from the sequence assembly was the presence of 11 super-scaffolds of a Thaumarchaeota-like genome associated with the highest coverage value (Fig. 2a; Additional file 1: Table S1). This coverage corresponds to a ratio of about 13 archaean cells per sponge cell (presumed diploid according to the few sponge karyotypes available [52]). This Thaumarchaeota species represents the main microorganism associated with O. minuta. Its global protein sequence similarity with the NR (non-redundant) databases and phylogenetic analyses strongly suggest that it is a new representative of the Cenarchaeum genus that we propose to name Candidatus Cenarchaeum massiliensis (Ca. C. massiliensis) (Fig. 2b; Additional file 1: Fig. S1).
The partially assembled genome sequence of Ca. C. massiliensis has a size of about 1.63 Mb (Megabases), a G + C content (guanine + cytosine content) of 37.3 ± 2.5%, an estimated completeness of 99.03% [53], and a relatively low coding density (83.9%) compared to other ammonium-oxidizing archaea (AOA) species (Additional file 1: Table S3). The orthology analysis of 23,013 predicted proteins from ten Thaumarcheota species found 3229 ortholog groups (gene families) using OrthoMCL algorithm and database (Additional file 1: Fig. S2a). Among them, 1517 (47%) were found only in one genus and a large part (1158 gene families) was found in only Nitrososphaera genus (representing 36% of total Thaumarcheota gene families). A large part of the Thaumarcheota pan genome is found in the Nitrososphaera genus that could be explained by its metabolic versatility and environmental adaptations [54,55,56,57].
We identified 908 gene families conserved across the five genera of Thaumarchaeota representing the core genome of Thaumarcheota (28% of total Thaumarcheota gene families). Among the conserved gene families, a large part corresponds to unknown function (184 (20%)) and another large part encodes ribosomal proteins, translation factors, or aminoacyl-tRNA synthetases (125 (14%)) (Supplementary Fig. 2b, Additional file 1). We also identified core gene families potentially involved in amino acid and nucleotide metabolism (74 (8%) and 39 (4%), respectively). In addition, we found 41 core gene families (4.5%) involved in transcription including transcription factors and RNA polymerases. The analysis identified 41 core families (4.5%) involved in transport including peptide transporters, aquaporins, cation/ion transporters, and ATPases. Unexpectedly, the metabolism of cofactors and vitamins represented a large part of core Thaumarchaeota gene families (66 (7%)) including essential genes for cobalamin biosynthesis (Additional file 1: Figs. S2 and S3). Among the 908 core gene families, the orthoMCL analysis identified 132 (14.5%) Thaumarchaeota-specific gene families (not found in another species in the OrthoMCL database) corresponding to the Thaumarcheota trademark protein set, including specific ribosomal proteins (presence of r-proteins s26e, s25e, and s30e but absence of the r-proteins L14e and L34e), DNA topoisomerase IB and subunits of ammonia monooxygenase (Amo) (Fig. 2b; Additional file 1: Table S4).
Interestingly, as much as 147 transposase encoding genes are present in this species. This number is among the largest reported to date in a thaumarcheotal genome [58, 59].
Using Card-FISH (catalyzed reporter deposition fluorescent in situ hybridization), we localized Ca. C. massiliensis in the tissues of O. minuta with a specific probe. This microorganism is present in the trabecular syncytium (Fig. 2c; Additional file 1: Fig. S4). Similar localizations were observed in different individuals sampled at different periods. Taken together, the abundance and the durability of the association suggest that Ca. C. massiliensis is a symbiont of O. minuta.
Our finding further supports the hypothesis that a stable Thaumarchaeota-Porifera relationships might be based on the ability of all Thaumarchaeota to oxidize ammonia in nitrite and to produce cobalamin anaerobically [58]. Such capabilities are absent in animals while cobalamin is essential for their life cycle [60, 61]. We propose that the symbiotic Ca. C. massiliensis may provide cobalamin to O. minuta while recycling the ammonia produced by its metabolism [62].
O. minuta genomic features
The O. minuta genome was assembled into 365 scaffolds. Its G + C content is 36 ± 2.1%. The quality and low fragmentation of the assembly is attested by a N50 value of 0.67 Mb comprising 31 sequences. The estimated total genome size is about 61 Mb, thus two- to six fold smaller compared to Demospongiae and Calcarea (Fig. 3a; Additional file 2: Table S5a). It is one of the smallest sponge genomes reported so far (the other being Oscarella pearsei: 57.7 Mb) [28]. Such differences in genome sizes are consistent with previous estimations of DNA content [63].

General features of the nuclear genome of Oopsacas minuta, compared to that of other sponges. a The assembled genome of O. minuta is one of the smallest so far described in sponges. b The O. minuta genome has fewer predicted proteins and is slightly more compact than the genomes of Amphimedon queenslandica and Ephydatia muelleri, both demosponges, which is the sister group to hexactinellids. Other genomic features are available in Additional file 2: Table S5, Table S6, and Fig. S5
The variation in genome sizes of closely related eukaryotic organisms can arise from the difference in their gene densities and/or numbers. The average number of introns per gene, the average intron size, and the average intergenic distance all govern gene density. In O. minuta, all of these are similar or only slightly lower than those in demosponges: 1.59 intron/gene, average intron size = 341 bp (base pairs), average intergenic distance = 2 kb (Fig. 3b; Additional file 2: Table S5b). Thus, the main reason O. minuta has the most compact sponge genome assembled so far is because it encodes far fewer proteins (16,413) than other sponge genomes: 39,245 for Ephydatia muelleri [30], 40,122 for Amphimedon queenslandica [19], and 37,416 for Tethya wilhelma [29]. This observation is validated by the high number of conserved core eukaryotic genes identified in the O. minuta draft sequence (91 to 93% using BUSCO (Benchmarking Universal Single-Copy Orthologs) [64]) attesting to its completeness. Through a first automated pass of functional annotation, 7737 protein-coding genes were attributed a Gene Ontology (GO) annotation (Additional file 2: Table S6 and Fig. S5).
The global orthology comparison between Oopsacas and other sponge species confirms this difference in gene content. Of the total number of genes, 86.6% were assigned to 24,721 orthogroups. For each species, the percentage of unassigned genes varies from 5.7% (A. queenslandica) to 20.3 (Sycon ciliatum). Oopsacas minuta shares only 36 to 40% of its orthologs with other sponges. It is further noticeable that 761 orthogroups shared by all other sponges are not present in Oopsacas (Additional file 2: Fig. S6a). These losses in glass sponges mainly concern enzymatic activities, biomolecule binding properties, and metabolic processes (Additional file 2: Fig. S6b). These findings suggest that Oopsacas differs from other sponges in its physiology and call for further experimental studies on this lineage. Other gene losses concern sensing processes, and regulation of cellular and developmental processes, some of which are detailed in the next sections. On the other hand, 637 orthogroups appear to be specific to the hexactinellid lineage as not being identified in the three other sponge lineages: again, GO terms point to probable specificities regarding enzymatic activities, metabolism, binding, cell communication, and signal transduction. We also noticed orthogroups related to transposition [65].
Interspersed repeats represent 34.10% of the genome, and these are mostly represented by unclassified repeats (18.01%) and DNA transposons (15.44%) (Additional file 2: Tables S7 and S8).
The epithelial gene set of a syncytial sponge
The unique syncytial organization of Hexactinellida [36, 66,67,68,69] raises exciting questions regarding the genes involved in the epithelial characteristics: cell polarity, cell junctions (CJs), and basement membrane (BM) [18, 70,71,72,73,74,75,76]. A previous survey [32] showed that Oopsacas possesses the whole set of genes encoding proteins involved in the polarity complexes (PAR, CRUMBS, SCRIBBLE), and in cadherin-catenin complexes (CCC) despite the apparent absence of conventional adherens junctions (AJs), suggesting that these proteins may have different functions in sponges [75]. Here we searched for proteins involved in other types of CJs and in the BM.
Three types of cell–cell junctions are defined, namely adherens, gap, and septate junctions (AJs, GJs, and SJs) [75, 77,78,79,80,81]. As for other sponge lineages, no innexin-related genes, involved in GJs, were found in this genome. This is in agreement with (i) the observation that glass sponge plugged junctions are specialized cytoplasmic structures that lie within a cytoplasmic bridge, in contrast to gap junctions which are channels within and between membranes of two apposed cells [67] and (ii) the fact that no gap junctions have been reported so far in any sponge lineage [67]. The content of the glass sponge proteinaceous plugged junction remains to be determined [36, 66]. Of SJ proteins, neither Claudin, Neuroglian, Neurexin IV, nor Contactin were found. Regarding Contactin (Cont), which belongs to the immunoglobulin superfamily (IgSF), neither our best blast hit (Additional file 3: Table S9) nor the sequence from Aphrocallites vastus annotated as Contactin [23] exhibit the characteristic glycosylphosphatidylinositol (GPI) anchor domain, supporting the absence of Cont in glass sponges (Fig. 4a). Finally, glass sponges have septae reminiscent of SJs [69] but do not encode the proteins involved in bilaterian SJ, suggesting these are convergent structures.

Survey of genes associated with tissue features of O. minuta. a Presence/absence in O. minuta, which has syncytial tissues, of genes involved in epithelial functions (in Bilateria) compared to sponges in the three other poriferan classes, which have cellular tissues. b Phylogenetic relationships between metazoan taxa where multiciliated cells (MCC) have been reported (red stars). c O. minuta larvae possess MCC, seen clearly in only one other group of deep-water demosponges (cladorhizids). The diagram of a larva (on the left) shows the position of MCC (red arrows) several cilia are visible in cross section in the TEM picture on the right (photo credits Sally Leys). d Survey of genes involved in MCC differentiation in vertebrates. e Survey of genes involved in ciliogenesis and basal body duplication in bilaterians
In cell-to-matrix junctions, the bilaterian focal adhesions (FAs) and hemidesmosomes (HDs) are based on interactions between integrins, proteins of the extracellular matrix (ECM), and the actin network [82,83,84,85]. We found a diversified set of integrins as in other sponges [20, 23, 30, 86, 87]. Five were assigned to the alpha chain family and three to the beta chain family. Their predicted domain structures are similar to those reported in other animals (Additional file 3: Fig. S7a).
Among the main components of the BM (laminins (Lam), type IV collagen, nidogen, and perlecan [71, 88,89,90,91]), only four laminins were found (Fig. 4a) with a shared characteristic domain architecture consisting in one α-like laminin-related protein, two chimeric Lamβ/γ-like chains as in A. queenslandica [92, 93] and a laminin γ-like chain with a characteristic single LamIVA domain (Additional file 3: Fig. S7b). Both our and Fahey’s surveys [92] suggest that α, β/γ, and γ-like laminin chain types were present in the last common ancestor of metazoans (LCAM). In contrast, a β-chain is identified neither in the demosponge Amphimedon nor in Hexactinellida, suggesting this type of laminin emerged later. When reassessing (by domain prediction) the sequence annotated as nidogen in Aphrocallistes vastus [23], we only identified the NIDO domain, which is not specific to nidogen. The absence of all components of the BM, except laminins, confirms the absence of BM in glass sponges, in agreement with morphological observations and previous transcriptomic analyses [18, 23, 71,72,73, 75, 94]. Type IV collagen was probably present in the LCAM and secondarily lost several times [20, 23, 30, 95]. In contrast, perlecan and nidogen probably emerged more recently [75].
Multiciliogenesis
Multiciliated cells (MCCs) have been described in Vertebrata [96], Spiralia, Ctenophora, and Porifera [97,98,99,100,101] (Fig. 4b). Vertebrates and protostomes share the same core set of proteins required for centriole formation and duplication (PLK4 (polo-like kinase 4), SAS6 (spindle assembly abnormal 6), and STIL (SCL/TAL1 interrupting locus)) but not the upstream regulatory network [102,103,104,105]. Three evolutionary scenarios are possible: (1) the upstream regulators of vertebrate MCCs are ancestral and were lost/replaced in protostomes, (2) the (unknown) upstream regulators of protostomes are ancestral and were lost/replaced in vertebrates, and (3) none of the upstream regulators are ancestral and MCCs emerged several times in Metazoa. Data from non-bilaterians are needed to assess these scenarios. O. minuta is one of the few sponge species having MCCs in the larva (Fig. 4c) [34, 66, 68, 94, 106, 107]. We therefore searched for genes involved in MCC differentiation and centriole duplication in bilaterians.
Except for the Notch signaling pathway (next section), neither the vertebrate upstream regulators (Mcidas (multiciliate differentiation and DNA synthesis associated cell), GMNC/GEMC1 (geminin coiled-coil domain containing) and the transcription factor E2F4) nor their targets (FoxJ1 (forkhead box J1), c-Myb (myeloblastosis proto-oncogen), Deup 1 (deuterosome assembly protein 1), Cyclin O (CCNO)) involved in MCC differentiation were found (Fig. 4d; Additional file 3: Table S10). The absence of FoxJ and Myb-related genes is unexpected, because they have an ancient origin [108, 109], and the latter has already been reported in sponges [110]. This suggests secondary losses of these two transcription factors in hexactinellids.
Among the key upstream proteins involved in bilaterian centriole duplication (Fig. 4e), we found genes encoding centrosome-associated proteins (CEP) CEP192 and CEP57, but not CEP63 and CEP152 (Additional file 3: Table S10), suggesting that centriole duplication may be initiated by a different proteomic network in Oopsacas than in bilaterians. In contrast, we found PLK4, SAS6, STIL, and CENPJ (Centromere Protein J), in agreement with their conserved function in centriole duplication across Eukaryota [111].
The above findings suggest that the formation of multiciliated cells in Oopsacas may involve common downstream terminal effectors, but that upstream regulators are different from those described in vertebrates and protostomes. In other words, sponge and bilaterian MCCs probably result from convergent evolution, as previously proposed on the basis of ultrastructural [105] and embryological [66] observations.
Signaling pathways
In metazoans, conserved signaling pathways are critical transduction cascades [112,113,114,115,116,117,118]. Surprisingly, previous transcriptomic analyses have suggested that key components of the canonical Wnt pathway are absent in glass sponges [23, 26, 119].
Our present analysis of Oopsacas whole genome confirms that neither wntless, wnt, nor dishevelled genes are present (Fig. 5a). In addition, frizzled (Fzd) A gene is absent, whereas frizzled B is present. In contrast, core members of two other key developmental pathways, namely Notch and TGF-β (transforming growth factor-beta), are present (Fig. 5b; Additional file 3: Table S11). So far, the absence of the Wnt pathway was only reported in myxozoans [120], a group of microscopic parasitic cnidarians. But, in contrast to myxozoans, glass sponges do not show a highly reduced body plan compared to other sponges (Fig. 1c), and so the absence of Wnt challenges the pivotal role often attributed to this pathway in the acquisition of multicellularity and axial patterning [121].

Survey of key genes involved in the metazoan developmental toolkit. a Presence/absence of genes involved in the canonical Wnt pathway: the absence of Wnt and dishevelled genes is an unusual feature suggesting the use of alternative pathways. b Survey of genes involved in three critical signaling pathways in O. minuta compared to the demosponge Amphimedon queenslandica: core members of Notch and TGF-β are present. c Inventory of basic Helix loop Helix (bHLH) transcription factors present in O. minuta compared to two other sponges. Numerous genes are absent in O. minuta. d Inventory of Homeobox transcription factors present in O. minuta compared to two other sponges. Several ancestral classes are absent in this species. Numbers in circles indicate the number of genes found, question mark indicates the absence of data in the literature
In search of a pathway that may “compensate” for such an absence, we surveyed genes encoding heteromeric G proteins and G protein-coupled receptors (GPCRs), which are known to play important roles in transducing a broad range of extracellular signals. Heterodimeric G proteins include Gα (Gs, Gi, Gq, G12, and Gv classes [122]), Gβ, and Gγ subunits. Here, we found a diverse set of 8 Gα, one belonging to Gs, G12, and Gv classes, two to the Gi class, and three to the Gq class (Additional file 3: Table S12) [123]. We suppose that the early development in Oopsacas could use a truncated Wnt pathway (no Wnt activation) and that the expanded Gq set found in Porifera (and Ctenophora), compared to most bilaterians, placozoans and cnidarians [123,124,125,126], suggests a broader involvement of Gq proteins in the development of these animals. Because recent studies have suggested the ability of Fzd5 to activate Gq [127], the FzdB copy found in glass sponges (the Fzd5-ortholog) might not be a remnant of a reduced Wnt pathway but might instead reflect its involvement in G protein signaling.
Transcription factors
Transcription factors (TFs) are also pivotal in animal body patterning. Here, we focused on basic helix loop helix (bHLH) and homeobox classes because (1) these TFs are significantly enriched in the animal TF repertoire [108] and (2) they have already been exhaustively surveyed in two other sponges thereby enabling comparisons [31].
There are six major groups (A, B, C, D, E, F) of bHLH TFs [128]. We identified 10 genes encoding proteins with a bHLH domain, two of which have an additional Per-Arnt-Sim (PAS) domain (Fig. 5c). Our phylogenetic analyses showed the presence of proteins in the AP4, MITF, SREBP, and E12/E47 families (class B bHLH) (Additional file 3: Table S13 and Fig. S8). In addition, one Oopsacas protein was found to cluster with several bHLH families constituting the Atonal-related superfamily [129,130,131]. The three remaining proteins are class C bHLH-PAS proteins: one of the Clock family, the two others clustering with ARNT (aryl hydrocarbon receptor nuclear translocator) and BMAL (brain and muscle Arnt-like) families [129] (Additional file 3: Fig. S9). Neither D, E, nor F class members were identified. There are clearly fewer bHLH homologs in Oopsacas than in S. ciliatum (30) and A. queenslandica (21). More than 10 families that are likely ancestral to sponges have been lost in this species (Fig. 5c).
Among the animal homeobox TFs classes, the Antennapedia (ANTP) and pair-ruled (PRD) are the largest [132,133,134]. In Oopsacas, we found 20 predicted proteins with a homeodomain (HD). According to both the identity of the best blast hits and domain analyses (Additional file 3: Table S14 and Fig. S10), only five sequences pertain to the ANTP class. Phylogenetic analyses show that there are members of the Msx (1), BarX (2), and NkX (2) families (Fig. 5d; Additional file 3: Fig. S11). Also present are six members of the super-class TALE, two members in the prd-like, LIM and Pou classes. We also identified one sequence assigned to the SIX (sine oculis homeobox) and the HNF (hepatocyte nuclear factor) classes (Fig. 5d; Additional file 3: Table S14). We found proteins containing a PAX domain, but none associating HD and PRD domains, suggesting that Prd class members are absent. We did not find any sequences from the ZF (zinc finger), Cut, Pros (Prospero), CerS (ceramid synthase), and MKX (Mohawk homeobox) classes.
Altogether, the number of ANTP TFs in Oopsacas (5) is lower than in other sponges (9–11) [31]. According to previous studies, some ANTP members were lost independently in sponge classes. The absence in Oopsacas of Hhex (haematopoietically expressed homeobox) and Prd genes, that are usually considered ancestral, is meaningful [31, 133, 135,136,137,138].
In summary, the TF complement of Oopsacas differs from that of other sponges studied so far and exhibits a markedly reduced number of the core metazoan TFs. Some authors have suggested that TF loss plays a major role in adaptation to environmental changes and in macroevolution [139]. However, the functional consequence of such TF losses on regulatory networks in glass sponges remains to be explored.
Photokinesis and signal transduction
Like unicellular eukaryotes, sponges lack neurons but respond to stimuli [140,141,142,143,144,145,146,147,148]. Photokinesis is the best studied sensory mechanism involved in sponge larval behavior; however, all sponge genomes examined so far lack opsins, as is also the case here. Nevertheless, O. minuta possesses two photolyase genes, also found in the demosponge A. queenslandica (Additional file 3: Table S15). It has been shown previously that, in a glass sponge, photolyases are not only involved in DNA repair but have also been found to be expressed regionally, which was suggested to be a response to light even at a depth of 30 m [149]. No experiments have been carried out to determine whether glass sponge larvae carry out photokinesis or whether the function of the photolyase genes in Oopsacas are similar to those described in Amphimedon [150, 151].
Hexactinellids are unusual among Porifera in terms of signal transduction because they coordinate arrests of their feeding current using action potentials that travel through syncytial tissues [143, 152, 153]. Although glass sponge syncytia are quite different than neurons, as they do propagate electrical signals, we searched for genes typically involved in chemical signaling in bilaterians. We found no evidence for conventional monoamine signaling receptors or biosynthesis pathway components (serotonin or dopamine). In contrast, components for glutamate and GABA (gamma-aminobutyric acid) synthesis and signaling were identified. Nitric oxide signaling is likely present as well as acetylcholinesterase (Additional file 3: Table S16).
In terms of voltage-gated ion channels (Additional file 3: Table S17), we found genes for voltage-gated calcium channels (two pore channels, sperm-associated calcium channels, and voltage-gated hydrogen channels) (Additional file 3: Fig. S12) but no genes for voltage-gated sodium channels (Nav) nor for obvious voltage-gated potassium channels (Kv) (Additional file 3: Fig. S13). We did not find ENaC (epithelial sodium channels) nor LEAK channels. Ionotropic glutamate receptors (iGluR) were also absent. In contrast, four genes encoding for anoctamins (voltage sensitive calcium activated chloride channels) and six chloride channels (H + /Cl-transporters) were identified. Oopsacas also has cyclic-gated nucleotide (CGN/HCN) channels as well as purinergic and ryanodine receptors. In addition, there were many hits for transient receptor potential (TRP) family proteins (including one TRP-ML and one TRP-A family protein).
The study of glass sponge conduction system performed on Rhabdocalyptus dawsoni [153] suggested that the action potential could be driven by calcium because it was blocked by calcium channel blockers. This may be consistent with the diversity of calcium channels identified here. While the return to resting potential is sensitive to a potassium channel blocker, Oopsacas does not appear to possess the voltage sensor region of K channels, so it is unknown which channels are responsible for resetting the membrane potential.
Finally, we also searched for genes involved in synapses in animals with neurons. Postsynaptic proteins include a wide range of scaffolding and vesicle fusion/transport proteins, and many are present in Oopsacas, among them synaptobrevin, syntaxin, Homer, and members of the SNARE (SNAP receptor) family SNAP 25 (soluble N-ethylmaleimide-sensitive factor-attachment protein), with the exception of neurexin and neuroligin. In contrast, most of conventional presynaptic proteins such as profilin, synaphin, synaptoporin, and synaptogamin were missing (Additional file 3: Tables S18 and S19). In summary, a complete synaptic machinery is absent in O. minuta. Although proteins involved in vesicle transport and ion channels were identified, they may play a variety of other roles.
The biosilicification toolkit
The typical 6-rayed spicules of glass sponges, hexactines, are made of silica. Although spiculogenesis has similarities across siliceous sponges, there are fundamental differences between hexactinellids and demosponges [154,155,156,157,158]. We therefore searched the genome of O. minuta for homologs of genes known to be involved in spicule formation in demosponges (Fig. 6a, b; Additional file 3: Table S20). We found no evidence of silicatein, silintaphin, or galectin genes, which are common in demosponges, but we identified a single glassin gene, 13 cathepsin L genes (Fig. 6c; Additional file 3: Tables S20 and S21), 1 ferretin, 4 silicases, and 1 chitin synthase. In addition, we found a homolog of conventional actin, which has been recently reported to be important for spicule scaffolding [159]. Shimizu and collaborators [156] tried unsuccessfully to isolate silicatein proteins from the spicules of the glass sponge Euplectella aspergillum and failed to recover any sequence belonging to the silicatein family in this species. Similarly, we did not find any silicatein ortholog (except cathepsin L). Transcripts for silicatein were also not found in the hexactinellid Aphrocallistes vastus, nor in any transcriptome of hexactinellids published to date, suggesting silicatein is only present in demosponges [23, 160]. While some authors claimed to have purified silicateins directly from the skeleton of other hexactinellid species [161,162,163,164,165], these reports most likely reflects either contamination or quite divergent sequences in other glass sponges. In light of our results, the most plausible explanation is that hexactinellids and demosponges, as well as homoscleromorphs, use different enzymes to build their skeleton, which is also supported by the fact that Homoscleromorpha (the third class with siliceous spicules) lack both silicatein and glassin [160]. This hypothesis has profound implications since it suggests a convergent evolution of the ability to produce siliceous spicules. However, it is also possible that the numerous cathepsin L genes (the family to which silicatein belongs) present in glass sponges (and also homoscleromorphs) are involved in spiculogenesis; functional studies would have to be performed to test this hypothesis.

Biosilicification toolkit in O. minuta. a Summary of major biosilicification proteins involved in demosponge siliceous skeleton production. b Schematic of the biomineralization process in the sclerosyncytia of Hexactinellida. CA carbonic anhydrase, CS chitin synthase, NBSCA NBC (Na+/HCO3− [Si(OH)4]) transporter, Si biosilica. c Phylogenetic hypothesis obtained by maximum likelihood of cathepsin evolution of the major sponge lineages. The sequences obtained from the genome of O. minuta are shown in blue. Bootstrap values less than 70 are not shown. The list of sequences used and their accession numbers are available in Additional file 3: Table S21, the alignment is provided at https://zenodo.org/communities/oops_13 [166]
For silicon transport into the glass sponge sclerosyncytia [167, 168], the O. minuta genome encodes a NBCSA (Na+/HCO3−[Si(OH)4]) cotransporter, four aquaporin 9 genes, and two ArsB (arsenite–antimonite) transporters. The genes encoding silicon transporters (SITs) used by other silicifying organisms [169] were not found in the Oopsacas genome. The exact mechanisms of interaction between the different enzymes are poorly known, but the identification of the largest complement of genes used for biosilicification in Oopsacas is definitely a fundamental step towards the full characterization of the process in Hexactinellida.
Discussion
The Oopsacas minuta whole genome is the first reported for a glass sponge. It differs significantly from all other available sponge genomes in having a far smaller complement of predicted protein-coding genes. At only 16,413 genes, it is among the smallest of non-parasitic metazoan genomes reported so far [120, 170,171,172].
The most striking and unexpected feature of the Oopsacas genome is the absence of many genes that are typically considered ancestral and essential for metazoan morphogenetic processes (e.g., a functional Wnt pathway and numerous transcription factors). Because these genes are present in the three other sponge lineages, their absence in Oopsacas most likely indicates secondary losses given that current phylogenetic relationships place Hexactinellida as sister to Demospongiae (Fig. 1a).
These losses are not associated with a highly reduced body size and/or complexity or a parasitic lifestyle but instead may reflect the peculiar syncytial organization typical of this group. However, given the large differences documented between demosponge species [30], it would be premature to infer that these characteristics are shared by the nearly 700 other described hexactinellid species [13]. Both the ongoing sequencing of two other glass sponge species, Aphrocallistes vastus and the “tulip sponge” (Sally Leys and Darrin Schultz, personal communications) and the use of powerful phylogenetic-based annotation pipelines, such as the OMA (Orthologous matrix) database [173], should allow further valuable comparisons.
Interestingly, our work further documents the important role of convergence during animal evolution, by suggesting the independent emergence of the different mineral skeletons found in sponges, of septate-like junctions, of electrical signaling, and of multiciliated cells at an early stage of animal evolution.
We also report, for the first time, the association of a new Thaumarchaeota species (named Candidatus Cenarchaeum massiliensis) with a hexactinellid sponge. Thaumarchaeota species were only previously known to associate with Demospongiae [58]. An unusual enrichment of transposases in the C. massiliensis genome points to a high potential for lateral gene transfer [58].
Conclusion
Sponges (Porifera) are ecologically important in aquatic ecosystems because of their high number of species (> 9500) and also for the habitat they provide for many other animals. They emerged and diversified over 600 Myrs ago into four distinct lineages, each having distinct features. Here we report on the first whole-genome analysis of a glass sponge (Hexactinellida) and provide new insights on sponge biology and evolution. Despite a filter feeding body plan similar to that of other sponges, Oopsacas has unusual genomic features and a very reduced gene content. More generally because sponges are part of one of the most ancient still extant animal lineages, the study of their genomes provides valuable insights into the evolution of animal body plans. Our findings reinforce the idea that gene losses, convergent evolution of similar body features, and tight relationships with microorganisms were important processes in the early diversification of animals.
Methods
Sample collections
Adult individuals (which brood embryos and pre-larvae all year long) were collected in the 3PP cave, La Ciotat (France, 43° 9.797′ N, 5° 36.000′ E). Because this population lives in a deep cave, it is considered to be isolated from other populations (from other caves and canyons), thereby the heterozygosity is expected to be low. Samples were brought back to the laboratory in a cooler, then immediately cut and cleaned of superficial sediment and organisms under a stereomicroscope with brush and forceps, and rinsed in 0.2 µm filtered sea water to reduce the chance of contamination. Samples were either used freshly or stored at − 80 °C for future use.
Genomic DNA and RNA extraction and sequencing
Because of the low quantity of tissues present in an adult, in order to obtain sufficient material, one different adult was used for each RNA or DNA extraction mentioned below.
For Illumina sequencing, gDNA extractions were performed on 22 mg of sample using the QIAamp® DNA Mini kit from QIAGEN according to manufacturer instructions. DNA quality and quantity were checked by electrophoresis, NanoDrop, and Qubit fluorometer.
RNA extractions were performed (Qiagen kits) by the ProfileXpert IBISA platform. Sequencing was performed using Illumina technology with RNA-seq paired-ends, DNA-seq paired-ends, and Nextera Mate Pair protocols on a HiSeq2500 sequencer with a read length of 150 bp (ProfileXpert-LCMT, Lyon, France, http://profilexpert.fr).
For PacBio sequencing, tissues (1.34 g) were cut in pieces and stirred in calcium and magnesium-free sea water together with 20 mM EDTA (following the protocol described for Oscarella lobularis [174]). The suspension was passed through a 40-µm cell strainer to remove spicules and then concentrated by centrifugation (400 g -3 min). DNA extraction was performed on the cell suspension with the MasterPure™ Complete DNA kit (Tebu Bio) according to the manufacturer instructions. DNA quantity and quality were checked by gel electrophoresis, NanoDrop, and Qubit. Long-read sequencing was performed on a Pacific Biosciences platform (University of Lausanne, Swiss, https://wp.unil.ch/gtf/).
Unless explicitly otherwise noted, all bioinformatic analyses, programs, and software used the default options.
Genome assembly
Raw assembly
Genome sequencing was first done using the Illumina platform as previously described [38]. In addition, complementary sequencing was carried out on a PacBio platform to give a total of 751,460 long reads. These reads were filtered based on their length and quality with Pacific Bioscience tools (SMRT Portal) then self-corrected with Canu [175]. All Illumina reads were mapped to the corrected PacBio reads with Bowtie 2.3.4.1 [176]. Selected Illumina reads and corrected PacBio reads were then assembled together with spades v3.9 using options careful and Kmers 21, 41, 61, 81, and 99 [177].
Scaffold selection
Scaffolds longer than 1 kb were retained and submitted to MetaGeneMark v3.26 [178] followed by a blastp v2.2.26 [179] strategy against NR (best hits with evalue < 10−5). The best hit taxonomy of each gene was used in a LCA-like method where the taxonomy of each scaffold was assigned if at least half of the annotated genes had the same annotation. Scaffolds were then attributed to 4 groups: Eukaryota, Bacteria, Archaea, and Not Annotated (also containing viruses).
Scaffold polishing
All Illumina reads selected by mapping on the corrected PacBio reads during the raw assembly process were also used for polishing. The Eukaryota group (936 scaffolds) and the Archaea group (50 scaffolds) were individually submitted to SSPACE v3.9 [171] with parameters -× 0 -z 0 -k 5 -g 0 -a 0.7 -n 15 -p 0. The resulting super-scaffolds were then submitted to pilon v1.21 [172] with default parameters and Gapfiller v1.10 [173] with parameters -m 30 -o 2 -r 0.7 -n 10 -d 50 -t 10 -g 0 -i 1 to obtain 11 Archaea and 365 Eukaryota polished super-scaffolds which were regarded as the two draft genomes.
Assembly metrics and comparisons
The 11 Archaea scaffolds were submitted to Checkm v1.07 [53] with a completeness score of 99.03% without contamination.
To compare the genome quality of O. minuta to that of other sponges, the 16,413 genes of O. minuta CDS (coding sequences) were submitted to BUSCO v2/3 [64] through the gVolante server (https://gvolante.riken.jp/analysis.html) using the 303 Eukaryota core genes set. The absence of potential contamination by human DNA was checked by blast against NR database, yielding no hit with similarities higher than 64%.
Gene prediction and annotation of the dominant Thaumarchaeota
We predicted 1990 Archaea genes using GeneMarkS v4.6b and the --prok option. Each sequence was submitted to multiple annotation strategies. A blastp (Basic Local Alignment Search Tool) against NR with an e-value < 10−5 and using the 10 best hits was used as the main information for functional annotation. A domain search was performed against Pfam 28.0, TIGRFAM 15.0, SMART 6.2, ProDom 2006.1, PANTHER 9.0, Prosite 20.113, Hamap 201502.04 using interproscan v5.14-53 [180]. A CD-search [181] was done against the conserved domain database at NCBI (National Center for Biotechnology Information). Potential (trans)membrane proteins were predicted using Phobius [182]. Specific repeat domains were assessed using HMM (Hidden Markov models) search from the hmmer suite v3.1b1 [183] on different hmm profiles (ankyrin repeat, BTB/POZ domain, CASC3/Barentsz eIF4AIII binding, collagen triple helix repeat, DUF3420, DUF3447, F-box, MORN repeat, pentapeptide repeat). All predicted proteins shorter than 100 amino acids without match to at least one of the above methods were discarded. These results were manually integrated to improve the functional annotation of the 1675 remaining Thaumarchaeota proteins. All 20 standard transfer RNAs except for tryptophan were predicted using tRNA-scan-SE v1.3.1 [184, 185]. Each 23S, 16S, and 5S ribosomal RNAs were predicted using barrnap v0.7 (https://github.com/tseemann/barrnap) for the archaeal kingdom.
Thaumarchaeota orthology analysis
We created a specific Thaumarchaeota protein database by merging predicted proteins from the new Thaumarchaeota genome with those from nine Thaumarchaeota genomes previously published [54, 55, 57, 62, 186,187,188,189,190]. Proteins were assigned to OrthoMCL-DB groups or to clusters into new ortholog groups (specific to Thaumarchaeota) using the OrthoMCL algorithm and database [191].
Repeat analysis and gene predictions in Oopsacas minuta
The 365 contigs were scanned and masked for repeats using RepeatModeler 2.0.4 and RepeatMasker 4.1.4 (http://www.repeatmasker.org) from the Dfam TE tool container (https://github.com/Dfam-consortium/TETools).
Four masked versions of the genome were obtained combining soft masking (-xsmall), hardmasking with or without low complexity regions masking (-nolow) [192].
Transcriptomic reads were mapped to all Eukaryota scaffolds using TopHat v2.0.12 [180] with the following options: -i 20 -I 5000 --b2-very-sensitive --library-type fr-unstranded --no-discordant. The genes were predicted using Braker v1.9 [181] and Augustus v3.2.3 [182] on the different versions of the genome, masked or not. An aberrant protein of around 48,000 amino acids was removed from the predictions before the submission to BUSCO v2/v3 [58] through the gVolante server (https://gvolante.riken.jp/analysis.html) using the 303 Eukaryota core genes set (OrthoDB v9). The results, summarized in Additional file 2: Table S7, show that for a comparable complete BUSCO score, the lowest duplication level was obtained with the predictions from the unmasked version of the genome.
The 17,059 predicted genes were then annotated and manually curated using different criteria. Genes shorter than 100 amino acids with a null transcriptomic coverage and alternative transcripts not supported by Augustus were removed. The 16,444 remaining genes were submitted to NCBI Tbl2asn and all discrepancies corrected (33 genes removed accordingly). Two genes manually detected during specific annotation were added, leading to a final dataset of 16,413 genes.
All proteins were annotated on the basis of the best hit using blastp against NR (evalue < 10−5). To infer Gene Ontology for each protein, a domain search was performed using InterProScan on the same databases as for Thaumarchaeota and the GO identifiers were used on WEGO (http://wego.genomics.org.cn/). Results on the first two levels are presented in the Additional file 1 for the 7737 genes that received a GO annotation (Additional file 1: Table S6 and Fig. S5) [193].
All 20 standard tRNAs as well as initiator methionyl-tRNA, selenocystein tRNA, and suppressor tRNA were predicted with at least one copy using tRNA-scan-SE for the eukaryotic model. 28S, 18S, 5.8S, and 5S ribosomal RNAs were identified using barrnap for the eukaryotes.
More detailed annotations were performed for a number of candidate genes (see “Result” subsections). For this purpose, blastP was used to search for specific proteins of interest in the predicted proteome of O. minuta. The returned hits with e-value < 10−2 were then checked by a reciprocal best-hit approach against NR database (NCBI). When needed, additional analyses were carried out: phylogenetic analyses and protein domain analyses were used for some sequences, and the specific methods used are indicated in the comments of the corresponding table or figure (Additional file 3). The list of query sequences and of best-hits obtained at the reciprocal best-hit step are provided in Additional file 3: Tables S7 through Table S18. Supporting phylogenetic analyses or domain prediction are provided in Additional file 3: Figs. S8 to S11 (for corresponding datasets see [166, 194,195,196,197]).
Global genome comparisons
A systematic orthology clustering was performed using orthofinder v2.3.12 [183] on 6 representative sponge proteomes: Oopsacas minuta (16,413 protein sequences from the present study), Oscarella pearsei (29,220 protein sequences), Sycon ciliatum (50,731 protein sequences), Tethya wilhelma (37,633 protein sequences), Amphimedon queenslandica (23,542 protein sequences), and Ephydatia muelleri (39,329 protein sequences). Corresponding source datasets used for comparison are available at:
Oscarella pearsei (previously named O. carmela): https://web.archive.org/web/20190531133238/http://www.compagen.org/datasets/OCAR_T-PEP_130911.zip
Sycon ciliatum: https://web.archive.org/web/20170108095659/http://compagen.org/datasets/SCIL_T-PEP_130802.zip
Tethya wilhelma: https://bitbucket.org/molpalmuc/tethya_wilhelma-genome/src/master/gene_sets/twilhelma_v01_augustus_prots.fasta.gz
Amphimedon queenslandica: https://ftp-ncbi-nlm-nih-gov.insb.bib.cnrs.fr/genomes/all/GCF/000/090/795/GCF_000090795.2_v1.1/GCF_000090795.2_v1.1_protein.faa.gz
Ephydatia muelleri: https://bitbucket.org/EphydatiaGenome/ephydatiagenome/downloads/Emu_v1_prots.fasta.gz
Localization of the main Thaumarchaeota species associated to O. minuta
O. minuta adults were collected from the 3PP cave in December 2014 (N = 3) and in August 2015 (N = 2) and preserved in 4% paraformaldehyde (PFA). After dehydration using ethanol, samples were embeded in paraffin wax. To obtain 8-µm sections, samples were desilicified in 5% hydrofluoric acid en-bloc for 8 min at room temperature; the block was rinsed with distilled water before sectioning.
After dewaxing with Neoclear®, endogenenous peroxydase was inhibited by adding 0.3% hydrogen peroxide in the first of 2 ethanol washes for 15 min, then sections were rehydrated and permeabilized with proteinase K (2 µg ml−1) for 45 min. Sections were post-fixed with 4% PFA for 15 min and Card-FISH (catalyzed reporter deposition fluorescent in situ hybridization) was carried out following protocols from [198, 199] (detailed protocols provided upon request).
The most abundant thaumarchaeotal species was localized using a specifically designed probe: THAUMOOPS840: CATTAGTACCGCTTCAGACC- HRP (horseradish peroxidase).
Negative controls consisted of the absence of probe (with or without TSA, tyramide signal amplification) or the use of a random sequence not 100% matching any sequence of the metagenome: NONOOPS02: GGTTCCTTAGTCACGCAGAA-HRP. We observed that the sponge tissue had an endogenous peroxidase activity (green background) that was incompletely abolished by 0.3% H2O2. Unfortunately, higher concentrations of hydrogen peroxide damaged the tissues to a point preventing localization.
Availability of data and materials
Most datasets supporting the conclusions of this article are included within the article and its additional file(s).
Sequences used to build HHMER profiles or alignments (phylogenetic analyses) are publicly available at https://zenodo.org/communities/oops_13/ [166, 194,195,196,197].
Transcriptomes and genomes are available at GenBank under the BioProject accession PRJNA761294 [200].
The Whole Genome Shotgun project of Oopsacas minuta is available under the accession JAKMXF000000000 [201].
The Whole Genome Shotgun project of the Thaumarchaeota species Candidatus Cenarchaeum massiliensis is available under the accession JAJIZT000000000 [202].
The reads dataset are available at the Sequence Read Archive (https://identifiers.org/insdc.sra:SRR23032961, https://identifiers.org/insdc.sra:SRR23032960, https://identifiers.org/insdc.sra:SRR23032959, https://identifiers.org/insdc.sra:SRR23032958, https://identifiers.org/insdc.sra:SRR23032957, https://identifiers.org/insdc.sra:SRR23032956, https://identifiers.org/insdc.sra:SRR23032955, https://identifiers.org/insdc.sra:SRR23032954, https://identifiers.org/insdc.sra:SRR23032953, https://identifiers.org/insdc.sra:SRR23032952).
Abbreviations
- AJ:
-
Adherens junctions
- AMO:
-
Ammonia monooxygenase
- ANTP:
-
Antennapedia
- AOA:
-
Ammonium-oxidizing archaea
- ARNT:
-
Aryl hydrocarbon receptor nuclear translocator
- ArsB:
-
Arsenite-antimonite (transporter)
- bHLH:
-
Basic helix loop helix
- BLAST:
-
Basic local alignment search tool
- BM:
-
Basement membrane
- BMAL:
-
Brain and muscle Arnt-like
- bp:
-
Base pair
- BUSCO:
-
Benchmarking Universal Single-Copy Orthologs
- Ca. C. massiliensis :
-
Candidatus Cenarchaeum massiliensis
- Card-FISH:
-
Catalyzed reporter deposition fluorescent in situ hybridization
- CCC:
-
Cadherin–catenin complexes
- CDS:
-
Coding sequences
- CEP:
-
Centrosome-associated protein
- CerS:
-
Ceramid synthase
- CJ:
-
Cell junction
- Cont:
-
Contactin
- CGN:
-
Cyclic-gated nucleotide
- CCNO:
-
Cyclin O
- DNA:
-
Deoxyribonucleic acid
- ECM:
-
Extracellular matrix
- ENaC:
-
Epithelial sodium channel
- FA:
-
Focal adhesion
- FoxJ1:
-
Forkhead box J1
- Fzd:
-
Frizzled
- GABA:
-
Gamma-aminobutyric acid
- G + C content:
-
Guanine + cytosine content
- GJ:
-
Gap junction
- GMNC:
-
Geminin coiled-coil domain containing
- GO:
-
Gene Ontology
- GPCR:
-
G protein-coupled receptor
- HD:
-
Homeodomain
- HDs:
-
Hemidesmosomes
- Hhex:
-
Hematopoietically expressed homeobox
- HMM:
-
Hidden Markov models
- HNF:
-
Hepatocyte nuclear factor
- HRP:
-
Horseradish peroxidase
- iGluR:
-
Ionotropic glutamate receptors
- IgSF:
-
Immunoglobulin superfamily
- Kb:
-
Kilo bases
- Kv:
-
Voltage-gated potassium channels
- Lam:
-
Laminins
- LCAM:
-
Last common ancestor of metazoans
- Mb:
-
Megabases
- MCC:
-
Multiciliated cell
- Mcidas:
-
Multiciliate differentiation and DNA synthesis associated cell
- MKX:
-
Mohawk homeobox
- Myb:
-
Myeloblastosis proto-oncogen
- Myrs:
-
Million years
- NAV:
-
Voltage gated sodium channels
- NBCSA:
-
(Na+/HCO3−[Si(OH)4]) co-transporter
- NCBI:
-
National Center for Biotechnology Information
- NR:
-
Non-redundant databases
- NO:
-
Nitric oxide
- PacBio:
-
Pacific Biosciences
- PA:
-
Per-Arnt-Sim domain
- PRD:
-
Pair-Ruled
- PLK4:
-
Polo-like kinase 4
- PROS:
-
Prospero
- SAS6:
-
Spindle assembly abnormal 6
- SIT:
-
Silicon transporters
- SIX:
-
Sine oculis homeobox
- SJ:
-
Septate junctions
- SNAP:
-
Soluble N-ethylmaleimide-sensitive factor-attachment protein
- SNARE:
-
SNAP receptor
- STIL:
-
SCL/TAL1 interrupting locus
- TF:
-
Transcription factors
- TGF-β:
-
Transforming growth factor-beta
- TRP:
-
Transient receptor potential
- TSA:
-
Tyramide signal amplification
- ZF:
-
Zinc finger
References
Dunn CW, Ryan JF. The evolution of animal genomes. Curr Opin Genet Dev. 2015;35:25–32.
Paps J, Holland PWH. Reconstruction of the ancestral metazoan genome reveals an increase in genomic novelty. Nat Commun. 2018;9(1):1730.
Renard E, Leys SP, Wörheide G, Borchiellini C. Understanding animal evolution: the added value of sponge transcriptomics and genomics. BioEssays. 2018;40(9):e1700237.
de Mendoza A, Hatleberg WL, Pang K, Leininger S, Bogdanovic O, Pflueger J, et al. Convergent evolution of a vertebrate-like methylome in a marine sponge. Nat Ecol Evol. 2019;3(10):1464–73.
Erives A, Fritzsch B. A screen for gene paralogies delineating evolutionary branching order of early metazoa. G3 (Bethesda). 2019;10(2):811–26.
Feuda R, Dohrmann M, Pett W, Philippe H, Rota-Stabelli O, Lartillot N, et al. Improved modeling of compositional heterogeneity supports sponges as sister to all other animals. Curr Biol. 2017;27(24):3864–3870.e4.
Laumer CE, Fernández R, Lemer S, Combosch D, Kocot KM, Riesgo A, et al. Revisiting metazoan phylogeny with genomic sampling of all phyla. Proc Biol Sci. 1906;2019(286):20190831.
Redmond AK, McLysaght A. Evidence for sponges as sister to all other animals from partitioned phylogenomics with mixture models and recoding. Nat Commun. 2021;12(1):1783.
Simion P, Philippe H, Baurain D, Jager M, Richter DJ, Di Franco A, et al. A large and consistent phylogenomic dataset supports sponges as the sister group to all other animals. Curr Biol. 2017;27(7):958–67.
Whelan NV, Kocot KM, Moroz TP, Mukherjee K, Williams P, Paulay G, et al. Ctenophore relationships and their placement as the sister group to all other animals. Nat Ecol Evol. 2017;1(11):1737–46.
Dunn CW, Leys SP, Haddock SHD. The hidden biology of sponges and ctenophores. Trends Ecol Evol. 2015;30(5):282–91.
Dohrmann M, Wörheide G. Dating early animal evolution using phylogenomic data. Sci Rep. 2017;7(1):3599.
van Soest R, Boury-Esnault N, Hooper J, Rützler K, de Voogd N, Alvarez B, et al. World Porifera database. 2022.
Borchiellini C, de Pao Mendonca K, Vernale A, Rocher C, Ereskovsky A, Vacelet J, et al. Porifera (sponges): recent knowledge and new perspectives. in : eLS, John Wiley & Sons, Ltd (Ed.): Chichester. 2021;2:1–10.
Colgren J, Nichols SA. The significance of sponges for comparative studies of developmental evolution. WIREs Dev Biol. 2020;9(2):e359.
Degnan BM, Adamska M, Richards GS, Larroux C, Leininger S, Bergum B, et al. Porifera. In: Wanninger A, editor., et al., Evolutionary developmental biology of invertebrates. Vienna: Springer Verlag; 2015:65–106.
Ereskovsky AV, Renard E, Borchiellini C. Cellular and molecular processes leading to embryo formation in sponges: evidences for high conservation of processes throughout animal evolution. Dev Genes Evol. 2013;223(1–2):5–22.
Fahey B, Degnan BM. Origin of animal epithelia: insights from the sponge genome: Evolution of epithelia. Evol Dev. 2010;12(6):601–17.
Fernandez-Valverde SL, Calcino AD, Degnan BM. Deep developmental transcriptome sequencing uncovers numerous new genes and enhances gene annotation in the sponge Amphimedon queenslandica. BMC Genomics. 2015;16(1):387.
Srivastava M, Simakov O, Chapman J, Fahey B, Gauthier MEA, Mitros T, et al. The Amphimedon queenslandica genome and the evolution of animal complexity. Nature. 2010;466(7307):720–6.
Adamska M, Degnan BM, Green K, Zwafink C. What sponges can tell us about the evolution of developmental processes. Zoology (Jena). 2011;114(1):1–10.
Gazave E, Lapébie P, Richards GS, Brunet F, Ereskovsky AV, Degnan BM, et al. Origin and evolution of the Notch signalling pathway: an overview from eukaryotic genomes. BMC Evol Biol. 2009;9:249.
Riesgo A, Farrar N, Windsor PJ, Giribet G, Leys SP. The analysis of eight transcriptomes from all poriferan classes reveals surprising genetic complexity in sponges. Mol Biol Evol. 2014;31(5):1102–20.
Schenkelaars Q, Fierro-Constain L, Renard E, Hill AL, Borchiellini C. Insights into Frizzled evolution and new perspectives. Evol Dev. 2015;17(2):160–9.
Schenkelaars Q, Fierro-Constain L, Renard E, Borchiellini C. Retracing the path of planar cell polarity. BMC Evol Biol. 2016;16:69.
Schenkelaars Q, Pratlong M, Kodjabachian L, Fierro-Constain L, Vacelet J, Le Bivic A, et al. Animal multicellularity and polarity without Wnt signaling. Sci Rep. 2017;7(1):15383.
Ereskovsky AV, Richter DJ, Lavrov DV, Schippers KJ, Nichols SA. Transcriptome sequencing and delimitation of sympatric Oscarella species (O. carmela and O. pearsei sp. nov) from California, USA. PLoS One. 2017;12(9):e0183002(9).
Nichols SA, Roberts BW, Richter DJ, Fairclough SR, King N. Origin of metazoan cadherin diversity and the antiquity of the classical cadherin/β-catenin complex. Proc Natl Acad Sci U S A. 2012;109(32):13046–51.
Francis WR, Eitel M, R SV, Adamski M, Haddock SH, Krebs S, et al. The genome of the contractile demosponge Tethya wilhelma and the evolution of metazoan neural signalling pathways. bioRxiv. 2017:120998.
Kenny NJ, Francis WR, Rivera-Vicéns RE, Juravel K, de Mendoza A, Díez-Vives C, et al. Tracing animal genomic evolution with the chromosomal-level assembly of the freshwater sponge Ephydatia muelleri. Nat Commun. 2020;11(1):3676.
Fortunato SAV, Adamski M, Ramos OM, Leininger S, Liu J, Ferrier DEK, et al. Calcisponges have a ParaHox gene and dynamic expression of dispersed NK homeobox genes. Nature. 2014;514(7524):620–3.
Belahbib H, Renard E, Santini S, Jourda C, Claverie JM, Borchiellini C, et al. New genomic data and analyses challenge the traditional vision of animal epithelium evolution. BMC Genomics. 2018;19:393.
Fierro-Constaín L, Schenkelaars Q, Gazave E, Haguenauer A, Rocher C, Ereskovsky A, et al. The conservation of the germline multipotency program, from sponges to vertebrates: a stepping stone to understanding the somatic and germline origins. Genome Biol Evol. 2017;9(3):474–88.
Boury-Esnault N, Vacelet J. Preliminary studies on the organization and development of a hexactinellid sponge from a Mediterranean cave, Oopsacas minuta. In: Van Soest R, Van Kempen ThV, Braekman J-C, editors. Sponges in time and space. Rotterdam: Balkema; 1994. p. 407–15.
Vacelet J, Boury-Esnault N, Harmelin JG. Hexactinellid cave, a unique deep-sea habitat in the scuba zone. Deep Sea Res Part I Oceanogr Res Pap. 1994;41(7):965–73.
Leys SP, Mackie GO, Reiswig HM. The biology of glass sponges. In: Advances in marine biology. Academic Press. 2007;52:1–145.
Overstreet RM, Lotz JM. Host–symbiont relationships: understanding the change from guest to pest. In: The Rasputin effect: when commensals and symbionts become parasitic. 2016;3:27–64.
Jourda C, Santini S, Rocher C, Le Bivic A, Claverie JM. Draft Genome sequence of an alphaproteobacterium associated with the mediterranean sponge Oscarella lobularis. Genome Announc. 2015;3(5):e00977-15.
Thomas T, Moitinho-Silva L, Lurgi M, Björk JR, Easson C, Astudillo-García C, et al. Diversity, structure and convergent evolution of the global sponge microbiome. Nat Commun. 2016;7:11870.
Roué M, Quévrain E, Domart-Coulon I, Bourguet-Kondracki ML. Assessing calcareous sponges and their associated bacteria for the discovery of new bioactive natural products. Nat Prod Rep. 2012;29(7):739–51.
Roux S, Brum JR, Dutilh BE, Sunagawa S, Duhaime MB, Loy A, et al. Ecogenomics and potential biogeochemical impacts of globally abundant ocean viruses. Nature. 2016;537(7622):689–93.
Hingamp P, Grimsley N, Acinas SG, Clerissi C, Subirana L, Poulain J, et al. Exploring nucleo-cytoplasmic large DNA viruses in Tara Oceans microbial metagenomes. ISME J. 2013;7(9):1678–95.
Winter C, Garcia JAL, Weinbauer MG, DuBow MS, Herndl GJ. Comparison of deep-water viromes from the atlantic ocean and the mediterranean sea. PLoS One. 2014;9(6):e100600.
Pascelli C, Laffy PW, Kupresanin M, Ravasi T, Webster NS. Morphological characterization of virus-like particles in coral reef sponges. PeerJ. 2018;6:e5625.
Claverie JM, Grzela R, Lartigue A, Bernadac A, Nitsche S, Vacelet J, et al. Mimivirus and Mimiviridae: giant viruses with an increasing number of potential hosts, including corals and sponges. J Invertebr Pathol. 2009;101(3):172–80.
Laffy PW, Wood-Charlson EM, Turaev D, Weynberg KD, Botté ES, van Oppen MJH, et al. HoloVir: a workflow for investigating the diversity and function of viruses in invertebrate holobionts. Front Microbiol. 2016;7:822.
Laffy PW, Wood-Charlson EM, Turaev D, Jutz S, Pascelli C, Botté ES, et al. Reef invertebrate viromics: diversity, host specificity and functional capacity. Environ Microbiol. 2018;20(6):2125–41.
Waldron FM, Stone GN, Obbard DJ. Metagenomic sequencing suggests a diversity of RNA interference-like responses to viruses across multicellular eukaryotes. PLoS Genet. 2018;14(7):e1007533.
Butina TV, Bukin YS, Khanaev IV, Kravtsova LS, Maikova OO, Tupikin AE, et al. Metagenomic analysis of viral communities in diseased Baikal sponge Lubomirskia baikalensis. Limnol Freshw Biol. 2019;1:155–62.
Soffer N, Brandt ME, Correa AM, Smith TB, Thurber RV. Potential role of viruses in white plague coral disease. ISME J. 2014;8(2):271–83.
Dennis TPW, Flynn PJ, de Souza WM, Singer JB, Moreau CS, Wilson SJ, et al. Insights into circovirus host range from the genomic fossil record. J Virol. 2018;92(16):e00145-18.
Ishijima J, Iwabe N, Masuda Y, Watanabe Y, Matsuda Y. Sponge cytogenetics - mitotic chromosomes of ten species of freshwater sponge. Zool Sci. 2008;25(5):480–6.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25(7):1043–55.
Spang A, Poehlein A, Offre P, Zumbrägel S, Haider S, Rychlik N, et al. The genome of the ammonia-oxidizing Candidatus Nitrososphaera gargensis: insights into metabolic versatility and environmental adaptations. Environ Microbiol. 2012;14(12):3122–45.
Zhalnina KV, Dias R, Leonard MT, Dorr de Quadros P, Camargo FAO, Drew JC, et al. Genome sequence of Candidatus Nitrososphaera evergladensis from group I.1b enriched from Everglades soil reveals novel genomic features of the ammonia-oxidizing archaea. PLoS One. 2014;9(7):e101648.
Kerou M, Offre P, Valledor L, Abby SS, Melcher M, Nagler M, et al. Proteomics and comparative genomics of Nitrososphaera viennensis reveal the core genome and adaptations of archaeal ammonia oxidizers. Proc Natl Acad Sci U S A. 2016;113(49):E7937–46.
Stieglmeier M, Klingl A, Alves RJE, Rittmann SKMR, Melcher M, Leisch N, et al. Nitrososphaera viennensis gen. nov., sp. nov., an aerobic and mesophilic, ammonia-oxidizing archaeon from soil and a member of the archaeal phylum Thaumarchaeota. Int J Syst Evol Microbiol. 2014;64(Pt 8):2738–52.
Haber M, Burgsdorf I, Handley KM, Rubin-Blum M, Steindler L. Genomic insights into the lifestyles of Thaumarchaeota inside sponges. Front Microbiol. 2021;11:622824.
Krupovic M, Makarova KS, Wolf YI, Medvedeva S, Prangishvili D, Forterre P, et al. Integrated mobile genetic elements in Thaumarchaeota. Environ Microbiol. 2019;21(6):2056–78.
Martens JH, Barg H, Warren MJ, Jahn D. Microbial production of vitamin B12. Appl Microbiol Biotechnol. 2002;58(3):275–85.
Roth J, Lawrence J, Bobik T. COBALAMIN (COENZYME B12): synthesis and biological significance. Annu Rev Microbiol. 1996;50(1):137–81.
Hallam SJ, Konstantinidis KT, Putnam N, Schleper C, Watanabe YI, Sugahara J, et al. Genomic analysis of the uncultivated marine crenarchaeote Cenarchaeum symbiosum. Proc Natl Acad Sci U S A. 2006;103(48):18296–301.
Jeffery NW, Jardine CB, Gregory TR. A first exploration of genome size diversity in sponges. Genome. 2013;56(8):451–6.
Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31(19):3210–2.
Santini S, Renard E. Gene Ontology at level 3 of orthogroups present in Oopsacas and absent in all other sponge genomes. Zenodo; 2023. https://zenodo.org/record/7774129.
Leys SP, Cheung E, Boury-Esnault N. Embryogenesis in the glass sponge Oopsacas minuta: formation of syncytia by fusion of blastomeres. Integr Comp Biol. 2006;46(2):104–17.
Leys SP. The significance of syncytial tissues for the position of the hexactinellida in the metazoa. Integr Comp Biol. 2003;43(1):19–27.
Leys SP, Zaman AK, Boury-Esnault N. Three-dimensional fate mapping of larval tissues through metamorphosis in the glass sponge Oopsacas minuta. Invertebr Biol. 2016;135(3):259–72.
Mackie GO, Singla CL. Studies on hexactinellid sponges - studies on hexactinellid sponges. I. Histology of Rhabdocalyptus dawsoni (Lambe, 1873). Phil Trans R Soc Lond B. 1983;301(1107):365–400.
Abedin M, King N. Diverse evolutionary paths to cell adhesion. Trends Cell Biol. 2010;20(12):734–42.
Fidler AL, Darris CE, Chetyrkin SV, Pedchenko VK, Boudko SP, Brown KL, et al. Collagen IV and basement membrane at the evolutionary dawn of metazoan tissues. Elife. 2017;6:e24176.
Leys SP, Riesgo A. Epithelia, an evolutionary novelty of metazoans. J Exp Zool (Mol Dev Evol). 2012;318(6):438–47.
Leys SP, Nichols SA, Adams EDM. Epithelia and integration in sponges. Integr Comp Biol. 2009;49(2):167–77.
Miller PW, Clarke DN, Weis WI, Lowe CJ, Nelson WJ. The evolutionary origin of epithelial cell-cell adhesion mechanisms. Curr Top Membr. 2013;72:267–311.
Renard E, Le Bivic A, Borchiellini C. Origin and evolution of epithelial cell types. Origin and evolution of metazoan cell types. In: Leys S, Hejnol A, editors. Origin and evolution of metazoan cell types. 1st ed. CRC Press Boca Raton. 2021;5:75–100.
Tyler S. Epithelium—the primary building block for metazoan complexity1. Integr Comp Biol. 2003;43(1):55–63.
Alexopoulos H, Böttger A, Fischer S, Levin A, Wolf A, Fujisawa T, et al. Evolution of gap junctions: the missing link? Curr Biol. 2004;14(20):R879–80.
Clarke DN, Miller PW, Lowe CJ, Weis WI, Nelson WJ. Characterization of the cadherin-catenin complex of the sea anemone Nematostella vectensis and implications for the evolution of metazoan cell-cell adhesion. Mol Biol Evol. 2016;33(8):2016–29.
Ganot P, Zoccola D, Tambutté E, Voolstra CR, Aranda M, Allemand D, et al. Structural molecular components of septate junctions in cnidarians point to the origin of epithelial junctions in eukaryotes. Mol Biol Evol. 2015;32(1):44–62.
Takaku Y, Hwang JS, Wolf A, Böttger A, Shimizu H, David CN, et al. Innexin gap junctions in nerve cells coordinate spontaneous contractile behavior in Hydra polyps. Sci Rep. 2014;4(1):3573.
Tucker RP, Adams JC. Chapter eight - adhesion networks of cnidarians: a postgenomic view. In: Jeon KW, editor. International Review of cell and molecular biology Tennessee. Academic Press; 2014;308:323–77.
De Pascalis C, Etienne-Manneville S. Single and collective cell migration: the mechanics of adhesions. Mol Biol Cell. 2017;28(14):1833–46.
Magie CR, Martindale MQ. Cell-cell adhesion in the cnidaria: insights into the evolution of tissue morphogenesis. Biol Bull. 2008;214(3):218–32.
Miller PW, Pokutta S, Mitchell JM, Chodaparambil JV, Clarke DN, Nelson WJ, et al. Analysis of a vinculin homolog in a sponge (phylum Porifera) reveals that vertebrate-like cell adhesions emerged early in animal evolution. J Biol Chem. 2018;293(30):11674–86.
Walko G, Castañón MJ, Wiche G. Molecular architecture and function of the hemidesmosome. Cell Tissue Res. 2015;360(2):363–78.
Mitchell JM, Nichols SA. Diverse cell junctions with unique molecular composition in tissues of a sponge (Porifera). EvoDevo. 2019;10:26.
Nichols SA, Dirks W, Pearse JS, King N. Early evolution of animal cell signaling and adhesion genes. Proc Natl Acad Sci USA. 2006;103(33):12451–6.
Fidler AL, Boudko SP, Rokas A, Hudson BG. The triple helix of collagens – an ancient protein structure that enabled animal multicellularity and tissue evolution. J Cell Sci. 2018;131(7):jcs203950.
Medwig TN, Matus DQ. Breaking down barriers: the evolution of cell invasion. Curr Opin Genet Dev. 2017;47:33–40.
Pozzi A, Yurchenco PD, Iozzo RV. The nature and biology of basement membranes. Matrix Biol. 2017;57–58:1–11.
Sekiguchi R, Yamada KM. Chapter four - basement membranes in development and disease. In: Litscher ES, Wassarman PM, editors. Current Topics in developmental biology. NY: Academic Press. 2018;130:143–91.
Fahey B, Degnan BM. Origin and evolution of laminin gene family diversity. Mol Biol Evol. 2012;29(7):1823–36.
Whittaker CA, Bergeron KF, Whittle J, Brandhorst BP, Burke RD, Hynes RO. The echinoderm adhesome. Dev Biol. 2006;300(1):252–66.
Ereskovsky A. The comparative embryology of sponges. Dordrecht: Springer; 2010.
Boute N, Exposito JY, Boury-Esnault N, Vacelet J, Noro N, Miyazaki K, et al. Type IV collagen in sponges, the missing link in basement membrane ubiquity. Biol Cell. 1996;88(1–2):37–44.
Boutin C, Kodjabachian L. Biology of multiciliated cells. Curr Opin Genet Dev. 2019;56:1–7.
Basquin C, Orfila AM, Azimzadeh J. Chapter 13 - The planarian Schmidtea mediterranea as a model for studying motile cilia and multiciliated cells. In: Basto R, Marshall WF, editors. Methods in cell biology. Academic Press. 2015;127:243–62.
Rink JC, Vu HTK, Sánchez AA. The maintenance and regeneration of the planarian excretory system are regulated by EGFR signaling. Development. 2011;138(17):3769–80.
Rompolas P, Azimzadeh J, Marshall WF, King SM. Chapter Twelve - analysis of ciliary assembly and function in planaria. In: Marshall WF, editor. Methods in enzymology. Academic Press. 2013;525:245–64.
Tamm S, Tamm SL. Development of macrociliary cells in Beroe. I. Actin bundles and centriole migration. J Cell Sci. 1988;89(1):67–80.
Thi-Kim VuH, Rink JC, McKinney SA, McClain M, Lakshmanaperumal N, Alexander R, et al. Stem cells and fluid flow drive cyst formation in an invertebrate excretory organ. eLife. 2015;4:e07405.
Tyler S. Development of cilia in embryos of the turbellarian Macrostomum. Hydrobiologia. 1981;84(1):231–9.
Hsu J, Sage J. Novel functions for the transcription factor E2F4 in development and disease. Cell Cycle. 2016;15(23):3183–90.
Arquint C, Nigg EA. The PLK4–STIL–SAS-6 module at the core of centriole duplication. Biochem Soc Trans. 2016;44(5):1253–63.
Azimzadeh J, Wong ML, Downhour DM, Alvarado AS, Marshall WF. Centrosome loss in the evolution of planarians. Science. 2012;335(6067):461–3.
Boury-Esnault N, Efremova S, Bézac C, Vacelet J. Reproduction of a hexactinellid sponge: first description of gastrulation by cellular delamination in the Porifera. Invertebr Reprod Dev. 1999;35(3):187–201.
Riesgo A, Taylor C, Leys SP. Reproduction in a carnivorous sponge: the significance of the absence of an aquiferous system to the sponge body plan. Evol Dev. 2007;9(6):618–31.
de Mendoza A, Sebé-Pedrós A, Šestak MS, Matejčić M, Torruella G, Domazet-Lošo T, et al. Transcription factor evolution in eukaryotes and the assembly of the regulatory toolkit in multicellular lineages. PNAS. 2013;110(50):E4858–66.
Vij S, Rink JC, Ho HK, Babu D, Eitel M, Narasimhan V, et al. Evolutionarily ancient association of the FoxJ1 transcription factor with the motile ciliogenic program. PLoS Genet. 2012;8(11):e1003019.
Biesalski HK, Doepner G, Tzimas G, Gamulin V, Schröder HC, Batel R, et al. Modulation of myb gene expression in sponges by retinoic acid. Oncogene. 1992;7(9):1765–74.
Nabais C, Peneda C, Bettencourt-Dias M. Evolution of centriole assembly. Curr Biol. 2020;30(10):R494-502.
Adamska M. Developmental signalling and emergence of animal multicellularity. In: Ruiz-Trillo I, Nedelcu AM, editors. Evolutionary transitions to multicellular life: principles and mechanisms. Advances in Marine Genomics. Springer, Dordrecht: Netherlands. 2015;2:425–50.
Babonis LS, Martindale MQ. Phylogenetic evidence for the modular evolution of metazoan signalling pathways. Philos Trans R Soc Lond B Biol Sci. 2017;372(1713):20150477.
Brunet T, King N. The origin of animal multicellularity and cell differentiation. Dev Cell. 2017;43(2):124–40.
Ispolatov I, Ackermann M, Doebeli M. Division of labour and the evolution of multicellularity. Proc Biol Sci. 2012;279(1734):1768–76.
Niklas KJ. The evolutionary-developmental origins of multicellularity. Am J Bot. 2014;101(1):6–25.
Richards GS, Degnan BM. The dawn of developmental signaling in the metazoa. Cold Spring Harb Symp Quant Biol. 2009;74:81–90.
Sebé-Pedrós A, de Mendoza A. Transcription factors and the origin of animal multicellularity. In: Ruiz-Trillo I, Nedelcu AM, editors. Evolutionary transitions to multicellular life: principles and mechanisms. Advances in Marine Genomics. Springer, Dordrecht, Netherlands. 2015;2:379–94.
Windsor Reid PJ, Matveev E, McClymont A, Posfai D, Hill AL, Leys SP. Wnt signaling and polarity in freshwater sponges. BMC Evol Biol. 2018;18(1):12.
Chang ES, Neuhof M, Rubinstein ND, Diamant A, Philippe H, Huchon D, et al. Genomic insights into the evolutionary origin of Myxozoa within Cnidaria. Proc Natl Acad Sci USA. 2015;112(48):14912–7.
Loh KM, van Amerongen R, Nusse R. Generating cellular diversity and spatial form: Wnt signaling and the evolution of multicellular animals. Dev Cell. 2016;38(6):643–55.
Oka Y, Saraiva LR, Kwan YY, Korsching SI. The fifth class of Galpha proteins. Proc Natl Acad Sci USA. 2009;106(5):1484–9.
Lokits AD, Indrischek H, Meiler J, Hamm HE, Stadler PF. Tracing the evolution of the heterotrimeric G protein α subunit in Metazoa. BMC Evol Biol. 2018;18(1):1–27.
Bastiani CA, Gharib S, Simon MI, Sternberg PW. Caenorhabditis elegans Galphaq regulates egg-laying behavior via a PLCbeta-independent and serotonin-dependent signaling pathway and likely functions both in the nervous system and in muscle. Genetics. 2003;165(4):1805–22.
Katanayeva N, Kopein D, Portmann R, Hess D, Katanaev VL. Competing activities of heterotrimeric G proteins in Drosophila wing maturation. PLoS One. 2010;5(8):e12331.
Macias-Muñoz A, Murad R, Mortazavi A. Molecular evolution and expression of opsin genes in Hydra vulgaris. BMC Genomics. 2019;20(1):992.
Wright SC, Cañizal MCA, Benkel T, Simon K, Le Gouill C, Matricon P, et al. FZD5 is a Gαq-coupled receptor that exhibits the functional hallmarks of prototypical GPCRs. Sci Signal. 2018;11(559):eaar5536.
Jones S. An overview of the basic helix-loop-helix proteins. Genome Biol. 2004;5(6):226.
Fortunato SAV, Vervoort M, Adamski M, Adamska M. Conservation and divergence of bHLH genes in the calcisponge Sycon ciliatum. EvoDevo. 2016;7:23.
Richards GS, Simionato E, Perron M, Adamska M, Vervoort M, Degnan BM. Sponge genes provide new insight into the evolutionary origin of the neurogenic circuit. Curr Biol. 2008;18(15):1156–61.
Simionato E, Ledent V, Richards G, Thomas-Chollier M, Kerner P, Coornaert D, et al. Origin and diversification of the basic helix-loop-helix gene family in metazoans: insights from comparative genomics. BMC Evol Biol. 2007;7(1):33.
Bürglin TR, Affolter M. Homeodomain proteins: an update. Chromosoma. 2016;125:497–521.
Ferrier DEK. Evolution of homeobox gene clusters in animals: the Giga-cluster and primary vs. secondary clustering. Front Ecol Evol. 2016;4:36.
Garcia-Fernàndez J. The genesis and evolution of homeobox gene clusters. Nat Rev Genet. 2005;6(12):881–92.
Fortunato SA, Leininger S, Adamska M. Evolution of the Pax-Six-Eya-Dach network: the calcisponge case study. EvoDevo. 2014;5(1):23.
Ryan JF, Pang K, Mullikin JC, Martindale MQ, Baxevanis AD, NISC Comparative Sequencing Program. The homeodomain complement of the ctenophore Mnemiopsis leidyi suggests that Ctenophora and Porifera diverged prior to the ParaHoxozoa. EvoDevo. 2010;1(1):9.
Larroux C, Fahey B, Degnan SM, Adamski M, Rokhsar DS, Degnan BM. The NK homeobox gene cluster predates the origin of Hox genes. Curr Biol. 2007;17(8):706–10.
Thomas-Chollier M, Martinez P. Origin of metazoan patterning systems and the role of ANTP-class homeobox genes. In: eLS. Chichester: John Wiley & Sons, Ltd (Ed.). 2016;1–10.
Zhao XW, Wu J, Kishino H, Chen L. Massive loss of transcription factors promotes the initial diversification of placental mammals. Int J Mol Sci. 2022;23(17):9720.
Elliott GRD, Leys SP. Coordinated contractions effectively expel water from the aquiferous system of a freshwater sponge. J Exp Biol. 2007;210(21):3736–48.
Leys SP. Elements of a “nervous system” in sponges. J Exp Biol. 2015;218(4):581–91.
Leys SP, Meech RW. Physiology of coordination in sponges. Can J Zool. 2006;84(2):288–306.
Leys SP, Mah JL, McGill PR, Hamonic L, De Leo FC, Kahn AS. Sponge behavior and the chemical basis of responses: a post-genomic view. Integr Comp Biol. 2019;59(4):751–64.
Mah JL, Leys SP. Think like a sponge: the genetic signal of sensory cells in sponges. Dev Biol. 2017;431(1):93–100.
Maldonado M. The ecology of the sponge larva. Can J Zool. 2006;84(2):175–94.
Nickel M. Evolutionary emergence of synaptic nervous systems: what can we learn from the non-synaptic, nerveless Porifera? Invertebr Biol. 2010;129(1):1–16.
Renard E, Vacelet J, Gazave E, Lapébie P, Borchiellini C, Ereskovsky AV. Origin of the neuro-sensory system: new and expected insights from sponges. Integr Zool. 2009;4(3):294–308.
Simpson TL. The cell biology of sponges. Springer-Verlag, New York; 1984.
Schröder HC, Krasko A, Gundacker D, Leys SP, Müller IM, Müller WEG. Molecular and functional analysis of the (6–4) photolyase from the hexactinellid Aphrocallistes vastus. Biochim Biophys Acta. 2003;1651(1):41–9.
Leys SP, Degnan BM. Cytological basis of photoresponsive behavior in a sponge larva. Biol Bull. 2001;201(3):323–38.
Rivera AS, Ozturk N, Fahey B, Plachetzki DC, Degnan BM, Sancar A, et al. Blue-light-receptive cryptochrome is expressed in a sponge eye lacking neurons and opsin. J Exp Biol. 2012;215(8):1278–86.
Leys SP, Mackie GO. Electrical recording from a glass sponge. Nature. 1997;387(6628):29–30.
Leys SP, Mackie GO, Meech RW. Impulse conduction in a sponge. J Exp Biol. 1999;202(Pt 9):1139–50.
Leys SP. Comparative study of spiculogenesis in demosponge and hexactinellid larvae. Microsc Res Tech. 2003;62(4):300–11.
Shimizu K, Amano T, Bari MR, Weaver JC, Arima J, Mori N. Glassin, a histidine-rich protein from the siliceous skeletal system of the marine sponge Euplectella, directs silica polycondensation. Proc Natl Acad Sci USA. 2015;112(37):11449–54.
Shimizu K, Kobayashi H, Nishi M, Tsukahara M, Bito T, Arima J. Exploration of genes associated with sponge silicon biomineralization in the whole genome sequence of the Hexactinellid Euplectella curvistellata. In: Endo K, Kogure T, Nagasawa H, editors. Biomineralization. Singapore: Springer; 2018:147–53.
Wang X, Schloßmacher U, Wiens M, Batel R, Schröder HC, Müller WEG. Silicateins, silicatein interactors and cellular interplay in sponge skeletogenesis: formation of glass fiber-like spicules. FEBS J. 2012;279(10):1721–36.
Woodland W. Memoirs: Studies in Spicule Formation: VIII–Some observations on the scieroblastic development of Hexactinellid and other siliceous sponge spicules. J Cell Sci. 1908;s2-52(205):139–57.
Ehrlich H, Luczak M, Ziganshin R, Mikšík I, Wysokowski M, Simon P, et al. Arrested in glass: actin within sophisticated architectures of biosilica in sponges. Adv Sci. 2022;9(11):2105059.
Riesgo A, Maldonado M, López-Legentil S, Giribet G. A proposal for the evolution of cathepsin and silicatein in sponges. J Mol Evol. 2015;80(5):278–91.
Müller WEG, Eckert C, Kropf K, Wang X, Schloßmacher U, Seckert C, et al. Formation of giant spicules in the deep-sea hexactinellid Monorhaphis chuni (Schulze 1904): electron-microscopic and biochemical studies. Cell Tissue Res. 2007;329(2):363–78.
Müller WEG, Schlossmacher U, Wang X, Boreiko A, Brandt D, Wolf SE, et al. Poly(silicate)-metabolizing silicatein in siliceous spicules and silicasomes of demosponges comprises dual enzymatic activities (silica polymerase and silica esterase). FEBS J. 2008;275(2):362–70.
Müller WEG, Wang X, Kropf K, Boreiko A, Schlossmacher U, Brandt D, et al. Silicatein expression in the hexactinellid Crateromorpha meyeri: the lead marker gene restricted to siliceous sponges. Cell Tissue Res. 2008;333(2):339–51.
Müller WEG, Wang X, Burghard Z, Bill J, Krasko A, Boreiko A, et al. Bio-sintering processes in hexactinellid sponges: fusion of bio-silica in giant basal spicules from Monorhaphis chuni. J Struct Biol. 2009;168(3):548–61.
Veremeichik GN, Shkryl YN, Bulgakov VP, Shedko SV, Kozhemyako VB, Kovalchuk SN, et al. Occurrence of a silicatein gene in glass sponges (Hexactinellida: Porifera). Mar Biotechnol. 2011;13(4):810–9.
Riesgo A. alignment of cathepsin and silicatein sequences. Zenodo; 2022. https://zenodo.org/record/7534980.
Schröder HC, Perović-Ottstadt S, Rothenberger M, Wiens M, Schwertner H, Batel R, et al. Silica transport in the demosponge Suberites domuncula: fluorescence emission analysis using the PDMPO probe and cloning of a potential transporter. Biochem J. 2004;381(Pt 3):665–73.
Maldonado M, López-Acosta M, Beazley L, Kenchington E, Koutsouveli V, Riesgo A. Cooperation between passive and active silicon transporters clarifies the ecophysiology and evolution of biosilicification in sponges. Sci Adv. 2020;6(28):eaba9322.
Marron AO, Ratcliffe S, Wheeler GL, Goldstein RE, King N, Not F, et al. The evolution of silicon transport in eukaryotes. Mol Biol Evol. 2016;33(12):3226–48.
Smith G, Manzano-Marín A, Reyes-Prieto M, Antunes CSR, Ashworth V, Goselle ON, et al. Human follicular mites: Ectoparasites becoming symbionts. Mol Biol Evol. 2022;39(6):msac125.
Yahalomi D, Atkinson SD, Neuhof M, Chang ES, Philippe H, Cartwright P, et al. A cnidarian parasite of salmon (Myxozoa: Henneguya) lacks a mitochondrial genome. Proc Natl Acad Sci U S A. 2020;117(10):5358–63.
Burke M, Scholl EH, Bird DM, Schaff JE, Colman SD, Crowell R, et al. The plant parasite Pratylenchus coffeae carries a minimal nematode genome. Nematology. 2015;17(6):621–37.
Altenhoff AM, Train CM, Gilbert KJ, Mediratta I, Mendes de Farias T, Moi D, et al. OMA orthology in 2021: website overhaul, conserved isoforms, ancestral gene order and more. Nucleic Acids Res. 2021;49(D1):D373-9.
Rocher C, Vernale A, Fierro-Constain L, Sejourne N, Chenesseau S, Marschal C, et al. The buds of Oscarella lobularis (Porifera): a new convenient model for sponge cell and developmental biology. bioRxiv. 2020:167296.
Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27(5):722–36.
Langmead B, Wilks C, Antonescu V, Charles R. Scaling read aligners to hundreds of threads on general-purpose processors. Bioinformatics. 2019;35(3):421–32.
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19(5):455–77.
Zhu W, Lomsadze A, Borodovsky M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 2010;38(12):e132.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–10.
Quevillon E, Silventoinen V, Pillai S, Harte N, Mulder N, Apweiler R, et al. InterProScan: protein domains identifier. Nucleic Acids Res. 2005;33: W116–120.
Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, et al. CDD: a conserved domain database for the functional annotation of proteins. Nucleic Acids Res. 2011;39(suppl_1):D225-9.
Käll L, Krogh A, Sonnhammer ELL. A combined transmembrane topology and signal peptide prediction method. J Mol Biol. 2004;338(5):1027–36.
Eddy SR. Accelerated profile HMM searches. PLoS Comput Biol. 2011;7(10):e1002195.
Chan PP, Lowe TM. tRNAscan-SE: searching for tRNA genes in genomic sequences. Methods Mol Biol. 2019;1962:1–14.
Lowe TM, Chan PP. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016;44(W1):W54-57.
Blainey PC, Mosier AC, Potanina A, Francis CA, Quake SR. Genome of a low-salinity ammonia-oxidizing archaeon determined by single-cell and metagenomic analysis. PLoS One. 2011;6(2):e16626.
Kim BK, Jung MY, Yu DS, Park SJ, Oh TK, Rhee SK, et al. Genome sequence of an ammonia-oxidizing soil archaeon, “Candidatus Nitrosoarchaeum koreensis” MY1. J Bacteriol. 2011;193(19):5539–40.
Park SJ, Kim JG, Jung MY, Kim SJ, Cha IT, Kwon K, et al. Draft genome sequence of an ammonia-oxidizing archaeon, “Candidatus Nitrosopumilus koreensis” AR1, from marine sediment. J Bacteriol. 2012;194(24):6940–1.
Santoro AE, Dupont CL, Richter RA, Craig MT, Carini P, McIlvin MR, et al. Genomic and proteomic characterization of “Candidatus Nitrosopelagicus brevis”: an ammonia-oxidizing archaeon from the open ocean. Proc Natl Acad Sci U S A. 2015;112(4):1173–8.
Walker CB, de la Torre JR, Klotz MG, Urakawa H, Pinel N, Arp DJ, et al. Nitrosopumilus maritimus genome reveals unique mechanisms for nitrification and autotrophy in globally distributed marine crenarchaea. Proc Natl Acad Sci U S A. 2010;107(19):8818–23.
Fischer S, Brunk BP, Chen F, Gao X, Harb OS, Iodice JB, et al. Using OrthoMCL to assign proteins to OrthoMCL-DB groups or to cluster proteomes into new ortholog groups. Curr Protoc Bioinformatics. 2011;6:6.12.1-6.12.19.
Santini S, Renard E, Claverie JM. Oopsacas minuta alternative masked genomes and predicted proteomes. Zenodo; 2023. https://zenodo.org/record/7505502.
SantiniS, Renard E, Claverie JM. Oopsacas minuta proteome with genbank id and locus tag. Zenodo; 2022. https://zenodo.org/record/7472271.
Vervoort M. Renard E. Alignment bHLH domains [Data set]. Zenodo; 2022. https://doi.org/10.5281/zenodo.7473983.
Renard E. Alignment of homeodomains of transcription factors of the ANTP family [Data set]. Zenodo; 2022. https://doi.org/10.5281/zenodo.7494952.
Leys SP. Alignment of potassium channels sequences [Data set]. Zenodo; 2023. https://doi.org/10.5281/zenodo.7534942.
Leys SP. Alignment used for voltage gated ion channels phylogenetic analysis [Data set]. Zenodo; 2023. https://doi.org/10.5281/zenodo.7534934.
Glöckner FO, Fuchs BM, Amann R. Bacterioplankton compositions of lakes and oceans: a first comparison based on fluorescence in situ hybridization. Appl Environ Microbiol. 1999;65(8):3721–6.
Teira E, Reinthaler T, Pernthaler A, Pernthaler J, Herndl GJ. Combining catalyzed reporter deposition-fluorescence in situ hybridization and microautoradiography to detect substrate utilization by bacteria and Archaea in the deep ocean. Appl Environ Microbiol. 2004;70(7):4411–4.
Santini S, Schenkelaars Q, Jourda C, Duschene M, Belahbib H, Rocher C, Selva M, Riesgo A, Vervoort M, Leys SP, Kodjabachian L, Le Bivic A, Borchiellini C, Claverie J-M, Renard E. Oopsacas minuta and associated symbionts. GenBank; 2022. https://identifiers.org/bioproject:PRJNA761294/.
Santini S, Schenkelaars Q, Jourda C, Duschene M, Belahbib H, Rocher C, Selva M, Riesgo A, Vervoort M, Leys SP, Kodjabachian L, Le Bivic A, Borchiellini C, Claverie J-M, Renard E. Oopsacas minuta isolate SPO-2, whole genome shotgun sequencing project. GenBank; 2022. https://www.ncbi.nlm.nih.gov/nuccore/JAKMXF000000000.
Santini S, Schenkelaars Q, Jourda C, Duschene M, Belahbib H, Rocher C, Selva M, Riesgo A, Vervoort M, Leys SP, Kodjabachian L, Le Bivic A, Borchiellini C, Claverie J-M, Renard E. Cenarchaeum symbiont of Oopsacas minuta SPO-1, whole genome shotgun sequencing project. GenBank. 2023 https://www.ncbi.nlm.nih.gov/nuccore/JAJIZT000000000.
Acknowledgements
The authors wish to pay tribute to their colleague and friend Pr. Michel Vervoort who passed away on December 8, 2022, for the excellence of his work in the evo-devo field. The authors are grateful to the diving staff of the OSU Pytheas (F. Zuberer, L. Vanbostal, D. Guillemain) and to the divers of the IMBE (P. Chevaldonné, T. Perez, C. Marschal, S. Chenesseau, J. Vacelet) for collecting samples. The molecular biology staff of the IMBE is acknowledged for providing reagents and equipment for high-quality DNA extraction development. We thank the ProfileXpert (Lyon) and Pacific Biosciences platforms for genome sequencing. We thank PACA Bioinfo for the computing support during assembly and analysis. We also thank Marc Garel (MIO, Marseille, France) for the kind advice on card-FISH experiments and IBDM France Bioimaging and IMBE morphology services for providing technical facilities and help. We also thank A. Ereskovsky and J. Vacelet for the helpful discussions on sponge anatomy and embryology.
Funding
This project was funded by the A*MIDEX foundation project (including CJ’s and HB’s salaries) (no. ANR-11-IDEX-0001-02) funded by the «Investissements d’Avenir» French Government program, managed by the French National Research Agency (ANR).
Author information
Authors and Affiliations
Contributions
All authors read and approved the final manuscript. ALB, CB, ER, and JMC designed the project. JMC, SS, and CJ chose sequencing and assembling strategy. AR, CB, CJ, CR, ER, HB, MD, MV, MS, QS, SL, and SS performed experiments and/or sequence analyses. ALB, AR, CB, CJ, ER, JMC, LK, MV, QS, SL, and SS interpreted the results and wrote and edited the text.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
No ethics approval was required for the invertebrates used in this study.
Consent for publication
For the Fig. S4 (Additional file 1), the authors obtained the permission of the editor Rob Van Soest to reuse the Fig. 1 page 409 published in Boury-Esnault, N.; Vacelet, J. (1994). Preliminary studies on the organization and development of a hexactinellid sponge from a Mediterranean cave, Oopsacas minuta. In: van Soest, R.W.M., van Kempen, Th.M.G. & Braekman, J.-C. (Eds), Sponges in Time and Space. (Balkema: Rotterdam): 1-515. 407-415.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Characterization of the microbiome of Oopsacas minuta. Table S1. Main features and taxonomic assignment of Oopsacas minuta’s metagenome assembly. Table S2. Taxonomic assignment of non-Eukaryotic and non-Archaeal Taxa. Figure S1. Phylogenetic positions of the main microbial species associated with O. minuta. Table S3. Genome features of Candidatus Cenarchaeum massiliensis compared to other Thaumarchaea. Table S4. Annotation of the predicted protein-coding genes of Ca. C. massilliensis. Figure S2. Characterization of the genome of Ca. C. massiliensis. Figure S3. The cobalamin synthesis genes in Thaumarchaeota. Figure S4. Localization of Ca. C. massiliensis in the tissues of O. minuta by Card-FISH.
Additional file 2:
Genomic characteristics of Oopascas minuta. Table S5. Genome features of the glass sponge O. minuta. Table S6. Respective number of predicted and annotated sequences in the genome of O. minuta. Figure S5. Gene ontology at the first two levels for the 7,737 genes that received GO annotations. Figure S6. a) Orthology clustering on predicted proteomes of six sponges. b) Gene Ontology at level3 for orthogroups absent in O. minuta and present in all other sponge lineages. Table S7. ORFING report, Comparison of protein sequence lengths and BUSCO scores between the different versions of the genome masked or unmasked. Table S8. Types and relative abundances of repeats in the genome of O. minuta.
Additional file 3:
Survey of candidate genes for epithelia, multicilliogenesis, signaling pathways, transcription factors, neuro-sensory functions, biosilicification. Table S9. Blastp search of proteins involved in bilaterian epithelial functions. Figure S7. Domain prediction of integrins and of laminins. Table S10. Blastp search concerning proteins involved in bilaterian multiciliogenesis. Table S11. Blast Search of core genes involved in three main ancestral signalling pathways. Table S12. Blastp search results concerning G-proteins. Table S13. Blastp search and protein domain analyses concerning basic Helix Loop Helixtranscription factors. Figure S8. Phylogenetic relationships among bHLH Transcription factors. Figure S9. Phylogenetic position of Oopsacas bHLH-PAS Transcription factors. Table S14. Blastp search, domain and phylogenetic analyses performed on various transcription factor types. Figure S10. Domain Prediction of HD transcription factors. Figure S11. Phylogenetic positions of Oopsacas Homeobox Transcription factors of the ANTP class. Table S15. Survey of candidate genes for photoreception. Table S16. Blastp search for proteins involved in chemical signaling. Table S17. HHMER searches in O. minuta for voltage gated ion channels. Figure S12. Diversity and phylogenetic positions of Oopsacas calcium channels. Figure S13. Phylogenetic positions of Oopsacas potassium channels. Table S18. Blastp search of proteins involved in bilaterian synapses. Table S19. Results of HMMER searches for SNARES proteins. Table S20. Blast P search of proteins involved in silica biogenesis. Table S21. List of sequences used for phylogenetic analyses of silicatein and cathepsin.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Santini, S., Schenkelaars, Q., Jourda, C. et al. The compact genome of the sponge Oopsacas minuta (Hexactinellida) is lacking key metazoan core genes. BMC Biol 21, 139 (2023). https://doi.org/10.1186/s12915-023-01619-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12915-023-01619-w