
Abstract
Plasma membrane integrity is essential for cellular homeostasis. In vivo, cells experience plasma membrane damage from a multitude of stressors in the extra- and intra-cellular environment. To avoid lethal consequences, cells are equipped with repair pathways to restore membrane integrity. Here, we assess plasma membrane damage and repair from a whole-body perspective. We highlight the role of tissue-specific stressors in health and disease and examine membrane repair pathways across diverse cell types. Furthermore, we outline the impact of genetic and environmental factors on plasma membrane integrity and how these contribute to disease pathogenesis in different tissues.
Similar content being viewed by others

Plasma membrane integrity
Confinement of a cell from its surrounding environment is a universal trait of microscopic life. The plasma membrane fulfills this role whereby its integrity is vital for cell function and survival. Accordingly, plasma membrane architecture and composition varies to provide resistance to injury in different cellular contexts. Despite this protection, various factors (herein referred to as stressors) present in the extra- and intra-cellular environment can induce chemical disruptions or physical breaches in the plasma membrane. Although not all wounds result in cell death, even sublytic damage can vastly change the intracellular landscape through cytosolic leakage and exposure to the outside environment.
In vivo, plasma membrane damage is encountered during normal physiological events such as muscle contraction and locomotion [1,2,3]. In these situations, sublytic damage can prove beneficial by stimulating paracrine signaling to shape the tissue environment. Alternatively, plasma membrane damage inflicted by microbial pathogens and immune cells can have deleterious consequences on cell fate during infection and inflammation [4, 5]. The prevalence of membrane damage across functionally distinct processes, from cell death to cancer cell migration, exemplifies how plasma membrane integrity is a fundamental aspect of cell biology [6, 7].
To deal with damage, cells are equipped with plasma membrane repair mechanisms. Ion imbalances “sound the alarm”, leading to cytoskeleton remodeling, repair factor recruitment, and vesicle trafficking, which cooperatively facilitate wound closure [8]. While several different repair pathways have been identified, our understanding of how these mechanisms cooperate to facilitate resealing remains unclear. Moreover, nearly all of the known repair mechanisms were identified based on studies of extracellular stressors, yet recent evidence highlights the importance of repair in response to internal stressors such as necroptotic pores and even endolysosomal damage [9, 10].
Plasma membrane integrity reflects resistance and repair capacity, both of which are influenced by host genetics and environmental factors. Imbalances in either of these determinants of membrane integrity can lead to disease pathogenesis. In the case of muscular dystrophies, genetic factors can dampen membrane resistance and repair whereas in other instances, such as traumatic brain injury, physical trauma can exceed a cell’s repair capacity and result in membrane lesions [11, 12]. Emerging evidence suggests that defective repair may even contribute to the pathogenesis of multifactorial diseases such as inflammatory bowel disease (IBD) [13]. Considering the plethora of factors that can dampen plasma membrane integrity, barrier maintenance should be regarded as an active cellular process that ultimately sustains tissue structure and function. While tissue damage is a common hallmark of disease, the role of plasma membrane integrity in disease initiation and progression remains understudied.
Plasma membrane damage and repair are often generalized across literature and experimental settings, which, despite advancing our mechanistic understanding of these processes, has made it difficult to infer the physiological relevance of these findings. Here, we summarize our current understanding of plasma membrane damage and repair from a whole-body perspective, including tissue-specific stressors and the influence of genetic and environmental factors on cell type-specific repair. We highlight that defects in plasma membrane resistance and repair exacerbate injury and contribute to a broad range of human diseases. Understanding wound resolution on a single-cell basis can lead to the identification of promising therapeutic targets to promote tissue regeneration in several pathophysiological states.
Types of plasma membrane damage
Plasma membrane damage is conventionally assessed by indirect means such as the entry of cell-impermeable molecules (e.g., dextran), calcium influx, or the detection of intracellular contents in the extracellular environment [14]. Direct measures of visualizing and characterizing plasma membrane wounds are challenged by technical constraints and the rapid speed of the repair process. Nonetheless, studies using such approaches have demonstrated that plasma membrane integrity can be compromised from two distinct types of damage: chemical disruptions and physical breaches.
Chemical disruptions
The biochemical nature of the plasma membrane renders it susceptible to several forms of chemical disruptions (Fig. 1a). In the presence of reactive oxygen species (ROS), the oxidation of polyunsaturated fatty acids (i.e., lipid peroxidation) can lead to the release of damaged lipid fragments and eventual loss of plasma membrane integrity [15]. Lipid peroxidation is accelerated by intracellular iron via the Fenton reaction [16] whereas membrane damage is countered by cholesterol, antioxidants, and glutathione peroxidase 4 (GPX4), a central lipid repair factor [15, 17, 18]. This form of membrane injury is often detected by end products [19]; however, these measures make it difficult to infer the extent of damage on a single cell level (i.e., transient or lytic) and the influence of redox status on cell fate. Alternatively, the plasma membrane is subject to enzymatic damage (e.g., phospholipases) that can alter membrane fluidity and render cells more prone to osmotic lysis—similar to what is observed upon cholesterol extraction [20,21,22]. Amphiphilic molecules (e.g., drugs, alcohol) can also disrupt membrane fluidity through direct interactions whereas at lower magnitudes they potentiate oxidative stress-induced damage [23, 24].

Plasma membrane damage is comprised of chemical disruptions and physical breaches. a Chemical disruptions of the plasma membrane can alter its biophysical properties and lead to a breach. Oxidative stress and intracellular iron promote lipid peroxidation of poly-unsaturated fatty acids leading to the removal of damaged fragments and destabilization of the plasma membrane. Membrane lipids are subject to enzymatic damage by host or foreign phospholipases. Alterations in membrane fluidity through interactions with amphipathic molecules or cholesterol extraction can weaken membrane resistance upon subsequent insult. b Depending on size and frequency, physical breaches elicit lytic or non-lytic damage and require active repair to restore membrane integrity. Nanoruptures result in ion imbalances and the leakage of small molecules, whereas membrane tears result in extensive leakage of cytosolic cargo. Larger membrane tears (> 100 nm) are broadly distinguished based on different repair requirements. Upon recognition of unique host receptors, pore-forming proteins can assemble into transmembrane pores that differ in terms of size, structure, and ion flux
Physical breaches
Physical breaches of varying size and nature can compromise plasma membrane integrity (Fig. 1b). Tiny punctures (< 1 nm) do not result in a permanent breach as it is energetically favorable for lipids surrounding the lesion to undergo spontaneous resealing [25]. However, larger injuries such as nanoruptures, tears, and pores are not self-limiting and result in a physical breach. Nanoruptures are characterized as small lesions (~ 1–10 nm wide) with exposed lipids around the wound edge that arise from mechanical force and sustained chemical disruptions [26, 27]. Larger membrane ruptures, herein referred to as tears, can be broadly categorized based on their size (i.e., greater or less than 100 nm) given differences in repair requirements [28]. Membrane tears are often encountered in muscle cells following exercise or can arise from physical trauma [12, 29]. Little is known about the topology of nanoruptures and tears, and whether some regions of plasma membrane are more prone to tearing than others (e.g., influence of local lipid composition or plasma membrane-organelle contact sites).
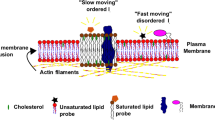
Pore formation is the most well-characterized type of physical breach given the prevalence of pore-forming proteins in immunity and infectious disease [4, 5]. As reviewed in great detail [30], pore formation entails the recognition of protein monomers with unique host surface receptors, which triggers oligomerization and the insertion of membrane pores that vary in terms of size and ion selectivity. In comparison to larger pores (~ 25–50 nm), small pores (~ 1–2 nm) generally persist longer on the plasma membrane which has been suggested to reflect lower extents of calcium influx, and consequently, delayed calcium-dependent repair mechanisms [31,32,33]. Contrary to tears, pores lack membrane edges which may explain why these wounds have different repair requirements [34]. Plasma membrane composition is a major determinant of cellular resistance against pore-induced damage. For instance, the availability of surface cholesterol and its distribution in microdomains can greatly influence the extent of membrane damage by cholesterol-dependent cytolysins [35, 36].
Extracellular and intracellular sources of plasma membrane damage
Within a tissue environment, a multitude of stressors can induce plasma membrane damage through chemical disruptions and physical breaches. Here, we categorize 5 major sources of plasma membrane damage: mechanical, chemical, microbial, immune, and intracellular stressors (Fig. 2). Notably, these sources of damage are not mutually exclusive, and in many cases, one form of injury predisposes the plasma membrane to subsequent insult (e.g., chemical damage exacerbates mechanical insult) [37].

Five major sources of plasma membrane damage. These sources of damage can be highly overlapping as one type of membrane injury can lead to another. (a) Cells experience mechanical stress from physiological events (e.g., locomotion), cell migration, and through interactions with inert substances in the local environment, all of which elicit membrane damage in the form of nanoruptures and tears. (b) Reactive oxygen species present in the extracellular environment or those generated from irradiation can promote lipid peroxidation. Additional sources of chemical disruptions include amphipathic molecules (e.g., NSAIDs and alcohol), which can compromise membrane integrity either through direct interactions or indirectly via oxidative stress. (c) Microbial species employ several strategies to induce plasma membrane damage. Virulence factors can inflict chemical disruptions (e.g., phospholipases) and physical breaches (e.g., pore-forming toxins); meanwhile, larger species can also exert brute force to damage host plasma membrane. (d) Immune cells elicit membrane damage, namely through pore-forming proteins and antimicrobials, under several unique contexts such as immune surveillance and neutrophil extracellular traps. (e) Intracellular sources of plasma membrane damage include oxidative stress, which can entice lipid peroxidation, and the leakage of cytotoxic enzymes from lysosomes. In the context of cell death, many pathways employ pore-induced damage (e.g., necroptosis, secondary necrosis, pyroptosis) whereas others are characterized by chemical disruptions (e.g., ferroptosis)
Mechanical
In vivo, physiological events such as locomotion can generate sufficient mechanical force to elicit plasma membrane damage in muscle, bone, and skin [2, 26, 29]. Alternatively, mechanical lesions can arise from physical trauma, thermal injury, and even exposure to penetrating noise and ultrasound [12, 38,39,40]. In all these contexts, our characterization of membrane damage is limited to the uptake of cell-impermeable dyes; however, one can speculate as to the general mode of disruption (e.g., stretch- or compression-induced tears). We also have a very limited understanding of the frequency and size of wounds caused by physiological force, although in the case of muscle contractions, damage is speculated to be a gradient of nanoruptures and tears on the scale of micrometers [1, 11, 29].
On a cellular level, physical breaches can arise from shear stress, such as during the transit of red blood cells and platelets in circulation [41,42,43]. Intriguingly, malignant cancer cells harness the abundance of platelets to bolster their resistance to such shear stress [44, 45]. Upon exiting circulation, cancer cells continue to face mechanical damage during cell migration which they combat through enhanced repair efforts [7, 46,47,48]. Moreover, it is apparent that cells bordering sites of tissue trauma also experience strain-induced plasma membrane damage [49] which seems likely to influence the rate of tissue repair.
Mechanical damage can also arise following the interaction of the plasma membrane with inert substances (e.g., protein aggregates, inhaled nanoparticles). For instance, intrinsically disordered proteins (e.g., β-amyloid) have the propensity to aggregate in the extra- and intra-cellular environment and form pore-like structures in the plasma membrane [50]. Similar modes of mechanical disruption contribute to the nanotoxicity of silica nanoparticles and multi-walled carbon nanotubes, albeit through different mechanisms [51, 52]. Understanding these forms of nanotoxicity is of critical importance in the development of safe medical solutions based on nanotechnology [53].
Chemical
Lipid peroxidation is a prominent stressor across tissue environments although the source of ROS varies. During hypoxia or ischemia/reperfusion (I/R) injury, inadequate blood supply leads to mitochondrial dysfunction and the generation of ROS which is further exacerbated upon reoxygenation [54]. Metabolically active tissues, such as kidney and liver, are particularly sensitive to this form of oxidative injury [55, 56]. Alternatively, lipid peroxidation can occur following irradiation [57, 58], exposure to extracellular ROS (e.g., cancer therapy) [59], and through interactions between intrinsically disordered proteins with the inner- and outer-leaflet of the plasma membrane [60, 61]. Notably, there appears to be a positive feedback cycle whereby oxidized membrane can foster protein aggregation at the cell surface to further exacerbate injury [61].
Early gastrointestinal studies revealed the ability of amphiphilic molecules such as bile acids, alcohol, and non-steroidal anti-inflammatory drugs (NSAIDs), to directly compromise plasma membrane integrity at high concentrations [62,63,64]. However at lower concentrations, these stressors promote oxidative stress through mitochondrial dysfunction [23, 65] and alter membrane lipid organization [66, 67]. The propensity for these stressors to induce damage is also influenced by nearby chemical species. For instance, while certain bile acids can alter membrane fluidity without an apparent breach [68], the simultaneous presence of NSAIDs can promote damaging extents of bile acid accumulation [69].
Microbial
Microbial species have several motives to induce plasma membrane damage such as entry into the host’s nutrient-rich intracellular environment, facilitating cellular escape and spread, or eliciting host damage [70, 71]. To breach the plasma membrane, chemical disruptions can be achieved by virulence factors such as secreted sphingomyelinases and phospholipases [72]. However, the use of mechanical stress to induce a breach is far more apparent across bacteria, fungi, protozoa, and viruses.
To facilitate invasion, larger species (e.g., fungi, protozoa) exert brute force onto the host plasma membrane. During the filamentous growth of yeast, hyphae protrusions damage the plasma membrane of epithelial cells [73, 74]. Similarly, Plasmodium sporozoites physically penetrate through the skin epithelium causing irreversible host cell damage [75]. The onset of necrotic death in both these instances argues in favor of large plasma membrane tears. However, such crude destruction is not always the case as parasites can nibble pieces of host plasma membrane through a process called “trogocytosis” [76]. Miraculously, the host can maintain plasma membrane integrity until the point of cell death, suggesting a role for active repair pathways during this process.
As cell death is not always the desired outcome, microbes are equipped with specialized machinery to induce smaller breaches. Bacteria utilize needle-like structures (termed secretion systems) to facilitate cargo delivery into host cells through a transmembrane pore (~ 1.2–5 nm in the case of type 3 secretion systems) [71, 77, 78]. Similarly, Microsporidia breach host membrane using a much larger “polar tube” (~ 0.1–0.15 μm diameter) although the insertion mechanism or type of wound this inflicts is unknown [79]. Viruses also exploit plasma membrane damage: for example, adenovirus relies on a single surface protein to induce damage and facilitate entry into the host cell [80].
Mechanical damage is also achieved by pore-forming toxins; the largest class of bacterial toxins and similar members are expressed by fungi and protozoan species [81,82,83,84]. While many of these toxins are capable of inducing cell lysis, sublytic damage provides initiating cues (e.g., ion imbalances) to alter the intracellular environment and, in some cases, initiate cell death pathways [4, 31, 85]. Although distinct from bacterial pore-forming toxins, viruses similarly compromise host membrane integrity through transmembrane channels termed viroporins [86, 87].
Immune
Plasma membrane damage is fundamental to several immune functions. Circulating complement factors can achieve cell lysis upon formation of a heteromeric pore (~ 12 nm wide) known as the membrane attack complex [88]. Under normal conditions, regulatory mechanisms protect host cells from complement-induced damage; however, autoantibodies or an overwhelming inflammatory response can promote this form of injury [89, 90]. Meanwhile, cytotoxic lymphocytes inflict sublytic damage at immune synapses through perforin pores (~ 13–20 nm wide) to enable the intracellular delivery of pro-apoptotic granzymes for discrete killing [91, 92]. Instead of pore-forming proteins, macrophages opt for the release of lysosomal cargo to induce heterolysis of cancer cells—likely by severe chemical disruptions [93]. Similar forms of plasma membrane damage can arise from neutrophil extracellular traps, which are web-like structures formed by the release or expulsion of neutrophil contents including antimicrobials and proteases [94,95,96]. Lastly, neutrophils alongside other immune cells have the propensity to engulf portions of plasma membrane from target cells as a means of intercellular communication (i.e., trogocytosis) [97] or cytotoxicity (i.e., trogoptosis) [98].
Intracellular
The intracellular environment contains an array of potentially cytotoxic stressors such as ROS, pore-forming proteins, and lysosomal contents, which under normal conditions are regulated by antioxidants, stringent activation requirements, and spatial confinement or zymogens, respectively [54, 99, 100]. When regulation goes awry, physical breaches induced by these stressors can result in, or accompany cell death. Many cell death pathways entail unique forms of plasma membrane damage [99]. In the case of ferroptosis, damage is achieved through iron-dependent lipid peroxidation [15] whereas transmembrane pores are a common feature of necroptosis, secondary necrosis, and pyroptosis [6]. Notably, these pores differ in composition and outcome, as insertion may favor osmotic rupture or weaken plasma membrane resistance against mechanical stress [101,102,103,104]. Lastly, the disruption and leakage of lysosomes can lead to detrimental enzyme activity and the onset of lysosomal-dependent cell death [105, 106].
Plasma membrane repair of physical breaches
Early observations in mechanically injured cells noted the ubiquitous requirement of extracellular calcium for membrane resealing [107,108,109]. Indeed, the steep calcium gradient between the extra- and intra-cellular environment (~ 2 mM and 100 nM, respectively) ensures that calcium entry is an ideal alarm for membrane damage [110]. Calcium influx through the wound site can be further amplified by voltage-gated channels and internal stores to bolster repair efforts [111, 112]. This leads to local activation of calcium-dependent proteases, such as calpains (CAPN), which promote disassembly of the cytoskeleton through cleavage of substrates including vimentin and cortactin [113, 114]. Alongside relieving membrane tension, this clears the landscape for incoming repair machinery, vesicle fusion events, and cytoskeleton remodeling to restore integrity.
It is convenient to conceptualize the requirements for cell repair by paralleling those of tissue healing, encompassing four complementary stages [115]: hemostasis (sealing off the breach), inflammation (removal of dangerous material that entered the cell), proliferation and migration (replacement of lost or damaged cell components), and remodeling (adaptive responses). In the context of membrane repair, these latter three steps are collectively referred to as “regeneration,” as they represent the steps needed to restore sites of damage to their original state. The means by which cells seal off a physical breach has received far more attention than the other steps of cell repair.
Sealing off the breach
While the tiniest of wounds can be healed by spontaneous lipid flow, more substantial injuries such as nanoruptures, tears, and pores require active repair by interior cell constituents. Both the size and nature of the wound (i.e., exposed lipid edge versus pore) are governing factors in mounting an appropriate repair response [28, 34]. As highlighted below, there are several mechanisms by which cells achieve plasma membrane repair (Fig. 3). Importantly, most of these pathways are not mutually exclusive and collaborate to seal off breaches.

Plasma membrane repair pathways to seal a physical breach. Depending on the extent of damage, several different pathways can cooperatively facilitate wound closure. (a) Exocytosis can relieve membrane tension at the wound site to promote wound closure. Vesicle recruitment to the cell surface occurs through kinesin- and myosin-dependent transport and may require additional support by proteins such as MG53. At the wound site, calcium-dependent fusion machinery such as synaptotagmins (SYT) or dysferlin (DYSF) mediate vesicle fusion. (b) Patching entails inter-vesicle fusion underneath the wound site to generate a membrane patch to seal large tears. (c) Lysosomal exocytosis can promote caveolar endocytosis upon the extracellular release of cathepsins and acid sphingomyelinase (ASM). The fate of endocytosed lesions is determined upon sorting in multivesicular bodies. (d) Concentric zones of actin regulators (e.g., RHOA, myosin-II, CDC42) can form around the wound site to facilitate closure by actomyosin contractions. (e) The accumulation of vesicles, calcium-sensitive proteins (e.g., ANXA1), and mitochondria at the wound site can form a temporary plug to limit diffusion of materials between the extra- and intra-cellular environment. (f) Different annexins can assemble along the wound edge to limit wound expansion (ANXA5), induce membrane curvature (ANXA4), and generate constriction force (ANXA6) to seal a breach. (g) The ANXA7-dependent recruitment of ESCRT-III machinery to the wound site can lead to membrane scission and the release of damaged membrane
Exocytosis
Exocytosis has been proposed to promote wound closure by lowering membrane tension to allow for membrane flow over wounds that would otherwise simply gape open [116]. This form of vesicle mediated-repair is calcium-dependent [117] and has been demonstrated to involve the fusion of vesicle compartments with the plasma membrane such as secretory granules [118,119,120], reserve granules [121], and lysosomes [122]. The importance of exocytosis in resealing is demonstrated by the lack of successful repair following manipulations that impair basic components of the exocytotic fusion machinery such as SNAREs and synaptotagmins (syt) [117, 123].
During repair, lysosomal fusion at the plasma membrane is mainly attributed to syt-VII activity; however, observations during astrocyte repair highlight cell type-specific differences in syt requirements [122, 124]. In the case of muscle repair, lysosomal exocytosis is also dependent on dysferlin, a calcium-dependent membrane-binding protein that can promote lysosome tethering to the cell surface [125, 126]. Dysferlin can be cleaved into a syt-like molecule which could render it able to directly promote fusion of intracellular vesicles with the plasma membrane [127].
Another potential facilitator of exocytosis is MG53, a ubiquitin ligase normally found in the cytosol, plasma membrane, and on intracellular vesicles [128]. Upon damage, MG53 accumulates at the wound site in an oxidation-dependent manner where it is thought to interact with caveolae-related proteins and phosphatidylserine [128,129,130,131]. Based on its localization to wound sites, it has been proposed that MG53 could help the recruitment of membranous compartments to facilitate repair [128].
Patching
The repair of larger tears has been proposed to occur through “patching,” whereby inter-vesicle fusion forms an underlying patch that fuses along the exposed edge of the wound. Whether integration of this patch at the wound site is directly achieved through exocytosis or fusion upon lipid disorder remains unclear [132]. Patching was initially proposed based on the observation that sea water injected into echinoderm eggs is walled off from the rest of the cytoplasm by what appears to be a membrane in electron micrographs [133]. More recently, patching has been directly observed in wounded Xenopus oocytes [120]. Importantly, both echinoderm eggs and Xenopus oocytes are unusually large cells and patching involves unique sources of vesicle (e.g., reserve granules and secretory granules) that are unspecified in other cell types. While evidence of patching in mammalian cells is scarce [134], it remains a solution for sealing larger tears given the accumulation of membranous compartments observed at wound sites [109, 128].
Endocytosis
The removal of smaller tears and transmembrane pores can be achieved through caveolar endocytosis [135, 136]. This process is facilitated through lysosomal exocytosis leading to the exofacial release of acid sphingomyelinase which can generate ceramide-enriched caveolae that are subsequently internalized [137]. Endocytosed lesions then undergo sorting in multivesicular bodies by members of the endosomal sorting complexes required for transport (ESCRT) family where they are destined for either lysosomal degradation or exosome release [138, 139]. Oxidative stress can also trigger lysosomal exocytosis [140], although whether this similarly triggers the removal of plasma membrane with chemical disruptions is unknown. Furthermore, it remains unclear how endocytosis could seal off larger tears (> 100 nm) which could not be accommodated by individual caveolae (50–100 nm) unless clustering allows for the processing of more extensive wounds [141, 142].
Contraction
A physical breach can be sealed through actomyosin contractions by cortical actin filaments (F-actin) and myosin-II that encircle the damage site [143]. Contraction-mediated repair has been observed in mechanically injured Xenopus oocytes and embryos [144, 145], Drosophila syncytium [146], and C. elegans epidermis [147, 148]. In these models, concentric zones of actin regulators (e.g., small GTPases such as RhoA and Cdc42) form around the wound site to facilitate the organization and contractile movement of actin rings [144, 146, 149, 150]. Such contractions have also been observed in transected invertebrate neurons [151], where it is thought to bring the edges of plasma membrane close enough for membrane fusion events to take over [152]. However, this contraction response is relatively slow, making it unlikely to account for those situations in which membrane resealing occurs within several seconds [153]. More broadly, the accumulation of actin and non-muscle myosin IIA at wound sites is critical for the delivery of repair machinery [154,155,156] and can indirectly promote resealing by controlling bleb dynamics in response to pore-induced damage [157].
Plugging
Plugging refers to the aggregation of vesicles and other material (e.g., mitochondria, proteins) at the wound site, forming the single cell equivalent of a clot [158,159,160]. Plug formation has been observed in response to small and large breaches, where it is proposed to limit exchange of material between the cytoplasm and the extracellular space while other forms of repair proceed. A role for plugging is supported by the accumulation of tightly opposed, membranous compartments at wound sites in essentially all cell types [109, 120, 152, 161]. Both annexins (ANXA) and dysferlin are promising candidates to promote plugging as they are calcium-dependent, membrane-binding proteins recruited to the wound site with the potential to crosslink vesicles to each other or the plasma membrane [110, 111, 120, 162, 163]. Regardless of the mechanism, because plugs fail to restore a uniform membrane at the damage site, they are necessarily temporary.
Constriction
Based on their known physical properties, certain annexins can facilitate wound closure through membrane constriction [164]. Upon recruitment to the damage site, trimers of ANXA4 bend the plasma membrane to promote constriction force dependent on ANXA6 [165]. This activity could potentially be assisted by ANXA5 arrays that suppress wound expansion, in addition to vesicle fusion events necessary to provide additional membrane required for constriction-mediated closure [166, 167].
Scission
Physical breaches situated on outward buds or extensions of the plasma membrane can be removed by ESCRT complexes [28]. Calcium influx promotes ANXA7-dependent recruitment of ESCRT-III members to the damage site, where they form a multi-subunit contractile lattice that snips off membrane containing a wound [28, 168]. With the exception of large tears (> 100 nm) [28], scission can facilitate the release of smaller breaches including membrane damaged by lipid peroxidation [9, 169,170,171]. Notably, the outward buds targeted by scission are distinct from larger, plasma membrane blebs that retract through actomyosin contractions [157, 172]. Exactly how the ESCRT-III system can discern between these membrane structures during repair is unclear.
Removal of dangerous material
Two major unwanted incursions from wounding are elevations in intracellular calcium and oxidation, which, if not terminated, will eventually kill the cell. A major source of calcium removal is through uptake by organelles such as the endoplasmic reticulum and mitochondria [173,174,175]. In the case of the latter, calcium uptake leads to mitochondrial fragmentation to promote greater calcium clearance and the release of local redox signals critical for repair [176, 177].
Similar to calcium, oxidative species are particularly high in the extracellular environment and enter the cell upon a breach [8]. During repair, cells rely on antioxidants (e.g., vitamin E, glutathione, GPX4) to prevent the onset of lipid peroxidation and irreparable damage, as evident in muscle cells depleted of these defenses [178]. In support of this notion, cells pretreated with lipid-directed antioxidants undergo faster rates of resealing [37, 179]. It remains unclear whether oxidant-induced damage solely exacerbates the initial breach or leads to the formation of new disruptions, which may spread repair efforts thin and lower the rate of successful resealing.
Replacement of lost or damaged cell components
A physical breach inevitably results in the leakage of cytosolic content into the extracellular environment. The loss of smaller molecules, such as ions and ATP, is common to most forms of breaches whereas larger wounds lead to substantial losses of protein [30, 180, 181]. Recovery of such material has mainly been studied in the context of potassium efflux during pore-induced damage. A decrease in intracellular potassium activates cytoprotective mitogen-associated protein kinase (MAPK) signaling-dependent events that partially restore levels in the subsequent hours following damage [182]. In addition, potassium loss triggers autophagy activation which likely recycles damaged cargo in the vicinity of the wound site [182]. Our current understanding of what seeps through a breach remains limited to only a handful of molecules, and exactly how and when lost material is replenished remains to be determined.
Upon damage, the sudden influx of calcium favors the local disassembly of the actin cytoskeleton, which, while necessary for vesicle fusion, is unfavorable for other steps of repair [154, 183]. Actin polymerization at the wound site is partially achieved upon the recruitment of ANXA2 and its binding partner S100A11, in addition to redox-dependent RhoA activity [7, 159]. These contributors of F-actin are critical for successful repair, which prevents further calcium influx and allows restoration of the actin cortex.
Membrane repair also leads to extensive remodeling of the plasma membrane and extracellular matrix [184]. Vesicle fusion events replenish plasma membrane that is directly lost from wound scission or endocytosis. However, an indirect consequence of removing plasma membrane is the accompanying loss of surface receptors and disruption of microdomains leading to impairments in cell signaling [185, 186].
Adaptive responses
Scars represent a “memory” of tissue damage; similarly, cells retain a “memory” of initial damage events which serve to enhance repair upon subsequent injury [187]. Such adaptive responses are most well understood in the context of purinergic signaling, whereby ATP leakage into the extracellular environment stimulates purinergic receptors (e.g., P2X7, P2Y) on both damaged and non-damaged cells [188]. This form of signaling triggers calcium influx which can promote cytoprotective blebbing even in non-damaged cells [189]. More importantly, calcium influx activates protein kinase activity which is required for priming vesicle-mediated repair to enhance the rate of resealing upon a second injury [123]. This protective response is evident in both the short-term (i.e., 5-min intervals between injury) [190] and long-term (i.e., 24 h) [191], albeit the latter is dependent on transcriptional changes. Other adaptive responses include tissue growth, as best described in muscle following the leakage of fibroblast growth factor upon exercise-induced damage [192]. Furthermore, cellular priming is not limited to soluble factors as damaged membrane, or vesicles shed during repair can be internalized by nearby cells to initiate signals including proliferation and even macrophage polarization [193,194,195].
Plasma membrane damage and repair: whole-body implications
In vivo, cellular architecture and composition are adapted to minimize plasma membrane damage by tissue-specific stressors. For example, muscle and endothelial cells have an abundance of caveolae to buffer frequent mechanical stress [196, 197] whereas metabolically active cells such as hepatocytes mitigate chemical disruptions through antioxidant defenses [198]. These forms of resistance are often compromised amidst disease and render the plasma membrane susceptible to damage (Table 1). While membrane repair pathways are generally conserved, cell type-specific adaptations arise from inherent differences in cell structure and the expression of repair machinery. Furthermore, genetic factors can have tissue-specific impacts on plasma membrane resistance and repair that may be causative of or exacerbate disease (Table 2). Here, we highlight our current understanding of cell type-specific damage and repair throughout the body (Figs. 4 and 5).

Plasma membrane damage and repair in the lung, gastrointestinal tract, skin, vasculature, and bone. Red arrows: sources of plasma membrane damage; black arrows: repair pathways; gray arrows: forms of cellular resistance. Human body was created with BioRender.com. (i) Pneumocytes: Mechanical stress during ventilation is typically alleviated by surfactant. Damage-induced ATP leakage promotes lysosomal exocytosis via P2Y2 receptors. MG53 facilitates repair in type I cells through caveolar endocytosis, although its protective role in type II cells remains unclear. Type II cells likely facilitate resealing through ANXA7-dependent fusion of surfactant granules. During S. aureus infection, pneumocytes evade damage from pore-forming toxin by releasing decoy exosomes enriched in host receptor ADAM10. (ii) Gastric Epithelium: Mucus integrity is compromised during H. pylori infection and by amphiphilic molecules such as NSAIDs and alcohol. Pore formation by VacA disrupts microvilli organization upon CAPN1-mediated cleavage of ezrin. Cholesterol extraction and lipid peroxidation are achieved by virulence factors including cholesterol-α-glucoside transferase (CGT), γ-glutamyl transpeptidase (GGT), and urease (via monochloramine, NH4Cl). Gastric repair includes lysosomal exocytosis and annexins, whereas HSP70 activity alleviates chemical disruptions although its exact role remains unclear. Meanwhile, NSAIDs and alcohol elicit damage through direct interactions with plasma membrane phospholipids or indirectly via oxidative stress. Cytoprotective factors include calpains and prostaglandin E2 (PGE2), the latter of which stimulates bicarbonate (HCO3-) release via SLC26A9 to alleviate acid-induced injury. (iii) Intestinal Epithelium: Enterocytes rid bacterial pore-forming toxins (~ 1–2 nm) through cytoplasm extrusion, preceded by oxidative stress as evident by lipid droplet formation and mitochondrial damage. Pores are also removed through vesicle trafficking events and microvilli shedding. Dietary lectins can lead to microvilli abnormalities and inhibit mucus secretion in goblet cells which is a form of membrane resealing. Other dietary molecules, such as poly-unsaturated fatty acids (PUFA) and undigested gliadin peptide, can promote damage through lipid peroxidation and pyroptosis, respectively. (iv) Keratinocytes: During S. aureus infection, keratinocytes internalize α-toxin pores and release them via exosomes. Resistance is achieved through the filaggrin (FLG)-dependent release of acid sphingomyelinase to reduce the availability of exofacial sphingomyelin, an alternative receptor of α-toxin. Ultraviolet A irradiation causes lipid peroxidation that is alleviated by NRF2-dependent antioxidant defenses. Phospholipase D (PLD) activity promotes vesicle fusion events such as lysosomal exocytosis. Alongside repair, caveolar endocytosis can result in caspase-8-mediated apoptosis. (v) Endothelium: Endothelial cells buffer hemodynamic force through caveolae. Advanced glycation end products (AGEs) entice lipid peroxidation whereas overexpression of receptor for AGEs (RAGE) prevents F-actin remodeling required for resealing. Complement-induced damage triggers the release of von Willebrand factor (VWF) which can limit further complement deposition. (vi) Osteoblasts, Osteocytes: Bone cells experience nanoruptures during locomotion that can be repaired through exocytosis with an apparent role for dietary Vitamin E in limiting further oxidative damage. ATP leakage from the wound site initiates calcium-dependent mechanotransduction in nearby, uninjured cells through P2 receptors

Plasma membrane damage and repair in the liver, pancreas, nervous system, kidney, and muscle. Red arrows: sources of plasma membrane damage; black arrows: repair pathways; gray arrows: forms of cellular resistance. Human body was created with BioRender.com. (i) Hepatocytes: Alcohol and drug metabolism or the accumulation of lipids and bile acids can promote lipid peroxidation which is alleviated by glutathione peroxidase 4 (GPX4) activity. Basolateral wounds can be removed through membrane scission whereas apical protrusions are prone to rupture. Biliary phospholipids confer protection by reducing the ability of bile acids to solubilize membrane. Ischemic-reperfusion injury (I/R injury) triggers dysferlin-mediated exocytosis which may involve ANXA6 activity given its role in hepatocyte vesicle trafficking. (ii) Pancreatic Cells: Acinar cell damage can indirectly arise following exposure to stressors such as alcohol, drugs, and bile. Abnormally high levels of intracellular calcium prompt the fusion of zymogen granules (ZG) with lysosomes (L), leading to the premature activation of zymogens (e.g., trypsin) which inflict membrane damage upon leakage into the cytosol. Pancreatic β cells experience membrane damage from amylin aggregates in the extracellular environment that are typically prevented by insulin co-secretion. In both cell types, repair likely involves exocytosis based on the abundance of granules and lysosomes underlying the plasma membrane. (iii) Neurons: Neuronal membrane damage can arise from exposure to protein aggregates (e.g., β-amyloid) in the extra- or intra-cellular environment which elicit mechanical damage and oxidative stress. Depending on the protein, resistance against intracellular aggregation may be achieved through multivesicular body sorting and lysosomal degradation or exocytosis. Lesions from β-amyloid aggregates may be removed through caveolar endocytosis and ESCRT-III activity as observed in other cell types. Oxidative stress can lead to nanoruptures in axonal membrane which is inherently protected by myelin sheath. Demyelination can exacerbate membrane damage upon the release of myelin basic protein (MBP). Neuronal repair entails calpain activity and vesicle trafficking events such as exocytosis, endocytosis, and plugging. (iv) Proximal Tubule Epithelium: Renal cells experience lipid peroxidation during I/R injury and exposure to nephrotoxins which are alleviated by antioxidants such as GPX4 and sirtuins (SIRT). Physical breaches are repaired through membrane remodeling events including microvilli shedding, caveolar endocytosis and MG53-mediated vesicle recruitment. (v) Myocytes: Mechanical stress is buffered through the dystrophin glycoprotein complex which connects the extracellular matrix to the actin cortex. This complex also anchors nitric oxide synthase (nNOS) at the cell surface to prevent ischemic injury. Upon damage, calcium influx is amplified by voltage-gated calcium channels (VGCC) and the release of lysosomal stores (MCOLN1). Calcium uptake required for successful repair is achieved by the endoplasmic reticulum and mitochondria, the latter of which promotes redox-dependent RhoA activity to drive F-actin assembly. GRAF1 promotes dysferlin at the plasma membrane where it can facilitate lysosomal exocytosis and patching. Vesicle fusion can also be achieved upon the calpain-dependent cleavage of dysferlin into a syt-like molecule. Vesicle recruitment to the wound site is promoted by MG53 and SIRT1 activity. Annexins (A) also promote wound closure by forming a highly organized repair cap which may drive constriction. Membrane remodeling is further achieved by the recruitment of regulators, such as EHD and BIN1, in addition to ESCRT-III-mediated scission
Lung
The alveolar epithelium is comprised of type 1 pneumocytes which control gas exchange and are replenished by surfactant-producing type 2 pneumocytes. During normal respiration, alveolar cells experience mechanical stress that is alleviated by surfactant production, basement membrane elasticity, and an abundance of caveolae on type I cells [240, 254, 255]. Exceedingly high tidal volumes (e.g., ventilation therapy) can result in mechanical damage as observed in ventilator-induced lung injury [199, 256]. This type of mechanical injury is exacerbated by dysfunctional surfactant, common to several disorders including acute respiratory distress syndrome and pulmonary fibrosis [257].
In type 1 pneumocytes, mechanical injury leads to actin depolymerization and endomembrane recruitment [258]. Lysosomal exocytosis partially accounts for these vesicle fusion events, and in pneumocytes, this process is dependent on purinergic signaling [259]. Caveolar endocytosis, promoted by MG53 activity, is another prominent form of repair in type I cells [240, 260] and while MG53 also fosters resealing in type II cells, its role in this context remains unclear [261]. Abnormalities in caveolae may accelerate ventilator-induced damage in pulmonary fibrosis patients who display reduced levels of caveolin-1 and mislocalization of MG53 [241, 261]. It is foreseeable that functional differences between these two cell types, such as surfactant secretion, may influence their resistance to injury and repair capacity. For instance, type II cells may trigger the release of specialized surfactant granules given this is a Ca2+-dependent process dependent on repair machinery such as ANXA7 [262, 263].
Pneumocytes are also exposed to microbial and chemical insults following inhalation. During bacterial respiratory infections, pathogenic microbes produce pore-forming toxins that elicit pneumocyte death by direct lysis or indirectly via necroptosis [204, 205]. In the case of smaller pores, such as α-toxin, pneumocytes generate decoy exosomes enriched in toxin receptor to circumvent membrane insult [206]. Meanwhile, the inhalation of inert particles (e.g., asbestos) and cigarette smoke can promote pneumocyte damage by lipid peroxidation [200,201,202,203].
Upper digestive tract
Cells within the oral cavity, such as the gingival epithelium surrounding teeth, can experience mechanical damage from orthodontic tooth movement and brushing [207, 208]. Chemical disruptions can arise from tooth whitening agents and lead to damaging oxidative stress in periodontal ligament cells [209]. Similarly, exposure to cigarette smoke and moist smokeless tobacco—known inducers of oral lesions—appear to induce both mechanical damage and lipid peroxidation, the latter of which is detectable in the saliva of smokers [210, 211].
Further down the digestive tract, the esophageal epithelium is susceptible to damage from contact with refluxed stomach contents during gastroesophageal reflux disease [264, 265]. Early studies demonstrated exposure of esophageal tissue to stomach acid and pepsin leads to intracellular acidification and the onset of cell necrosis [212, 266]. Exactly how cell acidification results in a physical breach remains elusive. Alternatively, acid and bile exposure can provoke ROS and cytokine production to create a damaging inflammatory milieu [213, 264]. Both mechanisms likely result in membrane damage through lipid peroxidation, considering the ability of lipid-directed antioxidants to ameliorate reflux severity in vivo [267]. Repair defects in the esophageal epithelium may contribute to the pathogenesis of Barrett’s esophagus which arises from prolonged damage [268].
Stomach
Mucus lining the gastrointestinal epithelium provides physical and biochemical protection against extracellular stressors [269]. Weakening of this mucus layer often precedes gastric cell injury during Helicobacter pylori infection and frequent NSAID exposure [23, 270]. Upon achieving close contact with the apical membrane of gastric cells, H. pylori inflicts damage through an arsenal of virulence factors [215]. Physical breaches caused by the pore-forming toxin, VacA, can disrupt microvilli and the function of acid-secreting Parietal cells [271, 272]. Alternatively, H. pylori effectors can induce chemical disruptions such as cholesterol extraction [273] and lipid peroxidation through the formation of monochloramine, a lipophilic oxidant [274]. Accordingly, heightened levels of lipid peroxidation are observed in gastric biopsies from H. pylori-infected patients and such damage is exacerbated by the bacterial-induced degradation of host glutathione [275, 276]. During H. pylori infection, membrane repair events such as annexin recruitment and lysosomal exocytosis have been observed, with an apparent role for HSP70 in alleviating chemical disruptions [243, 274]. Intriguingly, the overexpression of ANXA4 in H. pylori-associated gastric tumors may confer a survival advantage through enhanced resealing efforts [243, 277].
Non-steroidal anti-inflammatory drugs (e.g., aspirin) are commonly used for relieving inflammation and pain; however, frequent usage is associated with gastrointestinal lesions (e.g., peptic ulcers) [278]. Chemical disruptions can arise from direct interactions with plasma membrane phospholipids or as a consequence of mitochondrial dysfunction [279, 280]. NSAIDs can further exacerbate injury by inhibiting cyclooxygenase-dependent production of prostaglandin E2 (PGE2), a cytoprotective lipid mediator against gastric tissue damage [281, 282]. While its role in plasma membrane integrity remains elusive, PGE2 may enhance lysosomal exocytosis or alleviate acid-induced damage as previously described [283, 284].
Alcohol intake is another causative agent of gastritis and similar to NSAIDs, ethanol can compromise mucus integrity and promote lipid peroxidation [214, 285]. Early studies demonstrated the ability of absolute ethanol to cause gastric cell necrosis, presumably through extensive chemical disruptions [63, 281]. Rapid mucus secretion was observed in this injury model and later demonstrated to be an intrinsic response to membrane damage in surface mucus cells [63, 119]. In this context, repair may be achieved by gastric-specific calpains as mice deficient in CAPN8 or CAPN9 are more susceptible to ethanol-induced injury [242]. However, damage was only assessed on a histological level so the role of these calpains in gastric cell plasma membrane integrity remains unclear. Intriguingly, the authors highlighted missense variants in CAPN8 and CAPN9 that correspond to pathogenic mutations in CAPN3, which is relevant to muscular dystrophy [242, 250]. This raises the notion that genetic defects influencing gastric cell membrane integrity may similarly underlie gastropathies.
Intestine
The intestinal epithelium persists in a harsh environment and experiences membrane damage even at resting state [286]. Two major sources of damage include pathogenic microbes and ingested chemical stressors [182, 287]. Several enteric pathogens induce intestinal damage through pore-forming toxins that target apical microvilli and intercellular junctions [288, 289]. In C. elegans, enterocytes remove small pores (~ 1–2 nm) through endocytosis and microvilli shedding driven by exocytic events [288]. This leads to extensive disruptions in the underlying network of intermediate filaments, which serve to provide resistance against both chemical and mechanical stress [290]. Similar pore-induced damage in D. melanogaster enterocytes causes cytoplasm extrusion resulting in cell thinning, and this response is conserved in mammalian cells [291]. Exactly how such small breaches trigger this dramatic repair response is unclear, but oxidative stress is one potential candidate [182, 291].
In the small intestine, NSAIDs such as indomethacin can promote oxidative stress leading to plasma membrane tears and ulceration in vivo [292]. Similar to the stomach, prolonged NSAID exposure dampens intestinal mucus integrity which promotes enterocyte exposure to luminal aggressors including bile and bacteria [23, 69]. It appears that cells at the tips of villi are particularly susceptible to NSAID-induced lipid peroxidation [293] which implies inherent differences in epithelial resistance along the villus-crypt axis.
Dietary molecules, such as lectins, can lead to structural abnormalities in apical microvilli that are self-limiting [216, 294]. Distinct from pore-induced damage, lectin exposure appears to alter membrane fluidity and promote the formation of nanoruptures [295, 296]. Lectins also inhibit damage-induced mucus secretion in Goblet cells, which is a form of exocytosis-mediated repair [297]. Dietary molecules can also trigger membrane damage through indirect mechanisms as observed in celiac disease—certain peptides of gliadin, a driver of the disease, activate pyroptosis and the onset of pore-induced damage [298]. Meanwhile, excessive intake of iron and poly-unsaturated fatty acids can accelerate intestinal lipid peroxidation, the latter of which is particularly relevant to IBD [19, 217].
Skin
Skin is comprised of cellular diverse layers that form a barrier to the outside environment. Keratinocytes, which are abundant in the outermost layer known as the epidermis, experience mechanical damage during locomotion and pruritus (itch), the latter of which is common to many skin disorders [2, 219]. Exposed to a non-sterile environment, pathogens such as Staphylococcus aureus can directly inflict keratinocyte damage through α-toxin pores which are removed by endocytosis and subsequent release through exosomes [139]. Intriguingly, mature keratinocytes achieve resistance against pore-induced injury by secreting sphingomyelinase via lamellar bodies to limit the availability of α-toxin receptor, a strategy disrupted in atopic dermatitis patients with mutations in filaggrin (FLG) [221]. In the case of chronic intracellular S. aureus infections, keratinocytes are susceptible to lethal damage from the complement system to rid persistent bacteria by eliminating the host cell—highlighting a case where membrane damage is for the greater good [299].
Ultraviolet A (UVA) exposure from sunlight is a prominent chemical stressor of skin that promotes lipid peroxidation and in extreme cases, ferroptosis [300, 301]. Cell type-specific differences in the extent of UVA damage are highly dependent on intracellular levels of iron, as evident by dermal fibroblasts which are more prone to necrosis than keratinocytes [220]. Keratinocyte resistance against lipid peroxidation is also a reflection of NRF2 activity, a transcriptional regulator of antioxidants important for tissue repair in skin [302, 303]. In response to damaging UVA exposure, keratinocytes undergo lysosomal exocytosis to achieve membrane resealing [304]. In this case, however, the extracellular release of cathepsins was found to trigger caspase-8-dependent apoptosis which may serve to eliminate damaged cells in a discrete manner rather than lytic cell death. The importance of exocytosis for keratinocyte repair is supported by the involvement of phospholipase D, a regulator of vesicle fusion, in resealing mechanical wounds [305].
Vasculature
Endothelial cells line the vascular network and experience hemodynamic stress during increased cardiac output that can elicit damage, if not for caveolae [197, 306]. Stressors in circulation, such as bacterial pore-forming toxins and host complement, are a prominent source of membrane injury and sustained damage can lead to vascular dysfunction [226, 307]. In type 2 diabetes, hyperglycemia stimulates the formation of advanced glycation end products (AGEs) which can react with phospholipids to stimulate membrane oxidation [308]. Furthermore, AGEs can compromise endothelial repair through F-actin dysregulation, albeit indirectly, via the receptor for AGEs (RAGE) [237, 309]. The interspersed nature of the vasculature system also exposes the endothelium to tissue-specific stressors. For instance in Alzheimer’s disease, β-amyloid peptide can associate with endothelial membrane to elicit neurovascular damage with an emerging role for AGEs in antagonizing this process [222, 224, 310].
In response to complement-induced damage, endothelial cells facilitate repair through the calcium-dependent fusion of Weibel-Palade bodies (WPB) involving ANXA2 [225, 311,312,313]. The mobilization and fusion of these granules is critical for resealing as damage is exacerbated in cells from von Willebrand disease patients lacking WPBs [225]. Alongside relieving membrane tension, WPB fusion promotes the extracellular release of the clotting protein von Willebrand factor (VWF) to protect against further injury by complement [314]. Defects in this form of repair may contribute to vascular damage in Behcet’s disease considering the presence of ANXA2 autoantibodies [244, 315].
Bone
Bone formation is attributed to osteoblasts which eventually transition to signal-transducing osteocytes within mineralized tissue. During physiological loading or injury, mechanotransduction promotes bone remodeling through intercellular calcium waves which can be initiated by single plasma membrane damage events [26, 179, 316]. In weight-bearing bones, osteoblasts and osteocytes experience mechanical damage that triggers purinergic-dependent signaling and calcium uptake in nearby, non-wounded cells to stimulate biochemical signals [26, 179]. These lesions are predicted to be nanoruptures (~ 5 nm) based on the leakage of intracellular content and minimal requirement for calpain-dependent repair [26, 34, 179]. During aging, bone becomes less mechanoresponsive, and recent evidence suggests this is due to survival-based selection of osteocytes that display enhanced membrane repair [317].
Despite a clear role for damage in bone mechanotransduction, little is known about plasma membrane repair in these cell types. In osteoblasts, lesions are repaired through protein kinase-dependent exocytic events [26]; however, the identity of such vesicles and how their fusion facilitates wound closure is unknown. Similar forms of vesicle-mediated repair likely occur in osteocytes, which may also shed microvesicles as observed in response to mechanical deformation [318]. Furthermore, antioxidants have a defining role in promoting osteocyte repair as vitamin E-deficient mice display a higher frequency of damage events following acute exercise [319]..
Liver
Hepatocytes mediate metabolism and detoxification, both of which are energetically demanding processes that generate ROS. Under normal conditions, liver redox homeostasis is critical to prevent the onset of lipid peroxidation, as evident by the premature death of mice lacking GPX4 in hepatocytes [198]. Hepatocytes encounter a multitude of stressors that can promote damaging oxidative stress and contribute to liver disease [227]. For instance in alcoholic liver disease, ethanol can promote lipid peroxidation [320] whereas in non-alcoholic fatty liver disease, intracellular lipid accumulation instigates ROS production [321]. Depending on their concentration, bile acids can elicit hepatocyte membrane damage either directly by solubilizing lipids or indirectly through oxidative stress [322]. These forms of damage are prominent during cholestasis (i.e., bile acid accumulation) which can arise from biliary duct obstruction or impairments in bile acid homoeostasis [323]. Such impairments can be genetically linked as evident by mutations in MDR3, a translocator of biliary phospholipids which prevent the cytotoxic association of bile acids with the plasma membrane [228].
Our understanding of how hepatocytes facilitate resealing is limited. In response to damaging oxidative stress, hepatocytes form membrane protrusions on both apical and basolateral membrane [324, 325]. In vivo imaging revealed that basolateral structures can be released into circulation whereas apical blebs tend to rupture and indicate irreversible cell death. These forms of membrane remodeling likely coincide with dysferlin (DYSF)-dependent vesicle fusion events [326]. Annexin-mediated repair in hepatocytes is enticing given that ANXA6 comprises ~ 0.25% of total hepatic protein and facilitates apical vesicle trafficking [327]. These findings highlight an important role for hepatocyte repair against chemical damage although much remains to be determined.
Pancreas
The pancreas is an exocrine gland comprised of acinar and β cells which secrete zymogen and insulin granules, respectively. In acinar cells, physiological signals lead to zymogen granule fusion through the controlled release of calcium from internal stores [328]. However, abnormally high levels of intracellular calcium can lead to the premature activation of zymogens (e.g., trypsin) that leak into the cytosol and elicit damage. This form of acinar cell destruction encompasses acute pancreatitis (AP), and major initiators include alcohol abuse and biliary obstruction [328, 329]. These classical stressors promote cytotoxic calcium levels at the expense of internal calcium stores and/or activating plasma membrane calcium channels [330]. While plasma membrane damage can be observed at early timepoints of experimental AP [331, 332], in vitro evidence suggests that plasma membrane damage is an effect, rather than a cause of zymogen activation [230]. Nonetheless, acinar cells likely employ exocytosis-mediated repair given the abundance of lysosomes and secretory granules underlying the apical membrane and the involvement of ANXA6 in vesicle release [333].
Alongside insulin, amylin is released through β cell granules where it regulates glycemic control. In type 2 diabetes patients, there is a link between amylin aggregation and reduced β cells [334, 335]. Amylin is an intrinsically disordered protein with the propensity to aggregate into fibrils that can physically breach the plasma membrane and promote damaging oxidative stress [336,337,338]. Resistance against this form of injury is partially achieved through the co-secretion of insulin in much higher amounts to prevent amylin aggregation [229]. In contrast, other factors in the microenvironment such as dietary lipids can accelerate fibril formation and exacerbate damage [339, 340]. Considering apoptosis is the leading cause of β cell death [341], amylin-induced membrane damage may stimulate apoptotic cues such as cytotoxic calcium influx and CAPN2 hyperactivation [342, 343]. Membrane repair in β cells has yet to be studied, although granule exocytosis would be consistent with resealing in other secretory cell types [119].
Nervous system
While the nervous system is comprised of many different cell types, plasma membrane damage and repair are best described in neurons. Neurons transmit signals through 3 structurally diverse parts of the cell: dendrites, the cell body (i.e., soma), and myelin-sheath coated axons. Axonal damage is prevalent in a broad range of central nervous system disorders [344]. During traumatic brain injury, physical trauma can compromise axonal membrane, particularly in superficial layers of the brain [12]. If such trauma disrupts the blood-brain barrier or leads to occlusion (e.g., stroke), neurons are susceptible to secondary injuries such as lipid peroxidation during ischemia [232, 345]. Damaging levels of oxidative stress are suspected to cause nanoruptures in axons within neuroinflammatory lesions, common to multiple sclerosis and autoimmune encephalomyelitis [27, 233]. Axonal membrane resistance to mechanical and chemical damage is partially achieved through myelin sheath, as demyelinated axons or regions inherently exposed (e.g., Nodes of Ranvier) are more prone to injury [27, 233, 346]. Demyelination, common to several neurodegenerative disorders, can further exacerbate injury upon the release of myelin basic protein which has the propensity to breach neuronal membrane [347].
Protein aggregates are a commonality of several neurodegenerative disorders including Alzheimer’s (β-amyloid), Parkinson’s (α-synuclein), and Creutzfeldt-Jakob disease (prion) [231]. Upon processing or misfolding, these intrinsically disordered proteins can form aggregates in the extra- and intra-cellular environment that elicit mechanical damage (e.g., lipid extraction, pore-formation) and promote lipid peroxidation [61, 348, 349]. Neuronal resistance against intracellular aggregate formation is partially achieved through multivesicular body sorting, lysosomal degradation, and even lysosomal exocytosis [350,351,352]. In the case of β-amyloid, it appears aggregate-induced damage elicits a similar repair response as pore-forming toxins (e.g., caveolar endocytosis, ESCRT activity); however, confirmatory studies in neurons are required [353, 354]. While there is evidence to suggest plasma membrane composition is altered in neurodegenerative disease and through aging [355,356,357], whether such alterations impact neuronal resistance and repair against protein aggregate-induced damage remains to be determined.
Traditionally, axonal membrane repair has been studied following transection and is extensively reviewed elsewhere [358, 359]. In brief, calcium influx through the wound site is amplified by intracellular stores and voltage-gated calcium channels that lead to calpain activation and the recruitment of vesicles through microtubule transport [358, 360, 361]. At the wound site, immediate closure can be achieved through membrane-based plugs, whereas vesicle fusion events and contraction eventually lead to permanent wound closure [158, 358].
Kidney
The kidney facilitates blood filtration through renal epithelium organized into functional units termed nephrons that are further subdivided based on role. The proximal tubule is the main site of reabsorption and features high levels of oxidative metabolism to fuel transporter activity [362]. Consequently, the proximal tubule is a common site of damaging oxidative stress arising from I/R injury or exposure to nephrotoxins (e.g., chemotherapeutics) [362]. Proximal tubule cell injury encompasses many forms of acute kidney injuries (AKI) that can lead to chronic kidney disease [234]. Depending on location along the proximal tubule (i.e., segments 1–3), renal cells display differences in their resistance to chemical damage. For instance, ischemic damage leads to extensive necrosis of segment 3 whereas cells in the preceding segments repair successfully [55]. Such differences in membrane resistance are partially attributed to antioxidant production, and in the case of segment 1 cells, the absence of ROS-producing peroxisomes [363, 364].
In vivo, renal lipid peroxidation is actively repaired by GPX4 to prevent ferroptosis and the onset of AKI [365]. Renal protection against oxidative damage is also achieved by members of the sirtuin family (SIRT), a group of NAD+-dependent deacetylases that regulate redox signaling. For instance in response to cisplatin-induced AKI, SIRT1 and SIRT3 can alleviate oxidative stress generated by peroxisomes and mitochondria, respectively [366, 367]. Upon ischemic injury, proximal cells undergo extensive membrane remodeling events including caveolar endocytosis and microvilli shedding [55, 368]. These forms of repair may be facilitated by MG53 which was found to be protective against AKI in mice [245].
Muscle
During muscle function, myocytes buffer mechanical stress by stabilizing the plasma membrane (i.e., sarcolemma) through the dystrophin glycoprotein complex in addition to an abundance of caveolae [11, 196]. However, eccentric muscle contractions can lead to physical breaches in the sarcolemma that require active repair [369]. Genetic abnormalities in either myocyte resistance or repair encompass myopathies characterized by progressive muscle weakening, collectively referred to as muscular dystrophy (MD) [11]. Myocyte resistance against mechanical stress is weakened by mutations in dystrophin and dystroglycan that disrupt association of the sarcolemma with the cytoskeleton and extracellular matrix, respectively [246, 247]. In the case of Duchenne’s MD, dystrophin deficiency can indirectly result in ischemic damage due to the absence of nitric oxide activity at the cell surface to promote vasodilation [236]. Alternatively, other forms of MD can weaken myocyte resistance through defects in caveolae formation and improper muscle regeneration [250, 370].
Myocyte repair has been extensively studied in physiology and disease [371]. A physical breach in the sarcolemma leads to a rise in intracellular calcium due to entry through the wound site, voltage-gated calcium channels (VGCC), and lysosomal release through MCOLN1 [111, 112]. Cytotoxic calcium levels are partially prevented by organelle uptake as observed in mitochondria and endoplasmic reticulum via the mitochondrial calcium uniporter (MCU) and anoctamin-5 (ANO5), respectively [173, 174]. Calcium influx activates CAPN1 and CAPN2 activity essential for resealing tears in muscle [34, 127]. Dysferlin mediates vesicle fusion events in addition to promoting lysosomal exocytosis, and genetic abnormalities in this factor underlie defective repair in limb-girdle MD type 2B [125, 126]. Other potential regulators of exocytosis-mediated repair include GRAF1 and SIRT1, although the influence of the latter on vesicle dynamics remains unclear [372, 373]. Sarcolemma resealing is also facilitated by ESCRT-III activity and annexin recruitment, whereby annexins form an actin-dependent structure termed a repair cap [167, 374, 375]. Based on electron micrographs of this structure [161], the cap is likely equivalent to the complex network of membrane protrusions seen at wounds in other cell types [109, 153]. The accumulation of MG53, dysferlin, EHD1, and BIN1 adjacent to the repair cap suggests simultaneous vesicle fusion events and membrane remodeling to relieve tension during annexin-mediated closure [167, 376]. Intriguingly, a genetic variant of ANXA6 was identified as a modifier of MD in part through its ability to disrupt dysferlin recruitment to the repair cap [249, 377]. This reveals that inherent defects in plasma membrane repair can be polygenic in nature.
Heart
The heart is under continuous mechanical stress that potentiates plasma membrane damage in cardiomyocytes, aortic endothelium, and valve interstitial cells [3, 253, 306]. Following intensive cardiac output, membrane repair is critical to prevent the onset of heart pathologies such as cardiac arrest and ventricular injuries [238, 378]. As in skeletal muscle, cardiomyocyte resealing is dependent on calpain activity and dysferlin-dependent vesicle fusion [238, 378]. Defects in dysferlin-mediated repair can be genetically linked (e.g., LGMD2B) [379] or arise during viral myocarditis to exacerbate damage as a cell-cell spread tactic [239]. MG53 is also protective against mechanical damage in cardiomyocytes and this extends to I/R injury where it participates in ischemic preconditioning against subsequent stress [380,381,382].
Immune system
The dynamic nature of immune cells poses a challenge for identifying physiological stressors of plasma membrane integrity. However, it appears damage is cell type-specific given the highly specialized functions of immune cells. For instance, the release of neutrophil extracellular traps entails chromatin swelling that provides enough mechanical force to rupture the plasma membrane [383]. Meanwhile, phagocytes can experience damage following interactions with pathogens and unique stressors amidst inflammation (e.g., cholesterol crystals in atherosclerotic plaques) [71, 384]. Immune cell resistance against a particular stressor appears to be highly dependent on plasma membrane composition, as evident by the ability of cytotoxic T lymphocytes to repel perforin-induced damage destined for target cells [385]. Furthermore, certain stressors such as cholesterol-dependent pore-forming toxin display a preference for immune cell lineage (i.e., myeloid versus lymphoid) [35], highlighting the need for such fundamental comparisons when assessing damage.
Contrary to our understanding of how immune cells resolve infection, little is known about how they resolve plasma membrane wounds. In macrophages, lysosomal exocytosis serves a dual purpose: it alleviates membrane damage induced by intracellular pathogens and exports cytotoxic cargo to fend off extracellular bacteria [386, 387]. The membrane remodeling events that accompany repair are not always favorable given the importance of cell surface receptors for immune cell function. In B cells, lysosomal exocytosis can disrupt B cell receptor signaling by promoting the internalization of signaling domains which are required for its activation [185]. Moreover, pore-induced membrane shedding in macrophages can lead to the removal of cytokine receptors from the cell surface resulting in immune suppression [186]. While membrane repair can promote survival in immune cells, it appears to compromise function although how long these defects persist following wound resolution remains unclear.
Conclusions and perspectives
Plasma membrane integrity is a ubiquitous requirement of cells, regardless of tissue function. Tissue-specific stressors challenge plasma membrane resistance to which cells rely on active repair mechanisms. It is now apparent that cells encounter damage “in sickness and in health” and that membrane damage does not always lead to cell death. Instead, damage is a gradient that can dramatically alter the intracellular landscape and even facilitate intercellular signaling critical to tissue function.
Far more is known about what can damage cells as opposed to how they heal. While membrane repair pathways are generally conserved, it is apparent that cell type-specific adaptations can have broader effects depending on the tissue environment (e.g., mucus secretion in the gut, clotting factor release by endothelium). However, these unique mechanisms, let alone the ability to undergo repair, have yet to be studied in many cell types relevant to disease. Major technical strides towards resolving plasma membrane damage and repair in vivo hold great promise for understanding the importance of this process under physiological stress.
It is now apparent that defects in plasma membrane resistance and repair can cause human disease beyond muscular dystrophies. Genetic mutants may be causative of defects in membrane integrity, with additional genetic factors contributing to disease heterogeneity. Meanwhile in other conditions, environmental stress can be the dominating factor resulting in traumatic or acute injuries. Initial evidence suggests such gene-environment interactions contribute to the pathogenesis of multifactorial diseases which remains a promising avenue of study. Ultimately, understanding plasma membrane damage and repair on a single cell basis can provide therapeutic insight towards wound resolution on a tissue level.
Availability of data and materials
Not applicable.
References
Newham DJ, Jones DA, Edwards RHT. Plasma creatine kinase changes after eccentric and concentric contractions. Muscle Nerve. 1986;9:59–63 https://doi.org/10.1002/mus.880090109.
McNeil PL, Ito S. Molecular traffic through plasma membrane disruptions of cells in vivo. J Cell Sci. 1990;96:549–56.
Clarke MSF, Caldwell RW, Chiao H, Miyake K, McNeil PL. Contraction-induced cell wounding and release of fibroblast growth factor in heart. Circ Res. 1995;76:927–34 https://doi.org/10.1161/01.RES.76.6.927.
Brito C, Cabanes D, Sarmento Mesquita F, Sousa S. Mechanisms protecting host cells against bacterial pore-forming toxins. Cell Mol Life Sci. 2019;76:1319–39 https://doi.org/10.1007/s00018-018-2992-8.
Liu X, Lieberman J. Knocking ‘em dead: pore-forming proteins in immune defense. Annu Rev Immunol. 2020;38:455–85 https://doi.org/10.1146/annurev-immunol-111319-023800.
Zhang Y, Chen X, Gueydan C, Han J. Plasma membrane changes during programmed cell deaths. Cell Res. 2018;28:9–21 https://doi.org/10.1038/cr.2017.133.
Jaiswal JK, Lauritzen SP, Scheffer L, Sakaguchi M, Bunkenborg J, Simon SM, et al. S100A11 is required for efficient plasma membrane repair and survival of invasive cancer cells. Nat Commun. 2014;5:3795 https://doi.org/10.1038/ncomms4795.
Horn A, Jaiswal JK. Cellular mechanisms and signals that coordinate plasma membrane repair. Cell Mol Life Sci. 2018;75:3751–70 https://doi.org/10.1007/s00018-018-2888-7.
Gong Y-N, Guy C, Olauson H, Becker JU, Yang M, Fitzgerald P, et al. ESCRT-III acts downstream of MLKL to regulate necroptotic cell death and its consequences. Cell. 2017;169:286–300.e16 https://doi.org/10.1016/j.cell.2017.03.020.
Skowyra ML, Schlesinger PH, Naismith TV, Hanson PI. Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science. 2018;360:eaar5078 https://doi.org/10.1126/science.aar5078.
Cooper ST, Head SI. Membrane injury and repair in the muscular dystrophies. Neurosci. 2015;21:653–68 https://doi.org/10.1177/1073858414558336.
LaPlaca MC, Lessing MC, Prado GR, Zhou R, Tate CC, Geddes-Klein D, et al. Mechanoporation is a potential indicator of tissue strain and subsequent degeneration following experimental traumatic brain injury. Clin Biomech. 2019;64:2–13 https://doi.org/10.1016/j.clinbiomech.2018.05.016.
Tan JMJ, Mellouk N, Osborne SE, Ammendolia DA, Dyer DN, Li R, et al. An ATG16L1-dependent pathway promotes plasma membrane repair and limits Listeria monocytogenes cell-to-cell spread. Nat Microbiol. 2018;3:1472–85 https://doi.org/10.1038/s41564-018-0293-5.
Corrotte M, Castro-Gomes T, Koushik AB, Andrews NW. Approaches for plasma membrane wounding and assessment of lysosome-mediated repair responses. In: Methods in Cell Biology; 2015. p. 139–158. https://doi.org/10.1016/bs.mcb.2014.11.009.
Agmon E, Solon J, Bassereau P, Stockwell BR. Modeling the effects of lipid peroxidation during ferroptosis on membrane properties. Sci Rep. 2018;8:5155 https://doi.org/10.1038/s41598-018-23408-0.
Braughler JM, Duncan LA, Chase RL. The involvement of iron in lipid peroxidation. Importance of ferric to ferrous ratios in initiation. J Biol Chem. 1986;261:10282–9.
Zhang X, Barraza KM, Beauchamp JL. Cholesterol provides nonsacrificial protection of membrane lipids from chemical damage at air–water interface. Proc Natl Acad Sci. 2018;115:3255–60 https://doi.org/10.1073/pnas.1722323115.
Ayala A, Muñoz MF, Arguelles S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev. 2014;2014:1–31 https://doi.org/10.1155/2014/360438.
Mayr L, Grabherr F, Schwärzler J, Reitmeier I, Sommer F, Gehmacher T, et al. Dietary lipids fuel GPX4-restricted enteritis resembling Crohn’s disease. Nat Commun. 2020;11:1775 https://doi.org/10.1038/s41467-020-15646-6.
Adibhatla RM, Hatcher JF. Phospholipase A 2 , reactive oxygen species, and lipid peroxidation in CNS pathologies. BMB Rep. 2008;41:560–7 https://doi.org/10.5483/BMBRep.2008.41.8.560.
Mora-Obando D, Fernández J, Montecucco C, Gutiérrez JM, Lomonte B. Synergism between basic Asp49 and Lys49 phospholipase A2 myotoxins of viperid snake venom in vitro and in vivo. PLoS One. 2014;9:e109846 https://doi.org/10.1371/journal.pone.0109846.
Biswas A, Kashyap P, Datta S, Sengupta T, Sinha B. Cholesterol depletion by MβCD enhances cell membrane tension and its variations-reducing integrity. Biophys J. 2019;116:1456–68 https://doi.org/10.1016/j.bpj.2019.03.016.
Bjarnason I, Scarpignato C, Holmgren E, Olszewski M, Rainsford KD, Lanas A. Mechanisms of damage to the gastrointestinal tract from nonsteroidal anti-inflammatory drugs. Gastroenterology. 2018;154:500–14 https://doi.org/10.1053/j.gastro.2017.10.049.
Goldstein DB. Effect of alcohol on cellular membranes. Ann Emerg Med. 1986;15:1013–8 https://doi.org/10.1016/S0196-0644(86)80120-2.
Cooper ST, McNeil PL. Membrane repair: mechanisms and pathophysiology. Physiol Rev. 2015;95:1205–40 https://doi.org/10.1152/physrev.00037.2014.
Mikolajewicz N, Zimmermann EA, Willie BM, Komarova SV. Mechanically stimulated ATP release from murine bone cells is regulated by a balance of injury and repair. Elife. 2018;7:e37812 https://doi.org/10.7554/eLife.37812.
Witte ME, Schumacher A-M, Mahler CF, Bewersdorf JP, Lehmitz J, Scheiter A, et al. Calcium influx through plasma-membrane nanoruptures drives axon degeneration in a model of multiple sclerosis. Neuron. 2019;101:615–624.e5 https://doi.org/10.1016/j.neuron.2018.12.023.
Jimenez AJ, Maiuri P, Lafaurie-Janvore J, Divoux S, Piel M, Perez F. ESCRT machinery is required for plasma membrane repair. Science. 2014;343:1247136 https://doi.org/10.1126/science.1247136.
McNeil PL, Khakee R. Disruptions of muscle fiber plasma membranes: role in exercise-induced damage. Am J Pathol. 1992;140:1097–109.
Peraro MD, van der Goot FG. Pore-forming toxins: ancient, but never really out of fashion. Nat Rev Microbiol. 2016;14:77–92 https://doi.org/10.1038/nrmicro.2015.3.
Bouillot S, Reboud E, Huber P. Functional consequences of calcium influx promoted by bacterial pore-forming toxins. Toxins (Basel). 2018;10:387 https://doi.org/10.3390/toxins10100387.
Walev I, Martin E, Jonas D, Mohamadzadeh M, Müller-Klieser W, Kunz L, et al. Staphylococcal alpha-toxin kills human keratinocytes by permeabilizing the plasma membrane for monovalent ions. Infect Immun. 1993;61:4972–9 https://doi.org/10.1128/IAI.61.12.4972-4979.1993.
von Hoven G, Rivas AJ, Neukirch C, Meyenburg M, Qin Q, Parekh S, et al. Repair of a bacterial small β-barrel toxin pore depends on channel width. MBio. 2017;8 https://doi.org/10.1128/mBio.02083-16.
Piper A-K, Sophocleous RA, Ross SE, Evesson FJ, Saleh O, Bournazos A, et al. Loss of calpains-1 and -2 prevents repair of plasma membrane scrape injuries, but not small pores, and induces a severe muscular dystrophy. Am J Physiol Physiol. 2020;318:C1226–37 https://doi.org/10.1152/ajpcell.00408.2019.
Larpin Y, Besançon H, Iacovache M, Babiychuk VS, Babiychuk EB, Zuber B, et al. Bacterial pore-forming toxin pneumolysin: cell membrane structure and microvesicle shedding capacity determines differential survival of immune cell types. FASEB J. 2020;34:1665–78 https://doi.org/10.1096/fj.201901737RR.
Drücker P, Bachler S, Wolfmeier H, Schoenauer R, Köffel R, Babiychuk VS, et al. Pneumolysin-damaged cells benefit from non-homogeneous toxin binding to cholesterol-rich membrane domains. Biochim Biophys Acta Mol Cell Biol Lipids. 2018;1863:795–805 https://doi.org/10.1016/j.bbalip.2018.04.010.
Howard AC, McNeil AK, McNeil PL. Promotion of plasma membrane repair by vitamin E. Nat Commun. 2011;2:597 https://doi.org/10.1038/ncomms1594.
Mulroy MJ, Henry WR, McNeil PL. Noise-induced transient microlesions in the cell membranes of auditory hair cells. Hear Res. 1998;115:93–100 https://doi.org/10.1016/S0378-5955(97)00181-0.
Saito K, Miyake K, McNeil PL, Kato K, Yago K, Sugai N. Plasma membrane disruption underlies injury of the corneal endothelium by ultrasound. Exp Eye Res. 1999;68:431–7 https://doi.org/10.1006/exer.1998.0626.
Dalal ND, Lee RC. Treatment of burn injury by cellular repair. J Craniofac Surg. 2008;19:903–6 https://doi.org/10.1097/SCS.0b013e318175b541.
Vahidkhah K, Cordasco D, Abbasi M, Ge L, Tseng E, Bagchi P, et al. Flow-induced damage to blood cells in aortic valve stenosis. Ann Biomed Eng. 2016;44:2724–36 https://doi.org/10.1007/s10439-016-1577-7.
Kuypers FA. Red cell membrane damage. J Heart Valve Dis. 1998;7:389–95.
Cranmer SL, Ashworth KJ, Yao Y, Berndt MC, Ruggeri ZM, Andrews RK, et al. High shear–dependent loss of membrane integrity and defective platelet adhesion following disruption of the GPIbα-filamin interaction. Blood. 2011;117:2718–27 https://doi.org/10.1182/blood-2010-07-296194.
Barnes JM, Nauseef JT, Henry MD. Resistance to fluid shear stress is a conserved biophysical property of malignant cells. PLoS One. 2012;7:e50973 https://doi.org/10.1371/journal.pone.0050973.
Egan K, Cooke N, Kenny D. Living in shear: platelets protect cancer cells from shear induced damage. Clin Exp Metastasis. 2014;31:697–704 https://doi.org/10.1007/s10585-014-9660-7.
Lauritzen SP, Boye TL, Nylandsted J. Annexins are instrumental for efficient plasma membrane repair in cancer cells. Semin Cell Dev Biol. 2015;45:32–8 https://doi.org/10.1016/j.semcdb.2015.10.028.
Machado E, White-Gilbertson S, van de Vlekkert D, Janke L, Moshiach S, Campos Y, et al. Regulated lysosomal exocytosis mediates cancer progression. Sci Adv. 2015;1:e1500603 https://doi.org/10.1126/sciadv.1500603.
Leung C, Yu C, Lin MI, Tognon C, Bernatchez P. Expression of myoferlin in human and murine carcinoma tumors. Am J Pathol. 2013;182:1900–9 https://doi.org/10.1016/j.ajpath.2013.01.041.
Brock J, Midwinter K, Lewis J, Martin P. Healing of incisional wounds in the embryonic chick wing bud: characterization of the actin purse-string and demonstration of a requirement for Rho activation. J Cell Biol. 1996;135:1097–107 https://doi.org/10.1083/jcb.135.4.1097.
Sciacca MFM, Tempra C, Scollo F, Milardi D, La Rosa C. Amyloid growth and membrane damage: current themes and emerging perspectives from theory and experiments on Aβ and hIAPP. Biochim Biophys Acta Biomembr. 2018;1860:1625–38 https://doi.org/10.1016/j.bbamem.2018.02.022.
Nazemidashtarjandi S, Farnoud AM. Membrane outer leaflet is the primary regulator of membrane damage induced by silica nanoparticles in vesicles and erythrocytes. Environ Sci Nano. 2019;6:1219–32 https://doi.org/10.1039/C8EN01267A.
Hirano S, Kanno S, Furuyama A. Multi-walled carbon nanotubes injure the plasma membrane of macrophages. Toxicol Appl Pharmacol. 2008;232:244–51 https://doi.org/10.1016/j.taap.2008.06.016.
Chen KL, Bothun GD. Nanoparticles meet cell membranes: probing nonspecific interactions using model membranes. Environ Sci Technol. 2014;48:873–80 https://doi.org/10.1021/es403864v.
Zhou T, Prather E, Garrison D, Zuo L. Interplay between ROS and antioxidants during ischemia-reperfusion injuries in cardiac and skeletal muscle. Int J Mol Sci. 2018;19:417 https://doi.org/10.3390/ijms19020417.
Venkatachalam MA, Bernard DB, Donohoe JF, Levinsky NG. Ischemic damage and repair in the rat proximal tubule: differences among the S1, S2, and S3 segments. Kidney Int. 1978;14:31–49 https://doi.org/10.1038/ki.1978.87.
Poli G, Albano E, Dianzani MU. The role of lipid peroxidation in liver damage. Chem Phys Lipids. 1987;45:117–42 https://doi.org/10.1016/0009-3084(87)90063-6.
AlZahrani K, Al-Sewaidan HA. Nanostructural changes in the cell membrane of gamma-irradiated red blood cells. Indian J Hematol Blood Transfus. 2017;33:109–15 https://doi.org/10.1007/s12288-016-0657-z.
Bäumler W, Regensburger J, Knak A, Felgenträger A, Maisch T. UVA and endogenous photosensitizers – the detection of singlet oxygen by its luminescence. Photochem Photobiol Sci. 2012;11:107–17 https://doi.org/10.1039/C1PP05142C.
Yusupov M, Wende K, Kupsch S, Neyts EC, Reuter S, Bogaerts A. Effect of head group and lipid tail oxidation in the cell membrane revealed through integrated simulations and experiments. Sci Rep. 2017;7:5761 https://doi.org/10.1038/s41598-017-06412-8.
Gunn AP, Wong BX, Johanssen T, Griffith JC, Masters CL, Bush AI, et al. Amyloid-β peptide Aβ3pE-42 induces lipid peroxidation, membrane permeabilization, and calcium influx in neurons. J Biol Chem. 2016;291:6134–45 https://doi.org/10.1074/jbc.M115.655183.
Angelova PR, Choi ML, Berezhnov AV, Horrocks MH, Hughes CD, De S, et al. Alpha synuclein aggregation drives ferroptosis: an interplay of iron, calcium and lipid peroxidation. Cell Death Differ. 2020;27:2781–96 https://doi.org/10.1038/s41418-020-0542-z.
Billington D, Evans CE, Godfrey PP, Coleman R. Effects of bile salts on the plasma membranes of isolated rat hepatocytes. Biochem J. 1980;188:321–7 https://doi.org/10.1042/bj1880321.
Ito S, Lacy ER. Morphology of rat gastric mucosal damage, defense, and restitution in the presence of luminal ethanol. Gastroenterology. 1985;88:250–60 https://doi.org/10.1016/S0016-5085(85)80178-5.
Rainsford KD. Electronmicroscopic observations on the effects of orally administered aspirin and aspirin-bicarbonate mixtures on the development of gastric mucosal damage in the rat. Gut. 1975;16:514–27 https://doi.org/10.1136/gut.16.7.514.
Comporti M, Signorini C, Leoncini S, Gardi C, Ciccoli L, Giardini A, et al. Ethanol-induced oxidative stress: basic knowledge. Genes Nutr. 2010;5:101–9 https://doi.org/10.1007/s12263-009-0159-9.
Navarro-Tapia E, Querol A, Pérez-Torrado R. Membrane fluidification by ethanol stress activates unfolded protein response in yeasts. Microb Biotechnol. 2018;11:465–75 https://doi.org/10.1111/1751-7915.13032.
Zhou Y, Maxwell KN, Sezgin E, Lu M, Liang H, Hancock JF, et al. Bile acids modulate signaling by functional perturbation of plasma membrane domains. J Biol Chem. 2013;288:35660–70 https://doi.org/10.1074/jbc.M113.519116.
Jean-Louis S, Akare S, Ali MA, Mash EA, Meuillet E, Martinez JD. Deoxycholic acid induces intracellular signaling through membrane perturbations. J Biol Chem. 2006;281:14948–60 https://doi.org/10.1074/jbc.M506710200.
Zhou Y, Dial EJ, Doyen R, Lichtenberger LM. Effect of indomethacin on bile acid-phospholipid interactions: implication for small intestinal injury induced by nonsteroidal anti-inflammatory drugs. Am J Physiol Liver Physiol. 2010;298:G722–31 https://doi.org/10.1152/ajpgi.00387.2009.
Lam JGT, Vadia S, Pathak-Sharma S, McLaughlin E, Zhang X, Swanson J, et al. Host cell perforation by listeriolysin O (LLO) activates a Ca 2+ −dependent cPKC/Rac1/Arp2/3 signaling pathway that promotes Listeria monocytogenes internalization independently of membrane resealing. Mol Biol Cell. 2018;29:270–84 https://doi.org/10.1091/mbc.E17-09-0561.
Beckwith KS, Beckwith MS, Ullmann S, Sætra RS, Kim H, Marstad A, et al. Plasma membrane damage causes NLRP3 activation and pyroptosis during Mycobacterium tuberculosis infection. Nat Commun. 2020;11:2270 https://doi.org/10.1038/s41467-020-16143-6.
Flores-Díaz M, Monturiol-Gross L, Naylor C, Alape-Girón A, Flieger A. Bacterial sphingomyelinases and phospholipases as virulence factors. Microbiol Mol Biol Rev. 2016;80:597–628 https://doi.org/10.1128/MMBR.00082-15.
Westman J, Hube B, Fairn GD. Integrity under stress: host membrane remodelling and damage by fungal pathogens. Cell Microbiol. 2019;21:e13016 https://doi.org/10.1111/cmi.13016.
Allert S, Förster TM, Svensson C-M, Richardson JP, Pawlik T, Hebecker B, et al. Candida albicans-induced epithelial damage mediates translocation through intestinal barriers. MBio. 2018;9 https://doi.org/10.1128/mBio.00915-18.
Formaglio P, Tavares J, Ménard R, Amino R. Loss of host cell plasma membrane integrity following cell traversal by Plasmodium sporozoites in the skin. Parasitol Int. 2014;63:237–44 https://doi.org/10.1016/j.parint.2013.07.009.
Ralston KS, Solga MD, Mackey-Lawrence NM, Somlata, Bhattacharya A, Petri WA. Trogocytosis by Entamoeba histolytica contributes to cell killing and tissue invasion. Nature. 2014;508:526–30 https://doi.org/10.1038/nature13242.
Roy DA. Process for controlling intracellular bacterial infections induced by membrane injury. Science. 2004;304:1515–8 https://doi.org/10.1126/science.1098371.
Guignot J, Tran Van Nhieu G. Bacterial control of pores induced by the type III secretion system: mind the gap. Front Immunol. 2016;7:84 https://doi.org/10.3389/fimmu.2016.00084.
Xu Y, Weiss LM. The microsporidian polar tube: a highly specialised invasion organelle. Int J Parasitol. 2005;35:941–53 https://doi.org/10.1016/j.ijpara.2005.04.003.
Luisoni S, Suomalainen M, Boucke K, Tanner LB, Wenk MR, Guan XL, et al. Co-option of membrane wounding enables virus penetration into cells. Cell Host Microbe. 2015;18:75–85 https://doi.org/10.1016/j.chom.2015.06.006.
Moyes DL, Wilson D, Richardson JP, Mogavero S, Tang SX, Wernecke J, et al. Candidalysin is a fungal peptide toxin critical for mucosal infection. Nature. 2016;532:64–8 https://doi.org/10.1038/nature17625.
Nayak AP, Green BJ, Beezhold DH. Fungal hemolysins. Med Mycol. 2013;51:1–16 https://doi.org/10.3109/13693786.2012.698025.
Leippe M. Pore-forming toxins from pathogenic amoebae. Appl Microbiol Biotechnol. 2014;98:4347–53 https://doi.org/10.1007/s00253-014-5673-z.
Jimah JR, Salinas ND, Sala-Rabanal M, Jones NG, Sibley LD, Nichols CG, et al. Malaria parasite CelTOS targets the inner leaflet of cell membranes for pore-dependent disruption. Elife. 2016;5:e20621 https://doi.org/10.7554/eLife.20621.
Wolfmeier H, Schoenauer R, Atanassoff AP, Neill DR, Kadioglu A, Draeger A, et al. Ca2+−dependent repair of pneumolysin pores: a new paradigm for host cellular defense against bacterial pore-forming toxins. Biochim Biophys Acta Mol Cell Res. 2015;1853:2045–54 https://doi.org/10.1016/j.bbamcr.2014.09.005.
Carrasco L. Membrane leakiness after viral infection and a new approach to the development of antiviral agents. Nature. 1978;272:694–9 https://doi.org/10.1038/272694a0.
Hyser JM, Estes MK. Pathophysiological consequences of calcium-conducting viroporins. Annu Rev Virol. 2015;2:473–96 https://doi.org/10.1146/annurev-virology-100114-054846.
Dudkina NV, Spicer BA, Reboul CF, Conroy PJ, Lukoyanova N, Elmlund H, et al. Structure of the poly-C9 component of the complement membrane attack complex. Nat Commun. 2016;7:10588 https://doi.org/10.1038/ncomms10588.
Neher MD, Weckbach S, Flierl MA, Huber-Lang MS, Stahel PF. Molecular mechanisms of inflammation and tissue injury after major trauma-is complement the “bad guy”? J Biomed Sci. 2011;18:90 https://doi.org/10.1186/1423-0127-18-90.
Norris DA, Kissinger RM, Naughton GM, Bystryn JC. Evidence for immunologic mechanisms in human vitiligo: patients’ sera induce damage to human melanocytes in vitro by complement-mediated damage and antibody-dependent cellular cytotoxicity. J Invest Dermatol. 1988;90:783–9 https://doi.org/10.1111/1523-1747.ep12461505.
Keefe D, Shi L, Feske S, Massol R, Navarro F, Kirchhausen T, et al. Perforin triggers a plasma membrane-repair response that facilitates CTL induction of apoptosis. Immunity. 2005;23:249–62 https://doi.org/10.1016/j.immuni.2005.08.001.
Law RHP, Lukoyanova N, Voskoboinik I, Caradoc-Davies TT, Baran K, Dunstone MA, et al. The structural basis for membrane binding and pore formation by lymphocyte perforin. Nature. 2010;468:447–51 https://doi.org/10.1038/nature09518.
Hibbs JB. Heterocytolysis by macrophages activated by Bacillus Calmette-Guerin: lysosome exocytosis into tumor cells. Science. 1974;184:468–71 https://doi.org/10.1126/science.184.4135.468.
Marin-Esteban V, Turbica I, Dufour G, Semiramoth N, Gleizes A, Gorges R, et al. Afa/Dr diffusely adhering Escherichia coli strain C1845 induces neutrophil extracellular traps that kill bacteria and damage human enterocyte-like cells. Infect Immun. 2012;80:1891–9 https://doi.org/10.1128/IAI.00050-12.
Carmona-Rivera C, Zhao W, Yalavarthi S, Kaplan MJ. Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase-2. Ann Rheum Dis. 2015;74:1417–24 https://doi.org/10.1136/annrheumdis-2013-204837.
Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. 2018;18:134–47 https://doi.org/10.1038/nri.2017.105.
Ahmed KA, Munegowda MA, Xie Y, Xiang J. Intercellular trogocytosis plays an important role in modulation of immune responses. Cell Mol Immunol. 2008;5:261–9 https://doi.org/10.1038/cmi.2008.32.
Matlung HL, Babes L, Zhao XW, van Houdt M, Treffers LW, van Rees DJ, et al. Neutrophils kill antibody-opsonized cancer cells by trogoptosis. Cell Rep. 2018;23:3946–3959.e6 https://doi.org/10.1016/j.celrep.2018.05.082.
Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486–541 https://doi.org/10.1038/s41418-017-0012-4.
Messenger SW, Thomas DDH, Cooley MM, Jones EK, Falkowski MA, August BK, et al. Early to late endosome trafficking controls secretion and zymogen activation in rodent and human pancreatic acinar cells. Cell Mol Gastroenterol Hepatol. 2015;1:695–709 https://doi.org/10.1016/j.jcmgh.2015.08.002.
Xia B, Fang S, Chen X, Hu H, Chen P, Wang H, et al. MLKL forms cation channels. Cell Res. 2016;26:517–28 https://doi.org/10.1038/cr.2016.26.
Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun. 2017;8:14128 https://doi.org/10.1038/ncomms14128.
Chen X, He W, Hu L, Li J, Fang Y, Wang X, et al. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res. 2016;26:1007–20 https://doi.org/10.1038/cr.2016.100.
Davis MA, Fairgrieve MR, Den Hartigh A, Yakovenko O, Duvvuri B, Lood C, et al. Calpain drives pyroptotic vimentin cleavage, intermediate filament loss, and cell rupture that mediates immunostimulation. Proc Natl Acad Sci. 2019;116:5061–70 https://doi.org/10.1073/pnas.1818598116.
Ono K, Kim SO, Han J. Susceptibility of lysosomes to rupture is a determinant for plasma membrane disruption in tumor necrosis factor alpha-induced cell death. Mol Cell Biol. 2003;23:665–76 https://doi.org/10.1128/MCB.23.2.665-676.2003.
Wang F, Gómez-Sintes R, Boya P. Lysosomal membrane permeabilization and cell death. Traffic. 2018;19:918–31 https://doi.org/10.1111/tra.12613.
Heilbrunn LV. The surface precipitation reaction of living cells. Proc Am Philos Soc. 1930;69:295–301 http://www.jstor.org/stable/984364.
Bement WM, Capco DG. Analysis of inducible contractile rings suggests a role for protein kinase C in embryonic cytokinesis and wound healing. Cell Motil Cytoskeleton. 1991;20:145–57 https://doi.org/10.1002/cm.970200207.
Miyake K, McNeil PL. Vesicle accumulation and exocytosis at sites of plasma membrane disruption. J Cell Biol. 1995;131:1737–45 https://doi.org/10.1083/jcb.131.6.1737.
Cheng X, Zhang X, Yu L, Xu H. Calcium signaling in membrane repair. Semin Cell Dev Biol. 2015;45:24–31 https://doi.org/10.1016/j.semcdb.2015.10.031.
Lek A, Evesson FJ, Lemckert FA, Redpath GMI, Lueders A-K, Turnbull L, et al. Calpains, cleaved mini-dysferlinC72, and L-type channels underpin calcium-dependent muscle membrane repair. J Neurosci. 2013;33:5085–94 https://doi.org/10.1523/JNEUROSCI.3560-12.2013.
Cheng X, Zhang X, Gao Q, Ali Samie M, Azar M, Tsang WL, et al. The intracellular Ca2+ channel MCOLN1 is required for sarcolemma repair to prevent muscular dystrophy. Nat Med. 2014;20:1187–92 https://doi.org/10.1038/nm.3611.
Mellgren RL, Zhang W, Miyake K, McNeil PL, Calpain I. Required for the rapid, calcium-dependent repair of wounded plasma membrane. J Biol Chem. 2007;282:2567–75 https://doi.org/10.1074/jbc.M604560200.
Perrin BJ, Amann KJ, Huttenlocher A. Proteolysis of cortactin by calpain regulates membrane protrusion during cell migration. Mol Biol Cell. 2006;17:239–50 https://doi.org/10.1091/mbc.e05-06-0488.
Ellis S, Lin EJ, Tartar D. Immunology of wound healing. Curr Dermatol Rep. 2018;7:350–8 https://doi.org/10.1007/s13671-018-0234-9.
Togo T, Krasieva TB, Steinhardt RA. A decrease in membrane tension precedes successful cell-membrane repair. Mol Biol Cell. 2000;11:4339–46 https://doi.org/10.1091/mbc.11.12.4339.
Steinhardt R, Bi G, Alderton J. Cell membrane resealing by a vesicular mechanism similar to neurotransmitter release. Science. 1994;263:390–3 https://doi.org/10.1126/science.7904084.
Bi GQ, Alderton JM, Steinhardt RA. Calcium-regulated exocytosis is required for cell membrane resealing. J Cell Biol. 1995;131:1747–58 https://doi.org/10.1083/jcb.131.6.1747.
Miyake K, Tanaka T, McNeil PL. Disruption-induced mucus secretion: repair and protection. PLoS Biol. 2006;4:e276 https://doi.org/10.1371/journal.pbio.0040276.
Davenport NR, Sonnemann KJ, Eliceiri KW, Bement WM. Membrane dynamics during cellular wound repair. Mol Biol Cell. 2016;27:2272–85 https://doi.org/10.1091/mbc.E16-04-0223.
McNeil PL, Vogel SS, Miyake K, Terasaki M. Patching plasma membrane disruptions with cytoplasmic membrane. J Cell Sci. 2000;113:1891–902.
Reddy A, Caler EV, Andrews NW. Plasma membrane repair is mediated by Ca2+−regulated exocytosis of lysosomes. Cell. 2001;106:157–69 https://doi.org/10.1016/S0092-8674(01)00421-4.
Shen SS, Tucker WC, Chapman ER, Steinhardt RA. Molecular regulation of membrane resealing in 3T3 fibroblasts. J Biol Chem. 2005;280:1652–60 https://doi.org/10.1074/jbc.M410136200.
Sreetama SC, Takano T, Nedergaard M, Simon SM, Jaiswal JK. Injured astrocytes are repaired by Synaptotagmin XI-regulated lysosome exocytosis. Cell Death Differ. 2016;23:596–607 https://doi.org/10.1038/cdd.2015.124.
Bansal D, Miyake K, Vogel SS, Groh S, Chen C-C, Williamson R, et al. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature. 2003;423:168–72 https://doi.org/10.1038/nature01573.
Defour A, Van der Meulen JH, Bhat R, Bigot A, Bashir R, Nagaraju K, et al. Dysferlin regulates cell membrane repair by facilitating injury-triggered acid sphingomyelinase secretion. Cell Death Dis. 2014;5:e1306 https://doi.org/10.1038/cddis.2014.272.
Redpath GMI, Woolger N, Piper AK, Lemckert FA, Lek A, Greer PA, et al. Calpain cleavage within dysferlin exon 40a releases a synaptotagmin-like module for membrane repair. Mol Biol Cell. 2014;25:3037–48 https://doi.org/10.1091/mbc.e14-04-0947.
Cai C, Masumiya H, Weisleder N, Matsuda N, Nishi M, Hwang M, et al. MG53 nucleates assembly of cell membrane repair machinery. Nat Cell Biol. 2009;11:56–64 https://doi.org/10.1038/ncb1812.
Hwang M, Ko J, Weisleder N, Takeshima H, Ma J. Redox-dependent oligomerization through a leucine zipper motif is essential for MG53-mediated cell membrane repair. Am J Physiol Physiol. 2011;301:C106–14 https://doi.org/10.1152/ajpcell.00382.2010.
Zhu H, Lin P, De G, Choi K, Takeshima H, Weisleder N, et al. Polymerase transcriptase release factor (PTRF) anchors MG53 protein to cell injury site for initiation of membrane repair. J Biol Chem. 2011;286:12820–4 https://doi.org/10.1074/jbc.C111.221440.
Cai C, Weisleder N, Ko J-K, Komazaki S, Sunada Y, Nishi M, et al. Membrane repair defects in muscular dystrophy are linked to altered interaction between MG53, caveolin-3, and dysferlin. J Biol Chem. 2009;284:15894–902 https://doi.org/10.1074/jbc.M109.009589.
Andrews NW, Corrotte M. Plasma membrane repair. Curr Biol. 2018;28:R392–7 https://doi.org/10.1016/j.cub.2017.12.034.
Terasaki M, Miyake K, McNeil PL. Large plasma membrane disruptions are rapidly resealed by Ca2+−dependent vesicle–vesicle fusion events. J Cell Biol. 1997;139:63–74 https://doi.org/10.1083/jcb.139.1.63.
McDade JR, Michele DE. Membrane damage-induced vesicle–vesicle fusion of dysferlin-containing vesicles in muscle cells requires microtubules and kinesin. Hum Mol Genet. 2014;23:1677–86 https://doi.org/10.1093/hmg/ddt557.
Idone V, Tam C, Goss JW, Toomre D, Pypaert M, Andrews NW. Repair of injured plasma membrane by rapid Ca2+−dependent endocytosis. J Cell Biol. 2008;180:905–14 https://doi.org/10.1083/jcb.200708010.
Corrotte M, Almeida PE, Tam C, Castro-Gomes T, Fernandes MC, Millis BA, et al. Caveolae internalization repairs wounded cells and muscle fibers. Elife. 2013;2:e00926 https://doi.org/10.7554/eLife.00926.
Tam C, Idone V, Devlin C, Fernandes MC, Flannery A, He X, et al. Exocytosis of acid sphingomyelinase by wounded cells promotes endocytosis and plasma membrane repair. J Cell Biol. 2010;189:1027–38 https://doi.org/10.1083/jcb.201003053.
Corrotte M, Fernandes MC, Tam C, Andrews NW. Toxin pores endocytosed during plasma membrane repair traffic into the lumen of <scp>MVB</scp> s for degradation. Traffic. 2012;13:483–94 https://doi.org/10.1111/j.1600-0854.2011.01323.x.
Husmann M, Beckmann E, Boller K, Kloft N, Tenzer S, Bobkiewicz W, et al. Elimination of a bacterial pore-forming toxin by sequential endocytosis and exocytosis. FEBS Lett. 2009;583:337–44 https://doi.org/10.1016/j.febslet.2008.12.028.
Li X, Gulbins E, Zhang Y. Oxidative stress triggers Ca 2+ −dependent lysosome trafficking and activation of acid sphingomyelinase. Cell Physiol Biochem. 2012;30:815–26 https://doi.org/10.1159/000341460.
Nassoy P, Lamaze C. Stressing caveolae new role in cell mechanics. Trends Cell Biol. 2012;22:381–9 https://doi.org/10.1016/j.tcb.2012.04.007.
Yeow I, Howard G, Chadwick J, Mendoza-Topaz C, Hansen CG, Nichols BJ, et al. EHD proteins cooperate to generate caveolar clusters and to maintain caveolae during repeated mechanical stress. Curr Biol. 2017;27:2951–2962.e5 https://doi.org/10.1016/j.cub.2017.07.047.
Nakamura M, Dominguez ANM, Decker JR, Hull AJ, Verboon JM, Parkhurst SM. Into the breach: how cells cope with wounds. Open Biol. 2018;8:180135 https://doi.org/10.1098/rsob.180135.
Mandato CA, Bement WM. Contraction and polymerization cooperate to assemble and close actomyosin rings around Xenopus oocyte wounds. J Cell Biol. 2001;154:785–98 https://doi.org/10.1083/jcb.200103105.
Clark AG, Miller AL, Vaughan E, Yu H-YE, Penkert R, Bement WM. Integration of single and multicellular wound responses. Curr Biol. 2009;19:1389–95 https://doi.org/10.1016/j.cub.2009.06.044.
Nakamura M, Verboon JM, Parkhurst SM. Prepatterning by RhoGEFs governs Rho GTPase spatiotemporal dynamics during wound repair. J Cell Biol. 2017;216:3959–69 https://doi.org/10.1083/jcb.201704145.
Xu S, Chisholm AD. A Gαq-Ca2+ signaling pathway promotes actin-mediated epidermal wound closure in C. elegans. Curr Biol. 2011;21:1960–7 https://doi.org/10.1016/j.cub.2011.10.050.
Taffoni C, Omi S, Huber C, Mailfert S, Fallet M, Rupprecht JF, et al. Microtubule plus-end dynamics link wound repair to the innate immune response. Elife. 2020;9:e45047.
Benink HA, Bement WM. Concentric zones of active RhoA and Cdc42 around single cell wounds. J Cell Biol. 2005;168:429–39 https://doi.org/10.1083/jcb.200411109.
Vaughan EM, You J-S, Elsie Yu H-Y, Lasek A, Vitale N, Hornberger TA, et al. Lipid domain–dependent regulation of single-cell wound repair. Mol Biol Cell. 2014;25:1867–76 https://doi.org/10.1091/mbc.e14-03-0839.
Gallant P. Effects of the external ions and metabolic poisoning on the constriction of the squid giant axon after axotomy. J Neurosci. 1988;8:1479–84 https://doi.org/10.1523/JNEUROSCI.08-05-01479.1988.
Krause T, Fishman H, Ballinger M, Bittner G. Extent and mechanism of sealing in transected giant axons of squid and earthworms. J Neurosci. 1994;14:6638–51 https://doi.org/10.1523/JNEUROSCI.14-11-06638.1994.
Moe AM, Golding AE, Bement WM. Cell healing: calcium, repair and regeneration. Semin Cell Dev Biol. 2015;45:18–23 https://doi.org/10.1016/j.semcdb.2015.09.026.
McDade JR, Archambeau A, Michele DE. Rapid actin-cytoskeleton–dependent recruitment of plasma membrane–derived dysferlin at wounds is critical for muscle membrane repair. FASEB J. 2014;28:3660–70 https://doi.org/10.1096/fj.14-250191.
Lin P, Zhu H, Cai C, Wang X, Cao C, Xiao R, et al. Nonmuscle myosin IIA facilitates vesicle trafficking for MG53-mediated cell membrane repair. FASEB J. 2012;26:1875–83 https://doi.org/10.1096/fj.11-188599.
Meng X, Yang Q, Yu X, Zhou J, Ren X, Zhou Y, et al. Actin polymerization and ESCRT trigger recruitment of the fusogens syntaxin-2 and EFF-1 to promote membrane repair in C. elegans. Dev Cell. 2020;54:624–638.e5 https://doi.org/10.1016/j.devcel.2020.06.027.
Mesquita FS, Brito C, Mazon Moya MJ, Pinheiro JC, Mostowy S, Cabanes D, et al. Endoplasmic reticulum chaperone Gp96 controls actomyosin dynamics and protects against pore-forming toxins. EMBO Rep. 2017;18:303–18 https://doi.org/10.15252/embr.201642833.
Eddleman CS, Ballinger ML, Smyers ME, Godell CM, Fishman HM, Bittner GD. Repair of plasmalemmal lesions by vesicles. Proc Natl Acad Sci. 1997;94:4745–50 https://doi.org/10.1073/pnas.94.9.4745.
Horn A, Van der Meulen JH, Defour A, Hogarth M, Sreetama SC, Reed A, et al. Mitochondrial redox signaling enables repair of injured skeletal muscle cells. Sci Signal. 2017;10:eaaj1978 https://doi.org/10.1126/scisignal.aaj1978.
Babiychuk EB, Monastyrskaya K, Potez S, Draeger A. Blebbing confers resistance against cell lysis. Cell Death Differ. 2011;18:80–9 https://doi.org/10.1038/cdd.2010.81.
Croissant C, Gounou C, Bouvet F, Tan S, Bouter A. Annexin-A6 in membrane repair of human skeletal muscle cell: a role in the cap subdomain. Cells. 2020;9:1742 https://doi.org/10.3390/cells9071742.
Boye TL, Nylandsted J. Annexins in plasma membrane repair. Biol Chem. 2016;397:961–9 https://doi.org/10.1515/hsz-2016-0171.
Grill D, ALL M, de Vries WC, Kudruk S, Heflik M, Dörner W, et al. Bridging of membrane surfaces by annexin A2. Sci Rep. 2018;8:14662 https://doi.org/10.1038/s41598-018-33044-3.
Bendix PM, Simonsen AC, Florentsen CD, Häger SC, Mularski A, Zanjani AAH, et al. Interdisciplinary synergy to reveal mechanisms of annexin-mediated plasma membrane shaping and repair. Cells. 2020;9:1029 https://doi.org/10.3390/cells9041029.
Boye TL, Maeda K, Pezeshkian W, Sønder SL, Haeger SC, Gerke V, et al. Annexin A4 and A6 induce membrane curvature and constriction during cell membrane repair. Nat Commun. 2017;8:1623 https://doi.org/10.1038/s41467-017-01743-6.
Lin Y-C, Chipot C, Scheuring S. Annexin-V stabilizes membrane defects by inducing lipid phase transition. Nat Commun. 2020;11:230 https://doi.org/10.1038/s41467-019-14045-w.
Demonbreun AR, Quattrocelli M, Barefield DY, Allen MV, Swanson KE, McNally EM. An actin-dependent annexin complex mediates plasma membrane repair in muscle. J Cell Biol. 2016;213:705–18 https://doi.org/10.1083/jcb.201512022.
Sønder SL, Boye TL, Tölle R, Dengjel J, Maeda K, Jäättelä M, et al. Annexin A7 is required for ESCRT III-mediated plasma membrane repair. Sci Rep. 2019;9:6726 https://doi.org/10.1038/s41598-019-43143-4.
Wolfmeier H, Radecke J, Schoenauer R, Koeffel R, Babiychuk VS, Drücker P, et al. Active release of pneumolysin prepores and pores by mammalian cells undergoing a Streptococcus pneumoniae attack. Biochim Biophys Acta Gen Subj. 2016;1860:2498–509 https://doi.org/10.1016/j.bbagen.2016.07.022.
Rühl S, Shkarina K, Demarco B, Heilig R, Santos JC, Broz P. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDM0D activation. Science. 2018;362:956–60 https://doi.org/10.1126/science.aar7607.
Dai E, Meng L, Kang R, Wang X, Tang D. ESCRT-III–dependent membrane repair blocks ferroptosis. Biochem Biophys Res Commun. 2020;522:415–21 https://doi.org/10.1016/j.bbrc.2019.11.110.
Castro-Gomes T, Koushik AB, Andrews NW. ESCRT: nipping the wound in the bud? Trends Biochem Sci. 2014;39:307–9 https://doi.org/10.1016/j.tibs.2014.06.001.
Chandra G, Defour A, Mamchoui K, Pandey K, Mishra S, Mouly V, et al. Dysregulated calcium homeostasis prevents plasma membrane repair in Anoctamin 5/TMEM16E-deficient patient muscle cells. Cell Death Discov. 2019;5:118 https://doi.org/10.1038/s41420-019-0197-z.
Debattisti V, Horn A, Singh R, Seifert EL, Hogarth MW, Mazala DA, et al. Dysregulation of mitochondrial Ca2+ uptake and sarcolemma repair underlie muscle weakness and wasting in patients and mice lacking MICU1. Cell Rep. 2019;29:1274–1286.e6 https://doi.org/10.1016/j.celrep.2019.09.063.
Tang NH, Kim KW, Xu S, Blazie SM, Yee BA, Yeo GW, et al. The mRNA decay factor CAR-1/LSM14 regulates axon regeneration via mitochondrial calcium dynamics. Curr Biol. 2020;30:865–876.e7 https://doi.org/10.1016/j.cub.2019.12.061.
Horn A, Raavicharla S, Shah S, Cox D, Jaiswal JK. Mitochondrial fragmentation enables localized signaling required for cell repair. J Cell Biol. 2020;219:e201909154 https://doi.org/10.1083/jcb.201909154.
Fu H, Zhou H, Yu X, Xu J, Zhou J, Meng X, et al. Wounding triggers MIRO-1 dependent mitochondrial fragmentation that accelerates epidermal wound closure through oxidative signaling. Nat Commun. 2020;11:1050 https://doi.org/10.1038/s41467-020-14885-x.
Labazi M, McNeil AK, Kurtz T, Lee TC, Pegg RB, Angeli JPF, et al. The antioxidant requirement for plasma membrane repair in skeletal muscle. Free Radic Biol Med. 2015;84:246–53 https://doi.org/10.1016/j.freeradbiomed.2015.03.016.
Yu K, Sellman DP, Bahraini A, Hagan ML, Elsherbini A, Vanpelt KT, et al. Mechanical loading disrupts osteocyte plasma membranes which initiates mechanosensation events in bone. J Orthop Res. 2017;36:653–62 https://doi.org/10.1002/jor.23665.
Baaske R, Richter M, Möller N, Ziesemer S, Eiffler I, Müller C, et al. ATP release from human airway epithelial cells exposed to Staphylococcus aureus alpha-toxin. Toxins (Basel). 2016;8:365 https://doi.org/10.3390/toxins8120365.
Malet JK, Impens F, Carvalho F, Hamon MA, Cossart P, Ribet D. Rapid remodeling of the host epithelial cell proteome by the listeriolysin O (LLO) pore-forming toxin. Mol Cell Proteomics. 2018;17:1627–36 https://doi.org/10.1074/mcp.RA118.000767.
Gonzalez MR, Bischofberger M, Frêche B, Ho S, Parton RG, Van der Goot FG. Pore-forming toxins induce multiple cellular responses promoting survival. Cell Microbiol. 2011;13:1026–43.
Miyake K, McNeil PL, Suzuki K, Tsunoda R, Sugai N. An actin barrier to resealing. J Cell Sci. 2001;114:3487–94.
Castro-Gomes T, Corrotte M, Tam C, Andrews NW. Plasma membrane repair is regulated extracellularly by proteases released from lysosomes. PLoS One. 2016;11:e0152583 https://doi.org/10.1371/journal.pone.0152583.
Miller H, Castro-Gomes T, Corrotte M, Tam C, Maugel TK, Andrews NW, et al. Lipid raft–dependent plasma membrane repair interferes with the activation of B lymphocytes. J Cell Biol. 2015;211:1193–205 https://doi.org/10.1083/jcb.201505030.
Bhattacharjee P, Keyel PA. Cholesterol-dependent cytolysins impair pro-inflammatory macrophage responses. Sci Rep. 2018;8:6458 https://doi.org/10.1038/s41598-018-24955-2.
Togo T. Signaling pathways involved in adaptive responses to cell membrane disruption. Curr Top Membr. 2019;84:99–127.
Togo T. Short-term potentiation of membrane resealing in neighboring cells is mediated by purinergic signaling. Purinergic Signal. 2014;10:283–90 https://doi.org/10.1007/s11302-013-9387-y.
Schoenauer R, Atanassoff AP, Wolfmeier H, Pelegrin P, Babiychuk EB, Draeger A. P2X7 receptors mediate resistance to toxin-induced cell lysis. Biochim Biophys Acta Mol Cell Res. 2014;1843:915–22 https://doi.org/10.1016/j.bbamcr.2014.01.024.
Togo T, Alderton JM, Steinhardt RA. Long-term potentiation of exocytosis and cell membrane repair in fibroblasts. Mol Biol Cell. 2003;14:93–106 https://doi.org/10.1091/mbc.e02-01-0056.
Togo T. Long-term potentiation of wound-induced exocytosis and plasma membrane repair is dependant on cAMP-response element-mediated transcription via a protein kinase C- and p38 MAPK-dependent pathway. J Biol Chem. 2004;279:44996–5003 https://doi.org/10.1074/jbc.M406327200.
McNeil PL, Steinhardt RA. Loss, restoration, and maintenance of plasma membrane integrity. J Cell Biol. 1997;137:1–4 https://doi.org/10.1083/jcb.137.1.1.
Köffel R, Wolfmeier H, Larpin Y, Besançon H, Schoenauer R, Babiychuk VS, et al. Host-derived microvesicles carrying bacterial pore-forming toxins deliver signals to macrophages: a novel mechanism of shaping immune responses. Front Immunol. 2018;9:1688 https://doi.org/10.3389/fimmu.2018.01688.
Middel V, Zhou L, Takamiya M, Beil T, Shahid M, Roostalu U, et al. Dysferlin-mediated phosphatidylserine sorting engages macrophages in sarcolemma repair. Nat Commun. 2016;7:12875 https://doi.org/10.1038/ncomms12875.
Wäster P, Eriksson I, Vainikka L, Öllinger K. Extracellular vesicles released by melanocytes after UVA irradiation promote intercellular signaling via miR21. Pigment Cell Melanoma Res. 2020;33:542–55 https://doi.org/10.1111/pcmr.12860.
Lo HP, Nixon SJ, Hall TE, Cowling BS, Ferguson C, Morgan GP, et al. The caveolin–cavin system plays a conserved and critical role in mechanoprotection of skeletal muscle. J Cell Biol. 2015;210:833–49 https://doi.org/10.1083/jcb.201501046.
Cheng JPX, Mendoza-Topaz C, Howard G, Chadwick J, Shvets E, Cowburn AS, et al. Caveolae protect endothelial cells from membrane rupture during increased cardiac output. J Cell Biol. 2015;211:53–61 https://doi.org/10.1083/jcb.201504042.
Carlson BA, Tobe R, Yefremova E, Tsuji PA, Hoffmann VJ, Schweizer U, et al. Glutathione peroxidase 4 and vitamin E cooperatively prevent hepatocellular degeneration. Redox Biol. 2016;9:22–31 https://doi.org/10.1016/j.redox.2016.05.003.
Slutsky AS, Ranieri VM. Ventilator-induced lung injury. N Engl J Med. 2013;369:2126–36 https://doi.org/10.1056/NEJMra1208707.
Kosmider B, Messier EM, Chu HW, Mason RJ. Human alveolar epithelial cell injury induced by cigarette smoke. PLoS One. 2011;6:e26059 https://doi.org/10.1371/journal.pone.0026059.
Thelestam M, Curvall M, Enzell CR. Effect of tobacco smoke compounds on the plasma membrane of cultured human lung fibroblasts. Toxicology. 1980;15:203–17 https://doi.org/10.1016/0300-483X(80)90054-2.
Petruska JM, Leslie KO, Mossman BT. Enhanced lipid peroxidation in lung lavage of rats after inhalation of asbestos. Free Radic Biol Med. 1991;11:425–32 https://doi.org/10.1016/0891-5849(91)90160-5.
Bernareggi A, Conte G, Constanti A, Borelli V, Vita F, Zabucchi G. On the mechanism of the electrophysiological changes and membrane lesions induced by asbestos fiber exposure in Xenopus laevis oocytes. Sci Rep. 2019;9:2014 https://doi.org/10.1038/s41598-019-38591-x.
González-Juarbe N, Bradley KM, Shenoy AT, Gilley RP, Reyes LF, Hinojosa CA, et al. Pore-forming toxin-mediated ion dysregulation leads to death receptor-independent necroptosis of lung epithelial cells during bacterial pneumonia. Cell Death Differ. 2017;24:917–28 https://doi.org/10.1038/cdd.2017.49.
Rubins JB, Duane PG, Clawson D, Charboneau D, Young J, Niewoehner DE. Toxicity of pneumolysin to pulmonary alveolar epithelial cells. Infect Immun. 1993;61:1352–8 https://doi.org/10.1128/IAI.61.4.1352-1358.1993.
Keller MD, Ching KL, Liang F-X, Dhabaria A, Tam K, Ueberheide BM, et al. Decoy exosomes provide protection against bacterial toxins. Nature. 2020;579:260–4.
Orellana-Lezcano M, Major P, McNeil P, Borke J. Temporary loss of plasma membrane integrity in orthodontic tooth movement. Orthod Craniofac Res. 2005;8:106–13 https://doi.org/10.1111/j.1601-6343.2005.00306.x.
Amano K, Miyake K, Borke JL, McNeil PL. Breaking biological barriers with a toothbrush. J Dent Res. 2007;86:769–74 https://doi.org/10.1177/154405910708600816.
Kinomoto Y, Carnesjr D, Ebisu S. Cytotoxicity of intracanal bleaching agents on periodontal ligament cells in vitro. J Endod. 2001;27:574–7 https://doi.org/10.1097/00004770-200109000-00005.
Joyce AR, Hawkins W, Fariss MW, Sengupta TK. Role of plasma membrane disruption in reference moist smokeless tobacco-induced cell death. Toxicol Lett. 2010;198:191–9 https://doi.org/10.1016/j.toxlet.2010.06.014.
Kurtul N, Gökpınar E. Salivary lipid peroxidation and total sialic acid levels in smokers and smokeless tobacco users as Maraş powder. Mediators Inflamm. 2012;2012:1–8 https://doi.org/10.1155/2012/619293.
Tobey NA, Orlando RC. Mechanisms of acid injury to rabbit esophageal epithelium. Role of basolateral cell membrane acidification. Gastroenterology. 1991;101:1220–8.
Souza RF, Huo X, Mittal V, Schuler CM, Carmack SW, Zhang HY, et al. Gastroesophageal reflux might cause esophagitis through a cytokine-mediated mechanism rather than caustic acid injury. Gastroenterology. 2009;137:1776–84 https://doi.org/10.1053/j.gastro.2009.07.055.
Tamura M, Matsui H, Kaneko T, Hyodo I. Alcohol is an oxidative stressor for gastric epithelial cells: detection of superoxide in living cells. J Clin Biochem Nutr. 2013;53:75–80 https://doi.org/10.3164/jcbn.13-32.
Sgouras D, Tegtmeyer N, Wessler S. Activity and functional importance of Helicobacter pylori virulence factors. In: Advances in experimental medicine and biology; 2019. p. 35–56. https://doi.org/10.1007/5584_2019_358.
Weinman MD, Allan CH, Trier JS, Hagen SJ. Repair of microvilli in the rat small intestine after damage with lectins contained in the red kidney bean. Gastroenterology. 1989;97:1193–204 https://doi.org/10.1016/0016-5085(89)91690-9.
Lund EK, Fairweather-Tait SJ, Wharf SG, Johnson IT. Chronic exposure to high levels of dietary iron fortification increases lipid peroxidation in the mucosa of the rat large intestine. J Nutr. 2001;131:2928–31 https://doi.org/10.1093/jn/131.11.2928.
Sodhi CP, Fulton WB, Good M, Vurma M, Das T, Lai C-S, et al. Fat composition in infant formula contributes to the severity of necrotising enterocolitis. Br J Nutr. 2018;120:665–80 https://doi.org/10.1017/S0007114518001836.
Tivoli YA, Rubenstein RM. Pruritus: an updated look at an old problem. J Clin Aesthet Dermatol. 2009;2:30–6.
Zhong JL, Yiakouvaki A, Holley P, Tyrrell RM, Pourzand C. Susceptibility of skin cells to UVA-induced necrotic cell death reflects the intracellular level of labile iron. J Invest Dermatol. 2004;123:771–80 https://doi.org/10.1111/j.0022-202X.2004.23419.x.
Brauweiler AM, Bin L, Kim BE, Oyoshi MK, Geha RS, Goleva E, et al. Filaggrin-dependent secretion of sphingomyelinase protects against staphylococcal α-toxin–induced keratinocyte death. J Allergy Clin Immunol. 2013;131:421–427.e2 https://doi.org/10.1016/j.jaci.2012.10.030.
Fournet M, Bonté F, Desmoulière A. Glycation damage: a possible hub for major pathophysiological disorders and aging. Aging Dis. 2018;9:880 https://doi.org/10.14336/AD.2017.1121.
Madamanchi NR, Vendrov A, Runge MS. Oxidative stress and vascular disease. Arterioscler Thromb Vasc Biol. 2005;25:29–38 https://doi.org/10.1161/01.ATV.0000150649.39934.13.
Durán-Prado M, Frontiñán J, Santiago-Mora R, Peinado JR, Parrado-Fernández C, Gómez-Almagro MV, et al. Coenzyme Q10 protects human endothelial cells from β-amyloid uptake and oxidative stress-induced injury. PLoS One. 2014;9:e109223 https://doi.org/10.1371/journal.pone.0109223.
Riedl Khursigara M, Schlam D, Noone DG, Bruno V, Ortiz-Sandoval CG, Pluthero FG, et al. Vascular endothelial cells evade complement-mediated membrane injury via Weibel-Palade body mobilization. J Thromb Haemost. 2020;18:1484–94 https://doi.org/10.1111/jth.14767.
Lubkin A, Torres VJ. Bacteria and endothelial cells: a toxic relationship. Curr Opin Microbiol. 2017;35:58–63 https://doi.org/10.1016/j.mib.2016.11.008.
Li S, Tan H-Y, Wang N, Zhang Z-J, Lao L, Wong C-W, et al. The role of oxidative stress and antioxidants in liver diseases. Int J Mol Sci. 2015;16:26087–124 https://doi.org/10.3390/ijms161125942.
Srivastava A. Progressive familial intrahepatic cholestasis. J Clin Exp Hepatol. 2014;4:25–36 https://doi.org/10.1016/j.jceh.2013.10.005.
Sasahara K. Membrane-mediated amyloid deposition of human islet amyloid polypeptide. Biophys Rev. 2018;10:453–62.
Saluja AK, Bhagat L, Lee HS, Bhatia M, Frossard JL, Steer ML. Secretagogue-induced digestive enzyme activation and cell injury in rat pancreatic acini. Am J Physiol Liver Physiol. 1999;276:G835–42 https://doi.org/10.1152/ajpgi.1999.276.4.G835.
Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10:S10–7 https://doi.org/10.1038/nm1066.
Chodobski A, Zink BJ, Szmydynger-Chodobska J. Blood–brain barrier pathophysiology in traumatic brain injury. Transl Stroke Res. 2011;2:492–516 https://doi.org/10.1007/s12975-011-0125-x.
Nikić I, Merkler D, Sorbara C, Brinkoetter M, Kreutzfeldt M, Bareyre FM, et al. A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat Med. 2011;17:495–9 https://doi.org/10.1038/nm.2324.
Chevalier RL. The proximal tubule is the primary target of injury and progression of kidney disease: role of the glomerulotubular junction. Am J Physiol Physiol. 2016;311:F145–61 https://doi.org/10.1152/ajprenal.00164.2016.
Michailowsky V, Li H, Mittra B, Iyer SR, Mazála DAG, Corrotte M, et al. Defects in sarcolemma repair and skeletal muscle function after injury in a mouse model of Niemann-Pick type A/B disease. Skelet Muscle. 2019;9(1) https://doi.org/10.1186/s13395-018-0187-5.
Sander M, Chavoshan B, Harris SA, Iannaccone ST, Stull JT, Thomas GD, et al. Functional muscle ischemia in neuronal nitric oxide synthase-deficient skeletal muscle of children with Duchenne muscular dystrophy. Proc Natl Acad Sci. 2000;97:13818–23 https://doi.org/10.1073/pnas.250379497.
Howard AC, McNeil AK, Xiong F, Xiong W-C, McNeil PLA. Novel cellular defect in diabetes. Diabetes. 2011;60:3034–43 https://doi.org/10.2337/db11-0851.
Han R, Bansal D, Miyake K, Muniz VP, Weiss RM, McNeil PL, et al. Dysferlin-mediated membrane repair protects the heart from stress-induced left ventricular injury. J Clin Invest. 2007;117:1805–13 https://doi.org/10.1172/JCI30848.
Wang C, Wong J, Fung G, Shi J, Deng H, Zhang J, et al. Dysferlin deficiency confers increased susceptibility to coxsackievirus-induced cardiomyopathy. Cell Microbiol. 2015;17:1423–30 https://doi.org/10.1111/cmi.12473.
Nagre N, Wang S, Kellett T, Kanagasabai R, Deng J, Nishi M, et al. TRIM72 modulates caveolar endocytosis in repair of lung cells. Am J Physiol Cell Mol Physiol. 2016;310:L452–64 https://doi.org/10.1152/ajplung.00089.2015.
Wang XM, Zhang Y, Kim HP, Zhou Z, Feghali-Bostwick CA, Liu F, et al. Caveolin-1: a critical regulator of lung fibrosis in idiopathic pulmonary fibrosis. J Exp Med. 2006;203:2895–906 https://doi.org/10.1084/jem.20061536.
Hata S, Abe M, Suzuki H, Kitamura F, Toyama-Sorimachi N, Abe K, et al. Calpain 8/nCL-2 and calpain 9/nCL-4 constitute an active protease complex, G-calpain, involved in gastric mucosal defense. PLoS Genet. 2010;6:e1001040 https://doi.org/10.1371/journal.pgen.1001040.
Lin LL, Huang HC, Ogihara S, Wang JT, Wu MC, McNeil PL, et al. Helicobacter pylori disrupts host cell membranes, initiating a repair response and cell proliferation. Int J Mol Sci. 2012;13:10176–92 https://doi.org/10.3390/ijms130810176.
Chen P, Yan H, Tian Y, Xun Y, Shi L, Bao R, et al. Annexin A2 as a target endothelial cell membrane autoantigen in Behçet’s disease. Sci Rep. 2015;5:8162 https://doi.org/10.1038/srep08162.
Duann P, Li H, Lin P, Tan T, Wang Z, Chen K, et al. MG53-mediated cell membrane repair protects against acute kidney injury. Sci Transl Med. 2015;7:279ra36 https://doi.org/10.1126/scitranslmed.3010755.
Moens P, Baatsen PH, Marechal G. Increased susceptibility of EDL muscles from mdx mice to damage induced by contractions with stretch. J Muscle Res Cell Motil. 1993;14:446–51 https://doi.org/10.1007/BF00121296.
Han R, Kanagawa M, Yoshida-Moriguchi T, Rader EP, Ng RA, Michele DE, et al. Basal lamina strengthens cell membrane integrity via the laminin G domain-binding motif of α-dystroglycan. Proc Natl Acad Sci U S A. 2009;106:12573–9 https://doi.org/10.1073/pnas.0906545106.
Hayashi YK, Matsuda C, Ogawa M, Goto K, Tominaga K, Mitsuhashi S, et al. Human PTRF mutations cause secondary deficiency of caveolins resulting in muscular dystrophy with generalized lipodystrophy. J Clin Invest. 2009;119:2623–33 https://doi.org/10.1172/JCI38660.
Swaggart KA, Demonbreun AR, Vo AH, Swanson KE, Kim EY, Fahrenbach JP, et al. Annexin A6 modifies muscular dystrophy by mediating sarcolemmal repair. Proc Natl Acad Sci. 2014;111:6004–9 https://doi.org/10.1073/pnas.1324242111.
Hauerslev S, Sveen M-L, Duno M, Angelini C, Vissing J, Krag TO. Calpain 3 is important for muscle regeneration: evidence from patients with limb girdle muscular dystrophies. BMC Musculoskelet Disord. 2012;13:43 https://doi.org/10.1186/1471-2474-13-43.
Chakrabarti S, Kobayashi KS, Flavell RA, Marks CB, Miyake K, Liston DR, et al. Impaired membrane resealing and autoimmune myositis in synaptotagmin VII–deficient mice. J Cell Biol. 2003;162:543–9 https://doi.org/10.1083/jcb.200305131.
Boehler JF, Horn A, Novak JS, Li N, Ghimbovschi S, Lundberg IE, et al. Mitochondrial dysfunction and role of harakiri in the pathogenesis of myositis. J Pathol. 2019;249:215–26 https://doi.org/10.1002/path.5309.
Adesanya TMA, Russell M, Park KH, Zhou X, Sermersheim MA, Gumpper K, et al. MG53 protein protects aortic valve interstitial cells from membrane injury and fibrocalcific remodeling. J Am Heart Assoc. 2019;8 https://doi.org/10.1161/JAHA.118.009960.
Roan E, Waters CM. What do we know about mechanical strain in lung alveoli? Am J Physiol Cell Mol Physiol. 2011;301:L625–35 https://doi.org/10.1152/ajplung.00105.2011.
Diem K, Fauler M, Fois G, Hellmann A, Winokurow N, Schumacher S, et al. Mechanical stretch activates piezo1 in caveolae of alveolar type I cells to trigger ATP release and paracrine stimulation of surfactant secretion from alveolar type II cells. FASEB J. 2020;34:12785–804 https://doi.org/10.1096/fj.202000613RRR.
Albert RK, Smith B, Perlman CE, Schwartz DAI. Progression of pulmonary fibrosis due to ventilation-induced lung injury? Am J Respir Crit Care Med. 2019;200:140–51 https://doi.org/10.1164/rccm.201903-0497PP.
Han SH, Mallampalli RK. The role of surfactant in lung disease and host defense against pulmonary infections. Ann Am Thorac Soc. 2015;12:765–74 https://doi.org/10.1513/AnnalsATS.201411-507FR.
Godin LM, Vergen J, Prakash YS, Pagano RE, Hubmayr RD. Spatiotemporal dynamics of actin remodeling and endomembrane trafficking in alveolar epithelial type I cell wound healing. Am J Physiol Cell Mol Physiol. 2011;300:L615–23 https://doi.org/10.1152/ajplung.00265.2010.
Belete HA, Hubmayr RD, Wang S, Singh R-D. The role of purinergic signaling on deformation induced injury and repair responses of alveolar epithelial cells. PLoS One. 2011;6:e27469 https://doi.org/10.1371/journal.pone.0027469.
Kim SC, Kellett T, Wang S, Nishi M, Nagre N, Zhou B, et al. TRIM72 is required for effective repair of alveolar epithelial cell wounding. Am J Physiol Cell Mol Physiol. 2014;307:L449–59 https://doi.org/10.1152/ajplung.00172.2014.
Cong X, Nagre N, Herrera J, Pearson AC, Pepper I, Morehouse R, et al. TRIM72 promotes alveolar epithelial cell membrane repair and ameliorates lung fibrosis. Respir Res. 2020;21:132.
Vlahakis NE, Schroeder MA, Pagano RE, Hubmayr RD. Role of deformation-induced lipid trafficking in the prevention of plasma membrane stress failure. Am J Respir Crit Care Med. 2002;166:1282–9 https://doi.org/10.1164/rccm.200203-207OC.
Gerelsaikhan T, Vasa PK, Chander A. Annexin A7 and SNAP23 interactions in alveolar type II cells and in vitro: a role for Ca2+ and PKC. Biochim Biophys Acta - Mol Cell Res. 2012;1823:1796–806 https://doi.org/10.1016/j.bbamcr.2012.06.010.
Souza RF. Reflux esophagitis and its role in the pathogenesis of Barrett’s metaplasia. J Gastroenterol. 2017;52:767–76 https://doi.org/10.1007/s00535-017-1342-1.
Mise K, Capkun V, Jurcev-Savicevic A, Sundov Z, Bradaric A, Mladinov S. The influence of gastroesophageal reflux in the lung: a case-control study. Respirology. 2010;15:837–42 https://doi.org/10.1111/j.1440-1843.2010.01777.x.
Goldberg HI, Dodds WJ, Gee S, Montgomery C, Zboralske FF. Role of acid and pepsin in acute experimental esophagitis. Gastroenterology. 1969;56:223–30 https://doi.org/10.1016/S0016-5085(69)80121-6.
Rao CV, Vijayakumar M. Effect of quercetin, flavonoids and α-tocopherol, an antioxidant vitamin on experimental reflux oesophagitis in rats. Eur J Pharmacol. 2008;589:233–8 https://doi.org/10.1016/j.ejphar.2008.04.062.
Que J, Garman KS, Souza RF, Spechler SJ. Pathogenesis and cells of origin of Barrett’s esophagus. Gastroenterology. 2019;157:349–364.e1 https://doi.org/10.1053/j.gastro.2019.03.072.
Allen A, Flemström G. Gastroduodenal mucus bicarbonate barrier: protection against acid and pepsin. Am J Physiol Physiol. 2005;288:C1–19 https://doi.org/10.1152/ajpcell.00102.2004.
Celli JP, Turner BS, Afdhal NH, Keates S, Ghiran I, Kelly CP, et al. Helicobacter pylori moves through mucus by reducing mucin viscoelasticity. Proc Natl Acad Sci. 2009;106:14321–6 https://doi.org/10.1073/pnas.0903438106.
Szabo I. Formation of anion-selective channels in the cell plasma membrane by the toxin VacA of Helicobacter pylori is required for its biological activity. EMBO J. 1999;18:5517–27 https://doi.org/10.1093/emboj/18.20.5517.
Wang F, Xia P, Wu F, Wang D, Wang W, Ward T, et al. Helicobacter pylori VacA disrupts apical membrane-cytoskeletal interactions in gastric parietal cells. J Biol Chem. 2008;283:26714–25 https://doi.org/10.1074/jbc.M800527200.
Wunder C, Churin Y, Winau F, Warnecke D, Vieth M, Lindner B, et al. Cholesterol glucosylation promotes immune evasion by Helicobacter pylori. Nat Med. 2006;12:1030–8 https://doi.org/10.1038/nm1480.
Oyake J, Otaka M, Matsuhashi T, Jin M, Odashima M, Komatsu K, et al. Over-expression of 70-kDa heat shock protein confers protection against monochloramine-induced gastric mucosal cell injury. Life Sci. 2006;79:300–5 https://doi.org/10.1016/j.lfs.2006.01.013.
Santra A, Chowdhury A, Chaudhuri S, Das Gupta J, Banerjee PK, Mazumder DN. Oxidative stress in gastric mucosa in Helicobacter pylori infection. Indian J Gastroenterol Off J Indian Soc Gastroenterol. 2000;19:21–3.
Flahou B, Haesebrouck F, Chiers K, Van Deun K, De Smet L, Devreese B, et al. Gastric epithelial cell death caused by Helicobacter suis and Helicobacter pylori γ-glutamyl transpeptidase is mainly glutathione degradation-dependent. Cell Microbiol. 2011;13:1933–55 https://doi.org/10.1111/j.1462-5822.2011.01682.x.
Lin L-L, Chen C-N, Lin W-C, Lee P-H, Chang K-J, Lai Y-P, et al. Annexin A4: A novel molecular marker for gastric cancer withHelicobacter pylori infection using proteomics approach. Proteomics Clin Appl. 2008;2:619–34 https://doi.org/10.1002/prca.200780088.
Sostres C, Lanas A. Gastrointestinal effects of aspirin. Nat Rev Gastroenterol Hepatol. 2011;8:385–94 https://doi.org/10.1038/nrgastro.2011.97.
Tomisato W, Tsutsumi S, Rokutan K, Tsuchiya T, Mizushima T. NSAIDs induce both necrosis and apoptosis in guinea pig gastric mucosal cells in primary culture. Am J Physiol Liver Physiol. 2001;281:G1092–100 https://doi.org/10.1152/ajpgi.2001.281.4.G1092.
Nagano Y, Matsui H, Shimokawa O, Hirayama A, Tamura M, Nakamura Y, et al. Rebamipide attenuates nonsteroidal anti-inflammatory drugs (NSAID) induced lipid peroxidation by the manganese superoxide dismutase (MnSOD) overexpression in gastrointestinal epithelial cells. J Physiol Pharmacol. 2012;63:137–42.
Lacy ER, Ito S. Microscopic analysis of ethanol damage to rat gastric mucosa after treatment with a prostaglandin. Gastroenterology. 1982;83:619–25 https://doi.org/10.1016/S0016-5085(82)80198-4.
Starodub OT, Demitrack ES, Baumgartner HK, Montrose MH. Disruption of the Cox-1 gene slows repair of microscopic lesions in the mouse gastric epithelium. Am J Physiol Physiol. 2008;294:C223–32 https://doi.org/10.1152/ajpcell.00395.2006.
Divangahi M, Chen M, Gan H, Desjardins D, Hickman TT, Lee DM, et al. Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair. Nat Immunol. 2009;10:899–906 https://doi.org/10.1038/ni.1758.
Demitrack ES, Soleimani M, Montrose MH. Damage to the gastric epithelium activates cellular bicarbonate secretion via SLC26A9 Cl − /HCO 3 − exchange. Am J Physiol Liver Physiol. 2010;299:G255–64 https://doi.org/10.1152/ajpgi.00037.2010.
Qin X, Deitch EA. Dissolution of lipids from mucus: a possible mechanism for prompt disruption of gut barrier function by alcohol. Toxicol Lett. 2015;232:356–62 https://doi.org/10.1016/j.toxlet.2014.11.027.
McNeil PL, Ito S. Gastrointestinal cell plasma membrane wounding and resealing in vivo. Gastroenterology. 1989;96:1238–48 https://doi.org/10.1016/S0016-5085(89)80010-1.
Zeino Z, Sisson G, Bjarnason I. Adverse effects of drugs on small intestine and colon. Best Pract Res Clin Gastroenterol. 2010;24:133–41 https://doi.org/10.1016/j.bpg.2010.02.008.
Los FCO, Kao CY, Smitham J, McDonald KL, Ha C, Peixoto CA, et al. RAB-5- and RAB-11-dependent vesicle-trafficking pathways are required for plasma membrane repair after attack by bacterial pore-forming toxin. Cell Host Microbe. 2011;9:147–51.
Freedman J, Shrestha A, McClane B. Clostridium perfringens enterotoxin: action, genetics, and translational applications. Toxins (Basel). 2016;8:73 https://doi.org/10.3390/toxins8030073.
Geisler F, Coch RA, Richardson C, Goldberg M, Denecke B, Bossinger O, et al. The intestinal intermediate filament network responds to and protects against microbial insults and toxins. Development. 2019;146:e169482 https://doi.org/10.1242/dev.169482.
Lee K-Z, Lestradet M, Socha C, Schirmeier S, Schmitz A, Spenlé C, et al. Enterocyte purge and rapid recovery is a resilience reaction of the gut epithelium to pore-forming toxin attack. Cell Host Microbe. 2016;20:716–30 https://doi.org/10.1016/j.chom.2016.10.010.
Han YM, Park JM, Kang JX, Cha JY, Lee HJ, Jeong M, et al. Mitigation of indomethacin-induced gastrointestinal damages in fat-1 transgenic mice via gate-keeper action of ω-3-polyunsaturated fatty acids. Sci Rep. 2016;6:33992 https://doi.org/10.1038/srep33992.
Basivireddy J, Vasudevan A, Jacob M, Balasubramanian KA. Indomethacin-induced mitochondrial dysfunction and oxidative stress in villus enterocytes. Biochem Pharmacol. 2002;64:339–49 https://doi.org/10.1016/S0006-2952(02)01067-5.
Hart CA, Batt RM, Saunders JR, Getty B. Lectin-induced damage to the enterocyte brush border: an electron-microscopic study in rabbits. Scand J Gastroenterol. 1988;23:1153–9 https://doi.org/10.3109/00365528809090184.
Stutz K, Kaech A, Aebi M, Künzler M, Hengartner MO. Disruption of the C. elegans intestinal brush border by the fungal lectin CCL2 phenocopies dietary lectin toxicity in mammals. PLoS One. 2015;10:e0129381 https://doi.org/10.1371/journal.pone.0129381.
Gupta RK, Pande AH, Gulla KC, Gabius H-J, Hajela K. Carbohydrate-induced modulation of cell membrane. VIII. Agglutination with mammalian lectin galectin-1 increases osmofragility and membrane fluidity of trypsinized erythrocytes. FEBS Lett. 2006;580:1691–5 https://doi.org/10.1016/j.febslet.2006.02.006.
Miyake K, Tanaka T, McNeil PL. Lectin-based food poisoning: a new mechanism of protein toxicity. PLoS One. 2007;2:e687 https://doi.org/10.1371/journal.pone.0000687.
Gómez Castro MF, Miculán E, Herrera MG, Ruera C, Perez F, Prieto ED, et al. p31–43 gliadin peptide forms oligomers and induces NLRP3 inflammasome/caspase 1-dependent mucosal damage in small intestine. Front Immunol. 2019;10:31 https://doi.org/10.3389/fimmu.2019.00031.
Abu-Humaidan AH, Elvén M, Sonesson A, Garred P, Sørensen OE. Persistent intracellular Staphylococcus aureus in keratinocytes lead to activation of the complement system with subsequent reduction in the intracellular bacterial load. Front Immunol. 2018;9:396 https://doi.org/10.3389/fimmu.2018.00396.
Gaboriau F, Morlière P, Marquis I, Moysan A, Goegze M, Dubertret L. Membrane damage induced in cultured human skin fibroblasts by uva irradiation. Photochem Photobiol. 1993;58:515–20 https://doi.org/10.1111/j.1751-1097.1993.tb04924.x.
Vile GF, Tyrrell RM. Uva radiation-induced oxidative damage to lipids and proteins in vitro and in human skin fibroblasts is dependent on iron and singlet oxygen. Free Radic Biol Med. 1995;18:721–30 https://doi.org/10.1016/0891-5849(94)00192-M.
Tian F, Zhang F, Lai X, Wang L, Yang L, Wang X, et al. Nrf2-mediated protection against UVA radiation in human skin keratinocytes. Biosci Trends. 2011;5:23–9 https://doi.org/10.5582/bst.2011.v5.1.23.
Telorack M, Meyer M, Ingold I, Conrad M, Bloch W, Werner S. A glutathione-Nrf2-thioredoxin cross-talk ensures keratinocyte survival and efficient wound repair. PLoS Genet. 2016;12:e1005800 https://doi.org/10.1371/journal.pgen.1005800.
Appelqvist H, Wäster P, Eriksson I, Rosdahl I, Öllinger K. Lysosomal exocytosis and caspase-8-mediated apoptosis in UVA-irradiated keratinocytes. J Cell Sci. 2013;126:5578–84 https://doi.org/10.1242/jcs.130633.
Arun SN, Xie D, Howard AC, Zhong Q, Zhong X, McNeil PL, et al. Cell wounding activates phospholipase D in primary mouse keratinocytes. J Lipid Res. 2013;54:581–91 https://doi.org/10.1194/jlr.M027060.
Yu QC, McNeil PL. Transient disruptions of aortic endothelial cell plasma membranes. Am J Pathol. 1992;141:1349–60.
Ruan C-C, Gao P-J. Role of complement-related inflammation and vascular dysfunction in hypertension. Hypertension. 2019;73:965–71 https://doi.org/10.1161/HYPERTENSIONAHA.118.11210.
Bucala R, Makita Z, Koschinsky T, Cerami A, Vlassara H. Lipid advanced glycosylation: pathway for lipid oxidation in vivo. Proc Natl Acad Sci. 1993;90:6434–8 https://doi.org/10.1073/pnas.90.14.6434.
Xiong F, Leonov S, Howard AC, Xiong S, Zhang B, Mei L, et al. Receptor for advanced glycation end products (RAGE) prevents endothelial cell membrane resealing and regulates F-actin remodeling in a β-catenin-dependent manner. J Biol Chem. 2011;286:35061–70 https://doi.org/10.1074/jbc.M111.261073.
Butterfield DA, Drake J, Pocernich C, Castegna A. Evidence of oxidative damage in Alzheimer’s disease brain: central role for amyloid β-peptide. Trends Mol Med. 2001;7:548–54 https://doi.org/10.1016/S1471-4914(01)02173-6.
Hattori R, Hamilton KK, McEver RP, Sims PJ. Complement proteins C5b-9 induce secretion of high molecular weight multimers of endothelial von Willebrand factor and translocation of granule membrane protein GMP-140 to the cell surface. J Biol Chem. 1989;264:9053–60.
Chehab T, Santos NC, Holthenrich A, Koerdt SN, Disse J, Schuberth C, et al. A novel Munc13–4/S100A10/annexin A2 complex promotes Weibel–Palade body exocytosis in endothelial cells. Mol Biol Cell. 2017;28:1688–700 https://doi.org/10.1091/mbc.e17-02-0128.
Koerdt SN, Gerke V. Annexin A2 is involved in Ca (2+)-dependent plasma membrane repair in primary human endothelial cells. Biochim Biophys acta Mol cell Res. 2017;1864:1046–53.
Noone DG, Riedl M, Pluthero FG, Bowman ML, Liszewski MK, Lu L, et al. Von Willebrand factor regulates complement on endothelial cells. Kidney Int. 2016;90:123–34 https://doi.org/10.1016/j.kint.2016.03.023.
Lehner T, Almeida JD, Levinsky RJ. Damaged membrane fragments and immune complexes in the blood of patients with Behcet’s syndrome. Clin Exp Immunol. 1978;34:206–12.
Nomura S, Takano-Yamamoto T. Molecular events caused by mechanical stress in bone. Matrix Biol. 2000;19:91–6 https://doi.org/10.1016/S0945-053X(00)00050-0.
Hagan ML, Yu K, Zhu J, Vinson BN, Roberts RL, Montesinos Cartagena M, et al. Decreased pericellular matrix production and selection for enhanced cell membrane repair may impair osteocyte responses to mechanical loading in the aging skeleton. Aging Cell. 2020;19:13056 https://doi.org/10.1111/acel.13056.
Morrell AE, Brown GN, Robinson ST, Sattler RL, Baik AD, Zhen G, et al. Mechanically induced Ca2+ oscillations in osteocytes release extracellular vesicles and enhance bone formation. Bone Res. 2018;6(6) https://doi.org/10.1038/s41413-018-0007-x.
Hagan ML, Bahraini A, Pierce JL, Bass SM, Yu K, Elsayed R, et al. Inhibition of osteocyte membrane repair activity via dietary vitamin E deprivation impairs osteocyte survival. Calcif Tissue Int. 2019;104:224–34 https://doi.org/10.1007/s00223-018-0487-0.
Nourissat P, Travert M, Chevanne M, Tekpli X, Rebillard A, Le Moigne-Müller G, et al. Ethanol induces oxidative stress in primary rat hepatocytes through the early involvement of lipid raft clustering. Hepatology. 2007;47:59–70 https://doi.org/10.1002/hep.21958.
Bellanti F, Villani R, Facciorusso A, Vendemiale G, Serviddio G. Lipid oxidation products in the pathogenesis of non-alcoholic steatohepatitis. Free Radic Biol Med. 2017;111:173–85 https://doi.org/10.1016/j.freeradbiomed.2017.01.023.
Perez MJ, Briz O. Bile-acid-induced cell injury and protection. World J Gastroenterol. 2009;15:1677 https://doi.org/10.3748/wjg.15.1677.
Woolbright BL, Dorko K, Antoine DJ, Clarke JI, Gholami P, Li F, et al. Bile acid-induced necrosis in primary human hepatocytes and in patients with obstructive cholestasis. Toxicol Appl Pharmacol. 2015;283:168–77 https://doi.org/10.1016/j.taap.2015.01.015.
Gores GJ, Herman B, Lemasters JJ. Plasma membrane bleb formation and rupture: a common feature of hepatocellular injury. Hepatology. 1990;11:690–8 https://doi.org/10.1002/hep.1840110425.
Li F-C, Huang G-T, Lin C-J, Wang S-S, Sun T-L, Lo S-Y, et al. Apical membrane rupture and backward bile flooding in acetaminophen-induced hepatocyte necrosis. Cell Death Dis. 2011;2:e183 https://doi.org/10.1038/cddis.2011.68.
Yao W, Li H, Han X, Chen C, Zhang Y, Tai WL, et al. MG53 anchored by dysferlin to cell membrane reduces hepatocyte apoptosis which induced by ischaemia/reperfusion injury in vivo and in vitro. J Cell Mol Med. 2017;21:2503–13 https://doi.org/10.1111/jcmm.13171.
Enrich C, Rentero C, Grewal T. Annexin A6 in the liver: from the endocytic compartment to cellular physiology. Biochim Biophys Acta Mol Cell Res. 2017;1864:933–46 https://doi.org/10.1016/j.bbamcr.2016.10.017.
Gerasimenko JV, Peng S, Tsugorka T, Gerasimenko OV. Ca2+ signalling underlying pancreatitis. Cell Calcium. 2018;70:95–101 https://doi.org/10.1016/j.ceca.2017.05.010.
Wittel UA, Pandey KK, Andrianifahanana M, Johansson SL, Cullen DM, Akhter MP, et al. Chronic pancreatic inflammation induced by environmental tobacco smoke inhalation in rats. Am J Gastroenterol. 2006;101:148–59 https://doi.org/10.1111/j.1572-0241.2006.00405.x.
Gerasimenko JV, Gryshchenko O, Ferdek PE, Stapleton E, Hebert TOG, Bychkova S, et al. Ca2+ release-activated Ca2+ channel blockade as a potential tool in antipancreatitis therapy. Proc Natl Acad Sci. 2013;110:13186–91 https://doi.org/10.1073/pnas.1300910110.
Bockman DE, Guo J, Müller MW, Friess H, Büchler MW. Cell wounding in early experimental acute pancreatitis. Lab Investig. 2004;84:362–7 https://doi.org/10.1038/labinvest.3700064.
Müller MW, McNeil PL, Büchler P, Ceyhan GO, Wolf-Hieber E, Adler G, et al. Acinar cell membrane disruption is an early event in experimental acute pancreatitis in rats. Pancreas. 2007;35:e30–40 https://doi.org/10.1097/mpa.0b013e318120024c.
Thomas DDH, Kaspar KM, Taft WB, Weng N, Rodenkirch LA, Groblewski GE. Identification of annexin VI as a Ca 2+ −sensitive CRHSP-28-binding protein in pancreatic acinar cells. J Biol Chem. 2002;277:35496–502 https://doi.org/10.1074/jbc.M110917200.
Clark A, Wells CA, Buley ID, Cruickshank JK, Vanhegan RI, Matthews DR, et al. Islet amyloid, increased A-cells, reduced B-cells and exocrine fibrosis: quantitative changes in the pancreas in type 2 diabetes. Diabetes Res. 1988;9(4)151–9 http://www.ncbi.nlm.nih.gov/pubmed/3073901.
Janson J, Soeller WC, Roche PC, Nelson RT, Torchia AJ, Kreutter DK, et al. Spontaneous diabetes mellitus in transgenic mice expressing human islet amyloid polypeptide. Proc Natl Acad Sci. 1996;93:7283–8 https://doi.org/10.1073/pnas.93.14.7283.
Mirzabekov TA, Lin M, Kagan BL. Pore formation by the cytotoxic islet amyloid peptide amylin. J Biol Chem. 1996;271:1988–92 https://doi.org/10.1074/jbc.271.4.1988.
Sparr E, Engel MFM, Sakharov DV, Sprong M, Jacobs J, de Kruijff B, et al. Islet amyloid polypeptide-induced membrane leakage involves uptake of lipids by forming amyloid fibers. FEBS Lett. 2004;577:117–20 https://doi.org/10.1016/j.febslet.2004.09.075.
Pilkington EH, Gurzov EN, Kakinen A, Litwak SA, Stanley WJ, Davis TP, et al. Pancreatic β-cell membrane fluidity and toxicity induced by human islet amyloid polypeptide species. Sci Rep. 2016;6:21274 https://doi.org/10.1038/srep21274.
Mo X-D, Gao L-P, Wang Q-J, Yin J, Jing Y-H. Lipid accelerating the fibril of islet amyloid polypeptide aggravated the pancreatic islet injury in vitro and in vivo. Lipids Health Dis. 2018;17:42 https://doi.org/10.1186/s12944-018-0694-8.
Hull RL, Andrikopoulos S, Verchere CB, Vidal J, Wang F, Cnop M, et al. Increased dietary fat promotes islet amyloid formation and -cell secretory dysfunction in a transgenic mouse model of islet amyloid. Diabetes. 2003;52:372–9 https://doi.org/10.2337/diabetes.52.2.372.
Marrif HI, Al-Sunousi SI. Pancreatic β cell mass death. Front Pharmacol. 2016;7:83 https://doi.org/10.3389/fphar.2016.00083.
Casas S, Novials A, Reimann F, Gomis R, Gribble FM. Calcium elevation in mouse pancreatic beta cells evoked by extracellular human islet amyloid polypeptide involves activation of the mechanosensitive ion channel TRPV4. Diabetologia. 2008;51:2252–62 https://doi.org/10.1007/s00125-008-1111-z.
Huang C, Gurlo T, Haataja L, Costes S, Daval M, Ryazantsev S, et al. Calcium-activated calpain-2 is a mediator of beta cell dysfunction and apoptosis in type 2 diabetes. J Biol Chem. 2010;285:339–48 https://doi.org/10.1074/jbc.M109.024190.
Medana IM, Esiri MM. Axonal damage: a key predictor of outcome in human CNS diseases. Brain. 2003;126:515–30 https://doi.org/10.1093/brain/awg061.
Kaur P, Sharma S. Recent advances in pathophysiology of traumatic brain injury. Curr Neuropharmacol. 2018;16:1224–38 https://doi.org/10.2174/1570159X15666170613083606.
Maxwell WL. Histopathological changes at central nodes of Ranvier after stretch-injury. Microsc Res Tech. 1996;34:522–35 https://doi.org/10.1002/(SICI)1097-0029(19960815)34:6<522::AID-JEMT4>3.0.CO;2-L.
Zhang J, Sun X, Zheng S, Liu X, Jin J, Ren Y, et al. Myelin basic protein induces neuron-specific toxicity by directly damaging the neuronal plasma membrane. PLoS One. 2014;9:e108646 https://doi.org/10.1371/journal.pone.0108646.
Ambadi Thody S, Mathew MK, Udgaonkar JB. Mechanism of aggregation and membrane interactions of mammalian prion protein. Biochim Biophys Acta Biomembr. 2018;1860:1927–35 https://doi.org/10.1016/j.bbamem.2018.02.031.
Bode DC, Freeley M, Nield J, Palma M, Viles JH. Amyloid-β oligomers have a profound detergent-like effect on lipid membrane bilayers, imaged by atomic force and electron microscopy. J Biol Chem. 2019;294:7566–72 https://doi.org/10.1074/jbc.AC118.007195.
Annunziata I, Patterson A, Helton D, Hu H, Moshiach S, Gomero E, et al. Lysosomal NEU1 deficiency affects amyloid precursor protein levels and amyloid-β secretion via deregulated lysosomal exocytosis. Nat Commun. 2013;4:2734 https://doi.org/10.1038/ncomms3734.
Tsunemi T, Perez-Rosello T, Ishiguro Y, Yoroisaka A, Jeon S, Hamada K, et al. Increased lysosomal exocytosis induced by lysosomal Ca 2+ channel agonists protects human dopaminergic neurons from α-synuclein toxicity. J Neurosci. 2019;39:5760–72 https://doi.org/10.1523/JNEUROSCI.3085-18.2019.
Edgar JR, Willén K, Gouras GK, Futter CE. ESCRTs regulate amyloid precursor protein sorting in multivesicular bodies and intracellular amyloid-β accumulation. J Cell Sci. 2015;128:2520–8 https://doi.org/10.1242/jcs.170233.
Julien C, Tomberlin C, Roberts CM, Akram A, Stein GH, Silverman MA, et al. In vivo induction of membrane damage by β-amyloid peptide oligomers. Acta Neuropathol Commun. 2018;6:131 https://doi.org/10.1186/s40478-018-0634-x.
Fruhmann G, Marchal C, Vignaud H, Verduyckt M, Talarek N, De Virgilio C, et al. The impact of ESCRT on Aβ1–42 induced membrane lesions in a yeast model for Alzheimer’s disease. Front Mol Neurosci. 2018;11:406 https://doi.org/10.3389/fnmol.2018.00406.
Jones L, Holmans PA, Hamshere ML, Harold D, Moskvina V, Ivanov D, et al. Genetic evidence implicates the immune system and cholesterol metabolism in the aetiology of Alzheimer’s disease. PLoS One. 2010;5:e13950 https://doi.org/10.1371/journal.pone.0013950.
Sameni S, Malacrida L, Tan Z, Digman MA. Alteration in fluidity of cell plasma membrane in Huntington disease revealed by spectral phasor analysis. Sci Rep. 2018;8:734 https://doi.org/10.1038/s41598-018-19160-0.
Naudí A, Jové M, Ayala V, Portero-Otín M, Barja G, Pamplona R. Membrane lipid unsaturation as physiological adaptation to animal longevity. Front Physiol. 2013;4:372 https://doi.org/10.3389/fphys.2013.00372.
Mahar M, Cavalli V. Intrinsic mechanisms of neuronal axon regeneration. Nat Rev Neurosci. 2018;19:323–37 https://doi.org/10.1038/s41583-018-0001-8.
He Z, Jin Y. Intrinsic control of axon regeneration. Neuron. 2016;90:437–51 https://doi.org/10.1016/j.neuron.2016.04.022.
Kilinc D, Gallo G, Barbee KA. Mechanical membrane injury induces axonal beading through localized activation of calpain. Exp Neurol. 2009;219:553–61 https://doi.org/10.1016/j.expneurol.2009.07.014.
Bi G-Q, Morris RL, Liao G, Alderton JM, Scholey JM, Steinhardt RA. Kinesin- and myosin-driven steps of vesicle recruitment for Ca2+−regulated exocytosis. J Cell Biol. 1997;138:999–1008 https://doi.org/10.1083/jcb.138.5.999.
Ratliff BB, Abdulmahdi W, Pawar R, Wolin MS. Oxidant mechanisms in renal injury and disease. Antioxid Redox Signal. 2016;25:119–46 https://doi.org/10.1089/ars.2016.6665.
Kalakeche R, Hato T, Rhodes G, Dunn KW, El-Achkar TM, Plotkin Z, et al. Endotoxin uptake by S1 proximal tubular segment causes oxidative stress in the downstream S2 segment. J Am Soc Nephrol. 2011;22:1505–16 https://doi.org/10.1681/ASN.2011020203.
Vasko R. Peroxisomes and kidney injury. Antioxid Redox Signal. 2016;25:217–31 https://doi.org/10.1089/ars.2016.6666.
Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16:1180–91 https://doi.org/10.1038/ncb3064.
Hasegawa K, Wakino S, Yoshioka K, Tatematsu S, Hara Y, Minakuchi H, et al. Kidney-specific overexpression of Sirt1 protects against acute kidney injury by retaining peroxisome function. J Biol Chem. 2010;285:13045–56 https://doi.org/10.1074/jbc.M109.067728.
Morigi M, Perico L, Rota C, Longaretti L, Conti S, Rottoli D, et al. Sirtuin 3–dependent mitochondrial dynamic improvements protect against acute kidney injury. J Clin Invest. 2015;125:715–26 https://doi.org/10.1172/JCI77632.
Mahmoudi M, Willgoss D, Cuttle L, Yang T, Pat B, Winterford C, et al. In vivo and in vitro models demonstrate a role for caveolin-1 in the pathogenesis of ischaemic acute renal failure. J Pathol. 2003;200:396–405 https://doi.org/10.1002/path.1368.
Lieber RL, Friden J. Muscle damage is not a function of muscle force but active muscle strain. J Appl Physiol. 1993;74:520–6 https://doi.org/10.1152/jappl.1993.74.2.520.
Sinha B, Köster D, Ruez R, Gonnord P, Bastiani M, Abankwa D, et al. Cells respond to mechanical stress by rapid disassembly of caveolae. Cell. 2011;144:402–13 https://doi.org/10.1016/j.cell.2010.12.031.
Barthélémy F, Defour A, Lévy N, Krahn M, Bartoli M. Muscle cells fix breaches by orchestrating a membrane repair ballet. J Neuromuscul Dis. 2018;5:21–8 https://doi.org/10.3233/JND-170251.
Lenhart KC, O’Neill TJ, Cheng Z, Dee R, Demonbreun AR, Li J, et al. GRAF1 deficiency blunts sarcolemmal injury repair and exacerbates cardiac and skeletal muscle pathology in dystrophin-deficient mice. Skelet Muscle. 2015;5:27 https://doi.org/10.1186/s13395-015-0054-6.
Fujiwara D, Iwahara N, Sebori R, Hosoda R, Shimohama S, Kuno A, et al. SIRT1 deficiency interferes with membrane resealing after cell membrane injury. PLoS One. 2019;14:e0218329 https://doi.org/10.1371/journal.pone.0218329.
Scheffer LL, Sreetama SC, Sharma N, Medikayala S, Brown KJ, Defour A, et al. Mechanism of Ca2+−triggered ESCRT assembly and regulation of cell membrane repair. Nat Commun. 2014;5:5646 https://doi.org/10.1038/ncomms6646.
Carmeille R, Bouvet F, Tan S, Croissant C, Gounou C, Mamchaoui K, et al. Membrane repair of human skeletal muscle cells requires Annexin-A5. Biochim Biophys Acta - Mol Cell Res. 2016;1863:2267–79 https://doi.org/10.1016/j.bbamcr.2016.06.003.
Posey AD, Swanson KE, Alvarez MG, Krishnan S, Earley JU, Band H, et al. EHD1 mediates vesicle trafficking required for normal muscle growth and transverse tubule development. Dev Biol. 2014;387:179–90 https://doi.org/10.1016/j.ydbio.2014.01.004.
Demonbreun AR, Fallon KS, Oosterbaan CC, Bogdanovic E, Warner JL, Sell JJ, et al. Recombinant annexin A6 promotes membrane repair and protects against muscle injury. J Clin Invest. 2019;129:4657–70 https://doi.org/10.1172/JCI128840.
Taneike M, Mizote I, Morita T, Watanabe T, Hikoso S, Yamaguchi O, et al. Calpain protects the heart from hemodynamic stress. J Biol Chem. 2011;286:32170–7 https://doi.org/10.1074/jbc.M111.248088.
Sveen M-L, Thune JJ, Køber L, Vissing J. Cardiac involvement in patients with limb-girdle muscular dystrophy type 2 and Becker muscular dystrophy. Arch Neurol. 2008;65 https://doi.org/10.1001/archneur.65.9.1196.
Zhang C, Chen B, Wang Y, Guo A, Tang Y, Khataei T, et al. MG53 is dispensable for T-tubule maturation but critical for maintaining T-tubule integrity following cardiac stress. J Mol Cell Cardiol. 2017;112:123–30 https://doi.org/10.1016/j.yjmcc.2017.08.007.
Wang X, Xie W, Zhang Y, Lin P, Han L, Han P, et al. Cardioprotection of ischemia/reperfusion injury by cholesterol-dependent MG53-mediated membrane repair. Circ Res. 2010;107:76–83 https://doi.org/10.1161/CIRCRESAHA.109.215822.
Cao C-M, Zhang Y, Weisleder N, Ferrante C, Wang X, Lv F, et al. MG53 constitutes a primary determinant of cardiac ischemic preconditioning. Circulation. 2010;121:2565–74 https://doi.org/10.1161/CIRCULATIONAHA.110.954628.
Neubert E, Meyer D, Rocca F, Günay G, Kwaczala-Tessmann A, Grandke J, et al. Chromatin swelling drives neutrophil extracellular trap release. Nat Commun. 2018;9:3767 https://doi.org/10.1038/s41467-018-06263-5.
Shu F, Chen J, Ma X, Fan Y, Yu L, Zheng W, et al. Cholesterol crystal-mediated inflammation is driven by plasma membrane destabilization. Front Immunol. 2018;9:1163 https://doi.org/10.3389/fimmu.2018.01163.
Rudd-Schmidt JA, Hodel AW, Noori T, Lopez JA, Cho H-J, Verschoor S, et al. Lipid order and charge protect killer T cells from accidental death. Nat Commun. 2019;10:5396 https://doi.org/10.1038/s41467-019-13385-x.
Michelet X, Tuli A, Gan H, Geadas C, Sharma M, Remold HG, et al. Lysosome-mediated plasma membrane repair is dependent on the small GTPase Arl8b and determines cell death type in Mycobacterium tuberculosis infection. J Immunol. 2018;200:3160–9 https://doi.org/10.4049/jimmunol.1700829.
Miyatake Y, Yamano T, Hanayama R. Myoferlin-mediated lysosomal exocytosis regulates cytotoxicity by phagocytes. J Immunol. 2018;201:3051–7 https://doi.org/10.4049/jimmunol.1800268.
Acknowledgements
We extend gratitude to all frontline workers for their efforts to mitigate damage caused by this pandemic.
Funding
This work was supported by funding from the Canadian Institutes of Health Research (FDN#154329 to J.H.B.), National Institutes of Health (GM052932 to W.M.B.), and doctoral scholarships provided by NSERC and the Hospital for Sick Children’s Research Training Centre to D.A.A.
Author information
Authors and Affiliations
Contributions
D.A.A. wrote the manuscript under the supervision of W.M.B. and J.H.B. All three authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ammendolia, D.A., Bement, W.M. & Brumell, J.H. Plasma membrane integrity: implications for health and disease. BMC Biol 19, 71 (2021). https://doi.org/10.1186/s12915-021-00972-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12915-021-00972-y