
Abstract
Background
Rodent malaria parasites (RMP) are used extensively as models of human malaria. Draft RMP genomes have been published for Plasmodium yoelii, P. berghei ANKA (PbA) and P. chabaudi AS (PcAS). Although availability of these genomes made a significant impact on recent malaria research, these genomes were highly fragmented and were annotated with little manual curation. The fragmented nature of the genomes has hampered genome wide analysis of Plasmodium gene regulation and function.
Results
We have greatly improved the genome assemblies of PbA and PcAS, newly sequenced the virulent parasite P. yoelii YM genome, sequenced additional RMP isolates/lines and have characterized genotypic diversity within RMP species. We have produced RNA-seq data and utilised it to improve gene-model prediction and to provide quantitative, genome-wide, data on gene expression. Comparison of the RMP genomes with the genome of the human malaria parasite P. falciparum and RNA-seq mapping permitted gene annotation at base-pair resolution. Full-length chromosomal annotation permitted a comprehensive classification of all subtelomeric multigene families including the `Plasmodium interspersed repeat genes' (pir). Phylogenetic classification of the pir family, combined with pir expression patterns, indicates functional diversification within this family.
Conclusions
Complete RMP genomes, RNA-seq and genotypic diversity data are excellent and important resources for gene-function and post-genomic analyses and to better interrogate Plasmodium biology. Genotypic diversity between P. chabaudi isolates makes this species an excellent parasite to study genotype-phenotype relationships. The improved classification of multigene families will enhance studies on the role of (variant) exported proteins in virulence and immune evasion/modulation.
Similar content being viewed by others

Background
Rodent malaria parasites (RMP) are used extensively as models of human malaria [1],[2]. Four different species that infect African rodents have been adapted for laboratory use: Plasmodium berghei, P. yoelii, P. chabaudi and P. vinckei. Small differences exist in the biology of the different RMP in laboratory mice and this makes them particularly attractive models to investigate different aspects of human malaria. Specifically, P. chabaudi is a model to investigate mechanisms of drug resistances and immune evasion, in particular antigenic variation [3],[4]. It invades normocytes and reticulocytes and mostly produces chronic, non-lethal, infections. In contrast, P. berghei preferentially invades reticulocytes and usually produces infections in mice that induce severe pathology [2]. In combination with different mouse strains it has been used as a model to study immunopathology, experimental cerebral malaria, pregnancy-associated malaria and lung pathology [2]. P. yoelii is widely used in studies on the biology of liver stages and on innate and acquired immunity against liver stages [5],[6]. Blood stage P. yoelii parasites of some lines are restricted to reticulocytes whereas others can invade all red blood cells and have been used to study receptors for erythrocyte binding [7],[8]. The availability of efficient reverse genetics technologies for P. berghei and P. yoelii [9]-[11] and the ability to analyse these parasites throughout the complete life cycle have made these two species the preferred models for analysis of Plasmodium gene function [12]-[14]. For these two species more than 600 different genetically modified mutants have been reported [15].
The first draft RMP genome was published in 2002 for P. yoelii yoelii 17XNL [16]. This was followed by publication of draft genomes of P. berghei ANKA (PbA) and P. chabaudi chabaudi AS (PcAS) in 2005 [17]. Comparisons with the genome of the human parasite P. falciparum and other primate malaria species defined a large set of core genes that are shared between RMPs and primate malarias [18]-[20]. Although availability of draft RMP genomes made a significant impact in applying post-genomic technologies for understanding malaria biology [18] and were used in many follow-up functional genomics studies to analyse gene regulation and function [9],[10], these RMP genomes were highly fragmented and were annotated with little or no manual curation. The fragmented nature of the genomes has hampered genome wide analysis of gene regulation and function, especially of the (subtelomeric) multigene families. To utilise RMP models to their full potential, we therefore undertook production of high quality reference genomes: for PbA and PcAS large-scale improvement of their existing genomes, with re-sequencing, re-analysis and manual re-annotation, and for P. y. yoelii a genome sequence was produced de novo from the virulent YM line using the latest sequencing technologies and computational algorithms. In addition, we have utilised comprehensive RNA-seq data derived from a number of life-cycle stages to both improve gene model prediction and to provide genome-wide, quantitative data on gene expression. By sequencing additional isolates/lines of P. berghei, P. yoelii and P. chabaudi (including the subspecies P. c. adami) we have documented genotypic diversity that exists within different RMP species. The availability of RMP reference genomes in combination with the RNA-seq and genotypic diversity data will serve as excellent resources for gene-function and post-genomic analyses and, therefore, better interrogation of Plasmodium biology and development of anti-malaria interventions.
The genomes of RMP contain a number of multigene families located in the subtelomeric chromosomal regions. These include a large family of so-called `Plasmodium interspersed repeat genes' (pir) [16], that are present also in other human/primate Plasmodium species [20]-[23]. Most of these gene families are expressed in blood stages and these proteins show features that have been reported to contribute to immune evasion through antigenic variation [24]-[26] and may play a role in the sequestration of infected red blood cells and virulence [26],[27]. As a result of the improved annotation, we have been able to define all multigene families in the RMP genomes. Comparative phylogenetic analyses of the pir genes and analyses of pir expression patterns in blood stages of P. berghei provide evidence of functional diversification within this gene family. The improved classification of multigene families will enhance studies on the role of (variant) exported proteins in virulence and evasion and modulation of the immune system.
Results
Generation of high-quality RMP reference genomes
With a combination of Sanger and second generation sequencing (that is, Illumina and 454), automated scaffolding, gap closure, error correction and annotation transfer, followed by manual inspection, we obtained highly accurate and almost complete reference genomes of PbA, PcAS and P. y. yoelii YM (PyYM). This resulted in a significant reduction in contig number for the PbA and PcAS genomes (Table 1) compared with existing highly fragmented drafts [16],[17]. The new assemblies contain 4,979, 5,139 and 5,675 protein-coding genes for PbA, PcAS and PyYM, respectively, with 487 and 409 novel genes in the genomes of PbA and PcAS that were absent in the draft genomes (Table 1, Additional file 1). More than 98% of the predicted genes are now present as full-length gene models and we were able to ascribe putative functions (that is, they are not annotated as encoding hypothetical proteins of unknown function) to 56% to 61% of these. This percentage is comparable to the 60% of the P. falciparum genes that have annotated functions. As a result of eliminating incomplete gene models and merging multiple incorrect gene models into single gene models and by removing mouse DNA sequence contamination, only 63% and 77% of the previously annotated PbA and PcAS genes were mapped back to the new genomes. The RMP reference genomes have a size of 18.5 to 21.9 Mb (Table 1), confirming the smaller genome sizes of RMPs compared with primate malaria species but both the mitochondrial and apicoplast RMP genomes are highly comparable in size and gene content to those of P. falciparum (Table 1). The predicted proteomes were analysed for the presence of PEXEL-motifs, a characteristic of host-exported proteins, using ExportPred v2.0 [28]. Between 97 and 119 PEXEL-positive proteins were predicted for the different RMP. This indicates that, like P. berghei, the other RMP also contain three times more PEXEL-positive proteins than was previously predicted [29] (see Additional file 2).
Conserved chromosome organization and gene orthology between RMP and primate malaria parasites
The improved genomes confirmed the extensive conservation of the RMP genomes and the presence of only a single synteny breakpoint ([19]; Table 1). Despite the highly conserved internal regions of the 14 chromosomes, species-specific paralogous expansion and diversification of certain genes has occurred in each genome. Additional file 3A shows an example of such an expanded locus within a region of conserved synteny between PcAS and PyYM. In PbA only a single copy (PBANKA_091920) is present, whereas the genomes of PcAS and PyYM contain multiple copies (fam-d; Table 2, Additional file 4), organized as a single gene cluster on chromosome 9.
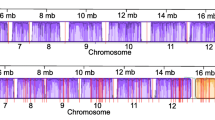
The re-annotation resulted in a better characterization of several non-coding and coding features of the chromosomes such as the centromeric and subtelomeric regions. As an example we show in Additional file 3B the size, location and GC-content of a positionally conserved centromere-containing region of chromosome 7 of PbA and PcAS. Figure 1A shows an example of the organization of RMP subtelomeric regions, visualizing the location of genes of multigene families. Although these regions contain members of multigene families that are shared between RMP, they are highly variable as a result of variation in gene copy number and the presence of species-specific genes and (non-coding) repeat sequences. For example, several PbA subtelomeric regions contain many copies of a large, 2.3 kb, repeat element that is P. berghei-specific (Figure 1B; [30]). Based on the coverage-depth of Illumina sequence data mapped onto the 2.3 kb repeats, we estimated that the PbA genome contains about 400 copies, representing approximately 4% of the total nucleotide content. In the PbA genome only 47 of these repeats have been assembled and the remaining copies (approximately 350) are either located as clusters in the sequence gaps that still exist in the PbA subtelomeric regions or are located within the existing current 2.3 kb repeat-arrays. These 2.3 kb repeats contain telomeric repeat sequences and many contain a copy of a highly degenerate pir pseudogene (Figure 1B; [30]) suggesting that the expansion of this repeat may have originally been driven by an expansion of pir gene numbers.

Organization of subtelomeric regions of RMP chromosomes. A) Organization of subtelomeric regions of chromosomes 13 of PyYM (left region), 5 of PcAS (left region) and 11 of PbA (left region). The order and orientation of the genes are shown, including genes belonging to the pir, fam-a, fam-b and fam-c gene families. Exons are shown in coloured boxes with introns as linking lines. As a comparison, a subtelomeric region of P. falciparum 3D7 chromosome 9 is shown. The shaded/grey areas mark the start of the conserved, syntenic regions. Black angular lines represent gaps. B) Artemis view showing a copy of the PbA-specific 2.3 kb repeat element containing a fragmented and `pseudogenised' bir gene (PBANKA_000720). Also shown is the location of three 27 bp telomeric repeat units. The HMM Pfam match `Cir_Bir_Yir PF06022' (grey box) spans amino acid 6 to 239 with an e-value of 2.5e-77. RPM, rodent malaria parasites.
We compared all predicted RMP protein-coding genes with those of three primate malaria species, P. falciparum, P. knowlesi and P. vivax using OrthoMCL and divided the predicted RMP proteome into three different categories: (1) RMP proteins with orthologs in any of the primate malarias; (2) RMP-specific proteins with no orthologs in primate malarias; and (3) primate malaria-specific proteins with no orthologs in any of the RMP (see Additional file 5). Between the predicted RMP proteomes (15,793 proteins in total) and primate malaria proteomes (15,853 proteins in total), approximately 87% of the RMP proteins had detectable orthologs in at least one of the primate malarias and only 2,104 proteins (13.3%) were predicted to be RMP-specific. Of those 2,104 proteins, 1,854 (88.1%) are from gene families, as defined in Additional file 4. For 2,306 primate malaria proteins (14.6%) no orthologs have been detected in the RMP. Of these primate malaria specific genes, approximately 1,635 (70.9%) are subtelomeric genes or members of subtelomeric gene families (see Additional file 5).
Genotypic diversity within RMP isolates: P. chabaudiisolates exhibit high level polymorphism amongst their genes
The availability of multiple isolates of RMP with different phenotypic traits offers the possibility of using genetics to study phenotype/genotype associations. To quantify the level of genotypic diversity across multiple RMP isolates we produced genome sequence data at a 79- to 437-fold coverage, from isolates of P. berghei (NK65, K173, SP11 and its pyrimethamine resistant descendant SP11 RLL), P. c. chabaudi (AJ, CB) and P. c. adami (DK, DS). In addition, we sequenced P. y. yoelii 17X (see Additional file 6 for parasite selection rationale and Additional file 7 for parasite origins). Single nucleotide polymorphisms (SNPs) in the genomes of these parasites were called by mapping the reads against their respective reference genomes (Table 3, Additional file 8) after excluding repetitive or low complexity regions of genes and members of multigene families (genes in Additional file 4). The level of polymorphism between the four P. berghei isolates is surprisingly low with SNPs detected in only 4 to 469 genes (Table 3, Additional file 8). In P. berghei the highest SNP numbers were found in two lines, SP11 RLL and K173cl1, which have been maintained in the laboratory for prolonged periods by mechanical blood passage between mice. Comparison of the genomes of SP11 and its pyrimethamine resistant descendant SP11 RLL, revealed the point mutation in the dihydrofolate reductase-thymidylate synthase gene, known to be involved in pyrimethamine resistance (see Additional file 8; [31]). Comparing the genomes of the non-lethal P. y. yoelii 17X isolate and its virulent descendant laboratory line YM showed limited polymorphism and revealed SNPs in the Duffy-binding protein that have been implicated in the different invasion and virulence phenotypes of these two lines (see Additional file 8; [7]). In contrast to the low numbers of P. y yoelii genes with SNPs (eight genes), large differences exist in gene copy number of subtelomeric multigene families (Table 3) which accounts for the difference in genome size between the two laboratory lines.
In contrast to the P. berghei isolates, the P. chabaudi isolates and subspecies have much higher SNP densities with 4,300 to 4,500 (out of 4,576) non-subtelomeric genes having at least one SNP (Table 3, Additional file 8). The high genotypic diversity is not only evident between the subspecies P. c. chabaudi and P. c. adami, but also between isolates of the same subspecies. For example, we found 94,668 and 71,074 unique SNPs (in 3,978 and 4,166 genes) in the P. c adami DK and DS isolates, respectively. Between different P. chabaudi isolates differences exist in virulence- and invasion phenotypes of blood stage infections (see Additional file 6). We detected multiple SNPs in P. chabaudi genes involved in binding to red blood cells (RBC) such as the Duffy-binding protein and reticulocyte binding proteins (see Additional file 8), genes that are associated with differences in virulence between P. y. yoelii lines. Isolate-specific protective immunity between P. c. chabaudi isolates has been linked to the merozoite surface protein 1 (MSP1; PCHAS_083130) [32],[33]. Our analyses revealed an excess of non-synonymous substitutions (reflected in high Ka/Ks values) in msp1 of all P. chabaudi isolates (see Additional file 8).
High resolution, genome-wide expression data from different RMP life cycle stages
To further improve gene annotation and to provide foundational data for gene-function studies, we generated RNA-seq data from several life-cycle stages. RNA was analysed from synchronised PbA asexual blood stages (ring forms, late trophozoites and schizonts) and from purified gametocytes and ookinetes. In addition, RNA-seq data was generated from multiple samples of blood stage trophozoites of PcAS and from blood stages of PyYM (see Additional file 9). To analyse the reproducibility of our RNA-seq data, we calculated Pearson correlations of the FPKM (fragments per kilo base of exon per million fragments mapped) values of RMP genes for which one-to-one ortholog relationships exist in the different RPM genomes (Figure 2A). Expression was highly correlated not only between biological replicates of the same species (r = 0.88 to 1.0), but also between comparable stages of different RMP, such as PbA and PcAS trophozoites (r = 0.77 to 0.86). Both the gametocyte and ookinete samples clustered separately from asexual blood stages, which reflects the different program of gene expression during sexual commitment and zygote development. Heat maps representing the expression of all PbA genes reveal clusters of genes with distinct expression patterns in the different life cycle stages (Figure 2B, left panel), consistent with both the morphological and functional differences between these stages. When PbA genes are ordered according to the expression levels of their P. falciparum orthologs [34], ring-, trophozoite- and schizont-expressed genes display the expected characteristic temporal cascade of gene expression (Figure 2B, right panel). Genome wide expression data of developing ookinetes have not been published before. We found that mature (24 hour) ookinetes have a distinct expression pattern compared to immature, developing ookinetes (16 hour). In Additional file 10 an overview is presented of all genes that are up- or down regulated in the two developmental stages of ookinetes. Genome ontology (GO)-annotation of differentially regulated genes reveals that genes encoding proteins involved in protein phosphorylation, inner membrane and myosin complex formation and ATP binding are most significantly up-regulated in 16 hour ookinetes compared to 24 hour ookinetes (see Additional file 10). In contrast, mature ookinetes show a strong up-regulation of genes encoding proteins involved in protein translation and ribosome formation (see Additional file 10), most likely in preparation for the rapid growth expansion of the oocyst after ookinete traversal of the mosquito midgut wall.

Gene expression (RNAseq) in multiple RMP life cycle stages. A) Spearman correlation of FPKM values of orthologous genes between life cycle stages of PbA, PcAS and PyYM. PbA: ring (RI), trophozoite (Tr), schizont (Sch), gametocyte (Gct) and 16 and 24 hour ookinetes (Ook). PcAS: trophozoites (trophozoites of blood (Pc-bl)-and vector-transmitted (Pc-vec) PcAS; PyYM; blood stages (2 lines PyYM_WT and PyYM_MUT). B) Heat maps of expression (FPKM normalized by gene) of PbA genes in different life cycle stages. Left panel, all PbA genes ordered based on P. berghei expression pattern (FPKM values >21; in total 4,733 genes). Right panel, 2,236 PbA genes with orthologs in P. falciparum and FPKM values >63, ordered according to the temporal expression levels (in asexual blood stages) of their P. falciparum orthologs as shown in [34]. FPKM, fragments per kilo base of exon per million fragments mapped; RMP, rodent malaria parasites.
To further improve the reference genomes we mapped the RNA-seq data onto the RMP genomes and visually inspected the alignments using the Artemis Comparison Tool (ACT), a genome viewing tool [35]. A comparative analysis with the P. falciparum 3D7 genome allowed us to determine gene structure at base-pair (bp) resolution for at least 89% of the genes. Of the 896 newly annotated protein-coding genes that were absent in the previous genome assemblies, 70% have primate malaria orthologs, 83% have expression evidence (RNA-seq FPKM values >21) and we could ascribe functions to 75% (see Additional file 1). The different RNA-seq data sets have also been used to confirm splice sites and to identify putative alternative splice sites (see Additional file 11). This analysis resulted in the identification of 839 alternative splicing events in a total of 567 RMP genes.
Characterization of RMP multigene families
As a result of having dramatically improved the annotation of the subtelomeric regions we were able to accurately define the RMP multigene families that are located there (Table 2, Additional file 4). For proteins of nearly all of these families experimental evidence exists that they are exported into the host RBC in the absence of a PEXEL motif [29]. The pir family is the most abundant multigene family (see next section) encoding exported proteins that lack a canonical PEXEL motif. The second largest gene family is the fam-a gene family, formerly identified as the pyst-a family in P. yoelii 17XNL and named as Pb-fam-1, Pc-fam-1 or fam-a [16],[17]. PbA fam-a proteins are exported into the host RBC and can be transported to the RBC surface membrane [29] but lack a PEXEL-motif. Single copy orthologs have been defined in all primate malarias and the expansion of this family is RMP-specific. Most members have a subtelomeric location (see Additional files 12, 13 and 14), but all three RMP have at least one internally located copy that is positionally conserved with the primate malaria orthologs and, therefore, likely to represent the ancestral copy of this family. In order to standardise the naming of orthologous multigene families in different RMP, we have renamed the two multigene families, pyst-b/pb-fam-3 and pyst-c genes [16],[17],[29] as fam-b and fam-c, respectively (Table 2; Additional file 4). The fam-b family is exclusively subtelomeric and is characterized by the presence of the pyst-b domain. Most members contain a transmembrane domain (58%), a signal peptide (75%) and PEXEL-motif (76%) (see Additional files 12, 13 and 14). PbA fam-b proteins are exported into the host RBC [29]. The fam-c is also exclusively found in the subtelomeric regions and is characterized by the presence of a pyst-c1 and/or pyst-c2 domain [16]. Most members have a transmembrane domain (60%) and a signal peptide (92%) (see Additional files 12, 13 and 14) and only a small percentage (24%) contain a predicted PEXEL-motif.
Other subtelomeric multigene families include the `early transcribed family of proteins' (ETRAMPs) and `putative reticulocyte binding proteins' (Table 2, Additional files 4, 12, 13 and 14). ETRAMPS are small exported proteins with a predicted signal peptide and transmembrane domain but without a PEXEL-motif. These proteins are mainly located in the parasitophorous vacuole membrane [36],[37]. The genes encoding putative reticulocyte binding proteins (RBP), that were first described in P. yoelii as Py235 and are expressed in merozoites [38], are clear orthologs of the reticulocyte binding proteins of P. vivax [39] and the RH proteins of P. falciparum [40]. These large proteins typically have a predicted signal sequence and at the C-terminus a transmembrane domain containing a rhomboid cleavage site and a cytoplasmic domain, although P. falciparum RH5 contains just the signal peptide and N-terminal ligand binding domain [41]. The RMPs have genes encoding two short RBPs reminiscent of P. falciparum RH5 (typified by PYYM_0101400 and PYYM_0701100) and six or more full length proteins (Table 2). Compared with PyYM, Py17X contains an additional full length RBP.
In PcAS several other expanded gene families are present in the subtelomeric regions. These include `putative lysophospholipases', `erythrocyte membrane antigen 1' (EMA1), and `putative haloacid dehalogenase-like hydrolases' (Table 2, Additional files 4, 12, 13 and 14). The genes encoding lysophospholipases are characterized by the `pst-a' domain [42] and all RMP have two copies with an internal chromosomal location that are syntenic with orthologs of primate malarias. For two of the five PbA lysophospholipases evidence exists that they are exported into the RBC [29] and again they lack a PEXEL-motif. In PcAS this family has expanded into 28 copies (Table 2, Additional file 4). In the genome of PyYM and PbA only a single gene encoding EMA1 is present whereas PcAS ema1 has expanded to more than 10 copies in the subtelomeric regions (Table 2, Additional file 4). These PEXEL-negative proteins, first described in P. chabaudi [43] are associated with the RBC membrane. The gene encoding the putative haloacid dehalogenase-like hydrolase has expanded only in PcAS, with eight subtelomeric copies.
A number of other genes are interspersed within the subtelomeric regions of RMP chromosomes. Many of these `other subtelomeric genes' (46 to 67 genes; Table 2, Additional file 4) encode proteins that are RMP-specific and more than 96% of these proteins contain a predicted signal peptide, transmembrane domain or PEXEL-motif and for several proteins experimental evidence exists for their export into the host RBC cytoplasm. Combined, these observations indicate that most, if not all, RMP subtelomeric genes (apart from the RBP family) encode exported proteins and most lack a PEXEL-motif. The presence of large numbers of PEXEL-negative exported proteins in RMP indicates alternative export mechanisms possibly common to all Plasmodium species and investigations with highly tractable RMP species can, therefore, be used to understand these mechanisms better.
The RMP pir multigene family: phylogeny and expression
We analysed the expression patterns of all members of the three largest multigene families, fam-a, fam-b and pir in the PbA life cycle stages using heat maps of the RNA-seq data. This revealed distinct transcription patterns both between the gene families and also between members within a family (Figure 3). All three families show strongly reduced transcription in ookinetes. Whereas most RMP-fam-a and RMP-fam-b members had reduced transcript levels in gametocytes compared to asexual blood stages, a large cluster of pir genes were up-regulated (at least a fold change of 2) in gametocytes. Expression patterns are not only different between asexual and sexual stages but also between different asexual stages, for example distinct pir gene clusters are up-regulated in schizonts. Distinct transcription patterns in different life cycle stages of gene clusters may indicate functional differences between members of a single gene family. With the new genome assemblies we were able to determine the total number of pirs and their structure and spatial organization more precisely (Table 2; Additional file 4). In PcAS and PbA the total number of pirs (excluding pseudogenes) is 194 and 100, respectively, whereas in PyYM this gene family is greatly expanded to 583 copies. In Additional files 12, 13 and 14 the chromosomal distribution of all pirs is shown. Most pir genes share a similar structure across the different species with a short first exon, long second exon and a third exon encoding a trans-membrane domain. They lack a PEXEL-motif and all pirs are chromosomally arranged such that they are transcribed in a centromere to telomere direction except for several members of PcAS (these are `long-form' pirs of clade L1; see below). A number of pirs have long low complexity regions in the predicted extracellular domain and a few Pc-pirs have a four exon structure (see Additional file 3C). Remarkably, as stated earlier, the Pb-pir genes include a large number (88 genes; 44%) of pseudogenes and nearly half (35 genes; 18%) of these Pb-pirs are contained within the 2.3 kb subtelomeric repeat described above (Figure 1B). To analyse whether the differential expression of groups of pir members was associated with definable sequence differences (and possibly functional differences) between pirs we undertook a detailed phylogenetic analysis of all RMP pirs (including predicted pseudogenes). Estimations of Maximum Likelihood (ML) phylogeny based on nucleotide sequences or amino acid sequences and an estimation of a Bayesian phylogeny resulted in a phylogenetic tree with a robust separation of `long-form' and `short form' pirs (Figure 4; Additional file 15). We identified 12 clades in the phylogeny that have robust support; four long-form clades (L1 to 4) with a mean pir length ranging from 1,062 to 2,369 aa and eight short-form clades (S1 to 8) with a mean length ranging from 786 to 952 aa. Most long-form pirs have an extended repetitive region located within the second exon, downstream of the core pir domain and upstream of the transmembrane region. All RMP species have both short- and long-form pirs, indicating that the presence or absence of an extended repetitive region has evolved once and defines a principle division in pir diversity. Many clades are dominated by pirs from one species, particularly PyYM (for example, clades S1, S4, S8; Figure 4). Yet, even such clades contain rare sequence types from PbA (for example, S1d, S2, S8) or PcAS (S1g) indicating that these lineages originate from the RMP ancestor and probably expanded after speciation. Both PcAS and PbA appear to have experienced their own specific expansions after speciation (for example, S4, S5, S7). The maintenance of orthology within clades, in the presence of frequent gene conversion (see Discussion section), may indicate that selection pressure maintains structural differences between pirs, for example diversifying selection under immune pressure or purifying selection on functional diversity. The observation that the ratio of the different pir clades is highly similar in the five, highly diverse, isolates of the two P. chabaudi subspecies (Figure 4) supports the presence of selective pressures that maintain the clade structure of pir genes. We next analysed whether the pir expression patterns in PbA blood stages were correlated with structural differences between pirs, by comparing the RNA-seq expression patterns with the phylogenetic clades. We observed that pirs that are predominantly expressed in gametocytes mainly belong to only two clades of the small-form pirs, S1 and S4, whereas genes up-regulated in schizonts are mainly long-forms of clades L1, L2 and L3 (see Additional file 16). The stage-specific up- or down-regulation of expression of clusters of structurally different pirs support the hypothesis of the existence of functional diversification within the pir family and is in agreement with other observations indicating that differences in pir sequences are associated with different functional properties [44],[45].

Expression of members of three large RMP multigene families in different life cycle stages. Temporal expression patterns of members of the three largest PbA multigene familes (pir, fam-a and fam-b) in different life cycle stages as visualized by heat maps of RNA-seq data. The expression (FKPM) values of genes over the life-cycle stages (yellow-red) are normalised per gene. The min/max column values are the log minimal and log maximal FPKM values for each gene. Only genes with an FPKM above 21, in all conditions, were included. FKPM, fragments per kilo base of exon per million fragments mapped; RMP, rodent malaria parasites.

Features of the RMP pir multigene family A) Phylogenetic tree of RMP pir s , showing the different clades (S1 to 8, L1 to 4) and separation of the `long' (L) and `short' form (S) pir s. B) Features of the different RMP pir clades. For each clade we show the total number of RMP pirs followed by their distribution (pie charts) in the three species and the distribution of gene lengths (box plots). In addition, pie charts show the relative abundance of clades in the different isolates/lines of P. chabaudi and P. yoelii. The expression bar plots (red bars) visualise the expression of the pirs of different clades in the different life cycle stages (except for the ookinete stage since expression/FPKM values are below the cut of level of 21). A pir is assigned to a life cycle stage based on the highest FPKM value. The height of the expression bar represents the percentage of all pirs in that clade. FKPM, fragments per kilo base of exon per million fragments mapped; RMP, rodent malaria parasites.
Discussion
By extensive re-sequencing and annotation we have generated three high quality RMP reference genomes with nearly all core genes as complete gene models and a much improved and almost complete representation of chromosomal subtelomeric regions. These reference genomes will greatly enhance the use of RMP as model organisms in malaria research. We provide full-length gene models for more than 98% of predicted protein-coding genes. The approximately 60% of genes with functional annotation is comparable to the percentage of functionally annotated genes in the P. falciparum 3D7 reference genome and a high percentage (approximately 90%) of the predicted RMP proteins have orthologs in primate malaria species. It is this high level of orthology between RMP and primate malaria genomes that strongly supports RMPs as models in experimental approaches to characterize the Plasmodium gene function. Similarly, the genome-wide RNA-seq data from different RMP developmental stages is a valuable resource to further analyse Plasmodium gene function and the regulatory networks underlying the multiple differentiation pathways of Plasmodium. The RNA-seq studies presented here provide information on gene expression at an unprecedented depth and breadth of coverage of multiple blood stages and ookinetes. Previously, only a few large-scale transcriptome (microarray) analyses of P. berghei blood stages and ookinetes had been performed [17],[46]. These studies were based on a highly fragmented draft P. berghei genome and, therefore, expression data were only generated for about half of all P. berghei genes. In addition, important/valuable large scale transcriptome studies have been performed on RMP life-cycle stages, such as sporozoites and liver stages [47]-[49]. These life-cycle stages would also benefit from re-examination using the latest RMP genome assemblies we provide in this study.
Our studies reveal that large scale changes in gene expression occur in ookinetes between 16 and 24 hours after fertilization, possibly required for the differentiation of (retort-form) zygotes into the mature ookinetes. The strong up-regulation in mature ookinetes of transcripts involved in ribosome biogenesis and protein translation suggest that the mature ookinete generates transcripts for proteins required after the ookinete has traversed the mosquito midgut wall and starts its rapid transition into the oocysts, possibly using mechanisms of translational repression similar to those in gametocytes [50],[51] and sporozoites [52],[53]. What these three stages have in common is that they are fully differentiated cells that will undergo rapid cellular differentiation and/or growth expansion upon entering a new environment. Whether mature ookinetes store repressed transcripts requires further investigation.
The additional sequence data from multiple RMP isolates will help to further unravel gene function and establish relationships between phenotypic traits and genotypic diversity. The near absence of polymorphisms within the genomes of P. berghei isolates was unexpected. Low sequence diversity of a limited number of genes of P. berghei isolates had been reported previously and it was proposed that this may result from cross-contamination of P. berghei isolates in the laboratory after isolation [54],[55]. However, this seems unlikely as one line would have needed to be mislabelled with the names of all other isolates, then all these mislabelled lines would have had to be sent to all the different laboratories worldwide replacing the `correct' isolates that may have existed in their recipient laboratories. However, sequencing of additional stocks from these isolates, which were frozen in different laboratories soon after isolation from the natural host, may reveal whether low sequence diversity is due to cross-contamination. The P. berghei isolates we have sequenced were obtained from other laboratories (SP11, NK65) and they also show a similar lack of sequence polymorphism. In contrast, the isolates of P. chabaudi exhibit considerable genotypic diversity. These P. chabaudi isolates exhibit differences in virulence, RBC invasion, growth rates and immunogenic profiles [7],[56]-[60] and further studies, for example using linkage or quantitative trait loci (QTL) analyses [33],[61]-[63], will facilitate identification of genes associated with defined phenotypes. For RMP species there is evidence that differences in virulence are associated with differences in RBC invasion [2],[7],[56],[60],[64]. For example, P. yoelii virulence has been associated with mutations in proteins involved in RBC invasion [7],[65],[66]. Interestingly, we found extensive sequence polymorphism in P. chabaudi genes encoding such proteins. While much attention is given to the role of exported proteins of multigene families and virulence in both human and RMP, further analysis of RMP proteins that regulate invasion phenotypes may reveal novel mechanisms that underlie virulence.
The new sequence data allowed for a much improved annotation of chromosomal subtelomeric regions and to better define the different subtelomeric multigene families. In addition to the large pir gene family, all three RMP contain an expanded gene family encoding exported proteins, fam-a, with orthology to a single-copy gene in primate malarias, which contains a START-domain (steroidogenic acute regulatory-related lipid transfer domain; [67]). START-containing proteins of eukaryotes are involved in the transfer of phospholipids, ceramide or fatty acids between membranes [68]. A START domain has also recently been identified in an exported, PEXEL-containing, P. falciparum protein that was shown to transfer phospholipids [69]. The single-copy RMP orthologs of this gene (PF3D7_0104200) also contain a PEXEL-motif, indicating that phospholipid-transporting proteins are exported into the RBC in both primate malarias and RMP. P. chabaudi contains an additional, highly expanded, gene family that contains domains involved in phospholipid/fatty acid metabolism. These genes, encoding putative lysophospholipases, lack a PEXEL motif; however, for several P. berghei orthologs as well as lysophospholipases of P. falciparum there is evidence for their export into the host RBC [29],[70]. Combined, these observations indicate the importance of phospholipid/fatty acid metabolism/transport mediated by Plasmodium proteins exported into the RBC cytosol. Why such genes have been differentially expanded into multigene families in different species remains to be investigated.
The pir family is the largest RMP multigene family and is shared with human and non-human primate species P. vivax, P. knowlesi and P. cynomolgi [20]-[23]. PIR proteins are exported into the RBC in the absence of a PEXEL-motif, and there is evidence that they are located on, or close to, the RBC surface or dispersed in the RBC cytoplasm [24]-[26],[29],[71],[72]. The function of pirs is unknown and no functional domains have been identified so far. Recently, it has been shown that in P. chabaudi a change in virulence was associated with differential expression of members of the pir multi-gene family [27]. It has been suggested that PIRs are transported to the surface of infected RBC and play a role in RBC sequestration comparable to the role of the Pfemp1 gene family of virulence factors in P. falciparum. However, for several P. berghei PIRs a direct role in RBC sequestration is unlikely since no evidence was found for their location on the RBC surface although they were exported into the RBC cytoplasm of both sequestering asexual blood stages and non-sequestering gametocytes [29]. For P. vivax PIRs it has been shown that different members have distinct subcellular locations in the infected RBC [26]. These observations indicate that functional differences may exist between members of the PIR family. Phylogenetic analyses support the possibility of functional differences between the PIRs. A recent phylogenetic analysis of the newly annotated PcAS pirs identified two distinct pir sub-families (A and B), which contain distinct amino acid sequence motifs [44]. Our phylogenetic analyses included pirs from all three RMP species and resulted in the identification of a number of different clades. The presence of clearly distinguishable clades indicates that structural differentiation exists among pirs and that this evolved prior to the separation of the RMP species. Our observations of the stage-specific up- or down-regulation of expression of clusters of structurally different pirs in different blood stages supports the hypothesis that there is functional diversification within the pir family and that purifying selection plays a role in shaping this family. By including multiple species in the pir phylogeny it is clear that this gene family is subject to rapid turnover, that is, gene gain and loss, indicating the absence of strong selective forces that would result in distinct orthologous groups/clades that are shared and maintained in different species for functional reasons. Gain of pir genes in different species is evident in the multiple species-specific expansions of clades. Assuming that the common ancestor had a pir family equal in abundance and diversity, the relatively limited instances of orthology (12 clades) indicates significant losses of ancestral sequence types. A plausible explanation for both the abundance of species-specific sequences and the paucity of ancestral sequences is a continual process of gene turnover driven by gene conversion, a mechanism that has been proposed for pirs of P. chabaudi [44] and which was evident in each of the clades revealed in this study (data not shown). The effect of frequent gene conversion is the replacement of ancestral sequence types with species-specific sequences, which results in distinct species-specific clades without orthology. Loss of orthology is only resisted when selective forces maintain structurally distinct pirs, which we propose, explains the presence of the (limited) orthology between pir clades of the different RMP species. The improved annotation and phylogeny demonstrating clusters of structurally different pirs in all RMP combined with expression profiles are powerful data that can help to further delineate function, the relationship of expression with virulence and how the (species-specific) expansion of the pirs is related to distinct selective pressures.
Conclusions
To maximise the utility of RMP we have greatly improved the genome assemblies of P. berghei and P. chabaudi, comprehensively sequenced the P. yoelii YM genome, sequenced multiple RMP isolates and generated in-depth expression data from multiple RMP life-cycle stages. Comparison of the RMP and P. falciparum genomes and RNA-seq mapping permitted gene annotation at base-pair resolution and has defined the level of orthology between RMP and human parasite genomes. The very high level orthology between RMP and human malarias (both in genome structure and gene content) supports the use of highly tractable RMPs as experimental models to characterize the function of the very many Plasmodium genes that remain uncharacterised.
Only a few large-scale transcriptome (microarray) analyses of different P. berghei life-cycle stages had previously been performed. Moreover, these studies were based on highly fragmented draft RMP genomes and consequently, for example, for one of the most well studied RMP, P. berghei, gene expression data was only mapped to about half of all P. berghei genes that have now been characterised. The RNA-seq studies we present provide information on gene expression, at an unprecedented depth and breadth of coverage, of multiple life cycle stages and provide the foundational data needed for the performance of large-scale analyses of gene regulatory networks that underlie cellular differentiation.
We show that extensive genotypic diversity exists between P. chabaudi isolates making this species an excellent organism to study genotype-phenotype relationships. Differences in virulence red blood cell (RBC) invasion, growth rates and immunogenic profiles exist between parasites of these isolates. Therefore, studies, such as linkage or quantitative trait loci analysis, are now possible to help identify genes associated with these defined phenotypes. For RMP species there is evidence that differences in virulence are associated with differences in RBC invasion, and, indeed, we find extensive sequence polymorphism in P. chabaudi genes encoding proteins involved in RBC invasion. Much attention is given to the role of exported proteins of multigene families and virulence in both human and RMP (for example, var, pirs), and analysis of differences between RMP proteins, that regulate invasion phenotypes, may reveal novel mechanisms that underlie virulence.
Full-length chromosomal annotation has permitted a comprehensive classification of all RMP subtelomeric multigene families. Our analyses indicate that most, if not all, RMP subtelomeric genes (apart from the RBP family) encode proteins exported out of the parasite; however, most lack a canonical PEXEL-motif. The presence of large numbers of PEXEL-negative exported proteins indicates alternative export mechanisms possibly common to all Plasmodium species. Investigations with highly tractable RMP species can therefore be used to understand these mechanisms better.
Our analyses of the phylogeny and expression of the largest RMP multi-gene family, the pirs, indicates functional diversification between members of the pir multigene family (this gene family is conserved between human/primate and RMP malaria species). Our new pir annotation and phylogeny demonstrates that clusters of structurally different pirs are differentially expressed. This is powerful data that can help to better understand their function, the relationship of pir expression with virulence and how the (species specific) pirs expansion is related to different selective pressures.
Methods
Animal experiments and parasites
All animal experiments performed in the Leiden malaria Research Group were approved by the Animal Experiments Committee of the Leiden University Medical Center (DEC 07171, DEC 10099). The Ethics Statement for P. yoelii YM and P. yoelii 17X: all animal work protocols were reviewed and approved by the Ethical Review Panel of the MRC National Institute for Medical Research and approved and licensed by the UK Home Office as governed by law under the Animals (Scientific Procedures) Act 1986 (Project license 80/1832, Malaria parasite- host interactions). Animals were handled in strict accordance with the `Code of Practice Part 1 for the housing and care of animals (21/03/05)' available at [73]. The numbers of animals used was the minimum consistent with obtaining scientifically valid data. The experimental procedures were designed to minimize the extent and duration of any harm and included predefined clinical and parasitological endpoints to avoid unnecessary suffering. The study of P. chabaudi DNA and RNA was carried out in strict accordance with the UK Animals (Scientific Procedures) Act 1986 and was approved by the Ethical Committee of the MRC National Institute for Medical Research, and the British Home Office (PPL: 80/2538).
For sequencing of the RMP reference genomes the following were used: for PbA the cloned reference line cl15cy1 of the ANKA isolate of P. berghei [11]; for PcAS the 2722 clone of the AS isolate of P. chabaudi chabaudi (cloned after mosquito-transmission in 1978 and obtained from D. Walliker, University of Edinburgh, Edinburgh, UK); for PyYM the cloned 17XYM line of the YM line of P. yoelii yoelii [74]. In Additional files 6 and 7 details are provided of the other RMP isolates/lines that have been sequenced.
Sequencing, assembly and annotation
Sequencing was performed using Sanger capillary, Illumina and 454 sequencing. Sequence assemblies were performed using different assemblers [75],[76], which were improved automatically using a number of configuration tools [77]-[82] and manual inspection. First pass annotation was performed through a combination of ab initio gene finding via Augustus [83] and transfer of annotation through orthology using RATT [80]. Gene models of the three reference genomes were corrected manually using RNA-Seq and orthologous information. Details of the assemblies and annotation are provided in Additional file 6. To define the orthologous and paralogous relationships between the predicted RMP proteins and those of human/primate malaria species OrthoMCL [84] was used. The presence of a PEXEL-motif was determined using the updated HMM algorithm ExportPred v2.0 with a cutoff value of 1.5 [28]. Classification of the RMP multigene families was done through manual inspection of conserved domains (Interpro) and gene structure. SNPs in the genomes of these parasites were called by mapping the reads against their respective reference genomes, ignoring low complexity and repetitive regions. From the SNPs the Ka/Ks ratio was calculated for the P. chabaudi isolates with the Bio::Align::DNAStatistics Perl module.
Transcriptomics
RNA was collected from multiple synchronized blood stages [85] and purified gametocytes and ookinetes [86] of PbA, from PcAS blood stages (late trophozoites), isolated from different mice as described [44] and from PyYM late blood stages of two parasite lines (the cloned YM line and mutant PY01365-KO) [8]. RNA was sequenced as described [8],[44],[87],[88]. To correct gene models and to compare the expression between samples, each sample was first mapped against its reference genome using TopHat [89] (version v2.0.6, parameter -g ). For the resulting output a custom Perl script was written to detect errors in the annotation and to find new or alternative splice sites. To determine transcript abundance FPKM values were calculated for all genes (FPKM: fragments per kilo base of exon per million fragments mapped) using Cufflinks [90]. Accepting 10% of the intron as real signal, a cut-off FPKM value of 21 over all RNA-seq samples was determined. See also Additional file 6 for a detailed description of the generation and analysis of the RNA-seq data.
Heatmaps were generated with FPKM values of each gene and condition, using the heatmap.2 function of the gplots package. Correlation plots were done in R (Foundation for Statistical Computing; [91]) and generated with the corrplot function of the corrplot R library. Only genes were included that had one to one orthology in the three rodent species. For differential expression we used cuffdiff [90] (v2.0.2, with parameters -u -q) to compensate for GC variation and repetitive regions. GO enrichment of differentially expressed genes was performed in R, using TopGO. As a GO-database the predicted GO-terms from the reference RMP genomes were used.
Phylogenetic analyses of pirs
All full-length RMP pir coding sequences, including predicted pseudogenes, were used. Translated nucleotide sequences for 1,160 genes were aligned in ClustalW [92]; all multiple alignments were manually edited to resolve all frame-shifts. Non-homologous positions at the N-terminus were removed by curtailing the alignment to the N-terminal-most conserved cysteine position. Non-homologous repetitive motifs were removed from `long-form' PIRs (that is, 188 proteins >1,200 amino acids in length). The resultant 1,266-character alignment constitutes the conserved core of all PIRs and almost the complete repertoire of `short-forms' (that is, <1,200 amino acids in length and 972/1,160 genes). A Maximum Likelihood phylogeny was estimated from the nucleotide sequence alignment using RAxML v7.0.4 [93] using a GTR + G model. Node support was assessed using 100 non-parametric bootstrap replicates [94]. A Bayesian phylogeny was estimated using MrBayes v3.2.1 [95] with a GTR + G model for a subsample of pir nucleotide sequences (MCMC settings: Nruns = 4, Ngen = 1,000,000, sample burnin = 1,000, and default prior distribution). See also Additional file 6 for a detailed description of the phylogenetic analyses.
Accession numbers
All the raw data used in the assemblies of the genome and the RNA-seq data have been deposited with accession numbers shown in Additional file 17. The reference genomes have the following accession numbers: P. chabaudi chromosomes: LK022878-LK022893 and scaffolds: LK022855-LK022877, P. berghei chromosomes: LK023116-LK023131 and scaffolds: LK022894-LK022977; P. yoelii YM chromosomes: LK934629-LK934644 and scaffolds: LK023132-LK023312 and P. yoelii 17X chromosomes: LM993655-LM993670 and scaffolds: LK022978-LK023115.
Authors' contributions
TDO, UB, MH, LR, SAO, CIN, AP, MB contributed to the genome assembly, annotation and analysis. TDO, BFF, WAMH, AAR, OB, SMK, HGS, APW, CJJ generated and analysed RNA-seq data of P. berghei. DC, JL, AAH provided materials for genome sequences and/or RNA-seq data of different isolates/lines of P. yoelii and P. chabaudi. AE provided materials for genomes of isolates of P. berghei. APJ performed the phylogenetic analysis. TDO, UB, APJ, BFF, WAMH, SMK, AAH, CIN, AP, MB, CJJ conceived the study, participated in its design and coordination and drafted the manuscript. All authors read and approved the final manuscript.
Additional files
References
Carter R, Diggs CL: Parasitic Protozoa, Volume 3. 1977, Academic, New York
Craig AG, Grau GE, Janse C, Kazura JW, Milner D, Barnwell JW, Turner G, Langhorne J: The role of animal models for research on severe malaria. PLoS Pathog. 2012, 8: e1002401-10.1371/journal.ppat.1002401.
Lamb TJ, Brown DE, Potocnik AJ, Langhorne J: Insights into the immunopathogenesis of malaria using mouse models. Expert Rev Mol Med. 2006, 8: 1-22. 10.1017/S1462399406010581.
Langhorne J, Ndungu FM, Sponaas AM, Marsh K: Immunity to malaria: more questions than answers. Nat Immunol. 2008, 9: 725-732. 10.1038/ni.f.205.
Prudencio M, Mota MM, Mendes AM: A toolbox to study liver stage malaria. Trends Parasitol. 2011, 27: 565-574. 10.1016/j.pt.2011.09.004.
Kappe SH, Vaughan AM, Boddey JA, Cowman AF: That was then but this is now: malaria research in the time of an eradication agenda. Science. 2010, 328: 862-866. 10.1126/science.1184785.
Otsuki H, Kaneko O, Thongkukiatkul A, Tachibana M, Iriko H, Takeo S, Tsuboi T, Torii M: Single amino acid substitution in Plasmodium yoelii erythrocyte ligand determines its localization and controls parasite virulence. Proc Natl Acad Sci U S A. 2009, 106: 7167-7172. 10.1073/pnas.0811313106.
Ogun SA, Tewari R, Otto TD, Howell SA, Knuepfer E, Cunningham DA, Xu Z, Pain A, Holder AA: Targeted disruption of py235ebp-1: invasion of erythrocytes by Plasmodium yoelii using an alternative Py235 erythrocyte binding protein. PLoS Pathog. 2011, 7: e1001288-10.1371/journal.ppat.1001288.
Carvalho TG, Menard R: Manipulating the Plasmodium genome. Curr Issues Mol Biol. 2005, 7: 39-55.
Janse CJ, Kroeze H, van Wigcheren A, Mededovic S, Fonager J, Franke-Fayard B, Waters AP, Khan SM: A genotype and phenotype database of genetically modified malaria-parasites. Trends Parasitol. 2011, 27: 31-39. 10.1016/j.pt.2010.06.016.
Janse CJ, Ramesar J, Waters AP: High-efficiency transfection and drug selection of genetically transformed blood stages of the rodent malaria parasite Plasmodium berghei. Nat Protoc. 2006, 1: 346-356. 10.1038/nprot.2006.53.
Silvie O, Mota MM, Matuschewski K, Prudencio M: Interactions of the malaria parasite and its mammalian host. Curr Opin Microbiol. 2008, 11: 352-359. 10.1016/j.mib.2008.06.005.
Sinden RE: Molecular interactions between Plasmodium and its insect vectors. Cell Microbiol. 2002, 4: 713-724. 10.1046/j.1462-5822.2002.00229.x.
Baker DA: Malaria gametocytogenesis. Mol Biochem Parasitol. 2010, 172: 57-65. 10.1016/j.molbiopara.2010.03.019.
RMgmDB - Rodent Malaria genetically modified Parasites. [], [http://www.pberghei.eu/]
Carlton JM, Angiuoli SV, Suh BB, Kooij TW, Pertea M, Silva JC, Ermolaeva MD, Allen JE, Selengut JD, Koo HL, Peterson JD, Pop M, Kosack DS, Shumway MF, Bidwell SL, Shallom SJ, van Aken SE, Riedmuller SB, Feldblyum TV, Cho JK, Quackenbush J, Sedegah M, Shoaibi A, Cummings LM, Florens L, Yates JR, Raine JD, Sinden RE, Harris MA, Cunningham DA, et al: Genome sequence and comparative analysis of the model rodent malaria parasite Plasmodium yoelii yoelii. Nature. 2002, 419: 512-519. 10.1038/nature01099.
Hall N, Karras M, Raine JD, Carlton JM, Kooij TW, Berriman M, Florens L, Janssen CS, Pain A, Christophides GK, James K, Rutherford K, Harris B, Harris D, Churcher C, Quail MA, Ormond D, Doggett J, Trueman HE, Mendoza J, Bidwell SL, Rajandream MA, Carucci DJ, Yates JR, Kafatos FC, Janse CJ, Barrell B, Turner CM, Waters AP, Sinden RE: A comprehensive survey of the Plasmodium life cycle by genomic, transcriptomic, and proteomic analyses. Science. 2005, 307: 82-86. 10.1126/science.1103717.
Kooij TW, Janse CJ, Waters AP: Plasmodium post-genomics: better the bug you know?. Nat Rev Microbiol. 2006, 4: 344-357. 10.1038/nrmicro1392.
Kooij TW, Carlton JM, Bidwell SL, Hall N, Ramesar J, Janse CJ, Waters AP: A Plasmodium whole-genome synteny map: indels and synteny breakpoints as foci for species-specific genes. PLoS Pathog. 2005, 1: e44-10.1371/journal.ppat.0010044.
Carlton JM, Escalante AA, Neafsey D, Volkman SK: Comparative evolutionary genomics of human malaria parasites. Trends Parasitol. 2008, 24: 545-550. 10.1016/j.pt.2008.09.003.
Neafsey DE, Galinsky K, Jiang RH, Young L, Sykes SM, Saif S, Gujja S, Goldberg JM, Young S, Zeng Q, Chapman SB, Dash AP, Anvikar AR, Sutton PL, Birren BW, Escalante AA, Barnwell JW, Carlton JM: The malaria parasite Plasmodium vivax exhibits greater genetic diversity than Plasmodium falciparum. Nat Genet. 2012, 44: 1046-1050. 10.1038/ng.2373.
Tachibana S, Sullivan SA, Kawai S, Nakamura S, Kim HR, Goto N, Arisue N, Palacpac NM, Honma H, Yagi M, Tougan T, Katakai Y, Kaneko O, Mita T, Kita K, Yasutomi Y, Sutton PL, Shakhbatyan R, Horii T, Yasunaga T, Barnwell JW, Escalante AA, Carlton JM, Tanabe K: Plasmodium cynomolgi genome sequences provide insight into Plasmodium vivax and the monkey malaria clade. Nat Genet. 2012, 44: 1051-1055. 10.1038/ng.2375.
Pain A, Bohme U, Berry AE, Mungall K, Finn RD, Jackson AP, Mourier T, Mistry J, Pasini EM, Aslett MA, Balasubrammaniam S, Borgwardt K, Brooks K, Carret C, Carver TJ, Cherevach I, Chillingworth T, Clark TG, Galinski MR, Hall N, Harper D, Harris D, Hauser H, Ivens A, Janssen CS, Keane T, Larke N, Lapp S, Marti M, Moule S, et al: The genome of the simian and human malaria parasite Plasmodium knowlesi. Nature. 2008, 455: 799-803. 10.1038/nature07306.
Cunningham D, Lawton J, Jarra W, Preiser P, Langhorne J: The pir multigene family of Plasmodium: antigenic variation and beyond. Mol Biochem Parasitol. 2010, 170: 65-73. 10.1016/j.molbiopara.2009.12.010.
Jemmely NY, Niang M, Preiser PR: Small variant surface antigens and Plasmodium evasion of immunity. Future Microbiol. 2010, 5: 663-682. 10.2217/fmb.10.21.
Bernabeu M, Lopez FJ, Ferrer M, Martin-Jaular L, Razaname A, Corradin G, Maier AG, del Portillo HA, Fernandez-Becerra C: Functional analysis of Plasmodium vivax VIR proteins reveals different subcellular localizations and cytoadherence to the ICAM-1 endothelial receptor. Cell Microbiol. 2012, 14: 386-400. 10.1111/j.1462-5822.2011.01726.x.
Spence PJ, Jarra W, Levy P, Reid AJ, Chappell L, Brugat T, Sanders M, Berriman M, Langhorne J: Vector transmission regulates immune control of Plasmodium virulence. Nature. 2013, 498: 228-231. 10.1038/nature12231.
Boddey JA, Carvalho TG, Hodder AN, Sargeant TJ, Sleebs BE, Marapana D, Lopaticki S, Nebl T, Cowman AF: Role of Plasmepsin V in export of diverse protein families from the Plasmodium falciparum Exportome. Traffic. 2013, 14: 532-550. 10.1111/tra.12053.
Pasini EM, Braks JA, Fonager J, Klop O, Aime E, Spaccapelo R, Otto TD, Berriman M, Hiss JA, Thomas AW, Mann M, Janse CJ, Kocken CH, Franke-Fayard B: Proteomic and genetic analyses demonstrate that Plasmodium berghei blood stages export a large and diverse repertoire of proteins. Mol Cell Proteomics. 2013, 12: 426-448. 10.1074/mcp.M112.021238.
Dore E, Pace T, Ponzi M, Picci L, Frontali C: Organization of subtelomeric repeats in Plasmodium berghei. Mol Cell Biol. 1990, 10: 2423-2427.
van Dijk MR, McConkey GA, Vinkenoog R, Waters AP, Janse CJ: Mechanisms of pyrimethamine resistance in two different strains of Plasmodium berghei. Mol Biochem Parasitol. 1994, 68: 167-171. 10.1016/0166-6851(94)00163-4.
Cheesman S, O’Mahony E, Pattaradilokrat S, Degnan K, Knott S, Carter R: A single parasite gene determines strain-specific protective immunity against malaria: the role of the merozoite surface protein I. Int J Parasitol. 2010, 40: 951-961. 10.1016/j.ijpara.2010.02.003.
Martinelli A, Cheesman S, Hunt P, Culleton R, Raza A, Mackinnon M, Carter R: A genetic approach to the de novo identification of targets of strain-specific immunity in malaria parasites. Proc Natl Acad Sci U S A. 2005, 102: 814-819. 10.1073/pnas.0405097102.
Bozdech Z, Llinas M, Pulliam BL, Wong ED, Zhu J, Derisi JL: The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum. PLoS Biol. 2003, 1: E5-10.1371/journal.pbio.0000005.
Carver T, Berriman M, Tivey A, Patel C, Bohme U, Barrell BG, Parkhill J, Rajandream MA: Artemis and ACT: viewing, annotating and comparing sequences stored in a relational database. Bioinformatics. 2008, 24: 2672-2676. 10.1093/bioinformatics/btn529.
Spielmann T, Gilberger TW: Protein export in malaria parasites: do multiple export motifs add up to multiple export pathways?. Trends Parasitol. 2010, 26: 6-10. 10.1016/j.pt.2009.10.001.
MacKellar DC, Vaughan AM, Aly AS, DeLeon S, Kappe SH: A systematic analysis of the early transcribed membrane protein family throughout the life cycle of Plasmodium yoelii. Cell Microbiol. 2011, 13: 1755-1767. 10.1111/j.1462-5822.2011.01656.x.
Keen J, Holder A, Playfair J, Lockyer M, Lewis A: Identification of the gene for a Plasmodium yoelii rhoptry protein: multiple copies in the parasite genome. Mol Biochem Parasitol. 1990, 42: 241-246. 10.1016/0166-6851(90)90167-K.
Galinski MR, Medina CC, Ingravallo P, Barnwell JW: A reticulocyte-binding protein complex of Plasmodium vivax merozoites. Cell. 1992, 69: 1213-1226. 10.1016/0092-8674(92)90642-P.
Khan SM, Jarra W, Preiser PR: The 235 kDa rhoptry protein of Plasmodium (yoelii) yoelii: function at the junction. Mol Biochem Parasitol. 2001, 117: 1-10. 10.1016/S0166-6851(01)00333-4.
Hayton K, Gaur D, Liu A, Takahashi J, Henschen B, Singh S, Lambert L, Furuya T, Bouttenot R, Doll M, Nawaz F, Mu J, Jiang L, Miller LH, Wellems TE: Erythrocyte binding protein PfRH5 polymorphisms determine species-specific pathways of Plasmodium falciparum invasion. Cell Host Microbe. 2008, 4: 40-51. 10.1016/j.chom.2008.06.001.
Fischer K, Chavchich M, Huestis R, Wilson DW, Kemp DJ, Saul A: Ten families of variant genes encoded in subtelomeric regions of multiple chromosomes of Plasmodium chabaudi, a malaria species that undergoes antigenic variation in the laboratory mouse. Mol Microbiol. 2003, 48: 1209-1223. 10.1046/j.1365-2958.2003.03491.x.
Favaloro JM, Kemp DJ: Sequence diversity of the erythrocyte membrane antigen 1 in various strains of Plasmodium chabaudi. Mol Biochem Parasitol. 1994, 66: 39-47. 10.1016/0166-6851(94)90034-5.
Lawton J, Brugat T, Yam XY, Reid AJ, Boehme U, Otto TD, Pain A, Jackson A, Berriman M, Cunningham D, Preiser P, Langhorne J: Characterization and gene expression analysis of the cir multi-gene family of Plasmodium chabaudi chabaudi (AS). BMC Genomics. 2012, 13: 125-10.1186/1471-2164-13-125.
Ebbinghaus P, Krucken J: Characterization and tissue-specific expression patterns of the Plasmodium chabaudi cir multigene family. Malar J. 2011, 10: 272-10.1186/1475-2875-10-272.
Vontas J, Siden-Kiamos I, Papagiannakis G, Karras M, Waters AP, Louis C: Gene expression in Plasmodium berghei ookinetes and early oocysts in a co-culture system with mosquito cells. Mol Biochem Parasitol. 2005, 139: 1-13. 10.1016/j.molbiopara.2004.03.003.
Kappe SH, Gardner MJ, Brown SM, Ross J, Matuschewski K, Ribeiro JM, Adams JH, Quackenbush J, Cho J, Carucci DJ, Hoffman SL, Nussenzweig V: Exploring the transcriptome of the malaria sporozoite stage. Proc Natl Acad Sci U S A. 2001, 98: 9895-9900. 10.1073/pnas.171185198.
Tarun AS, Peng X, Dumpit RF, Ogata Y, Silva-Rivera H, Camargo N, Daly TM, Bergman LW, Kappe SH: A combined transcriptome and proteome survey of malaria parasite liver stages. Proc Natl Acad Sci U S A. 2008, 105: 305-310. 10.1073/pnas.0710780104.
Zhou Y, Ramachandran V, Kumar KA, Westenberger S, Refour P, Zhou B, Li F, Young JA, Chen K, Plouffe D, Henson K, Nussenzweig V, Carlton J, Vinetz JM, Duraisingh MT, Winzeler EA: Evidence-based annotation of the malaria parasite's genome using comparative expression profiling. PLoS One. 2008, 3: e1570-10.1371/journal.pone.0001570.
Mair GR, Braks JA, Garver LS, Wiegant JC, Hall N, Dirks RW, Khan SM, Dimopoulos G, Janse CJ, Waters AP: Regulation of sexual development of Plasmodium by translational repression. Science. 2006, 313: 667-669. 10.1126/science.1125129.
Mair GR, Lasonder E, Garver LS, Franke-Fayard BM, Carret CK, Wiegant JC, Dirks RW, Dimopoulos G, Janse CJ, Waters AP: Universal features of post-transcriptional gene regulation are critical for Plasmodium zygote development. PLoS Pathog. 2010, 6: e1000767-10.1371/journal.ppat.1000767.
Silvie O, Briquet S, Muller K, Manzoni G, Matuschewski K: Post-transcriptional silencing of UIS4 in Plasmodium berghei sporozoites is important for host switch. Mol Microbiol. 2014, 91: 1200-1213. 10.1111/mmi.12528.
Annoura T, van Schaijk BC, Ploemen IH, Sajid M, Lin JW, Vos MW, Dinmohamed AG, Inaoka DK, Rijpma SR, van Gemert GJ, Chevalley-Maurel S, Kielbasa SM, Scheltinga F, Franke-Fayard B, Klop O, Hermsen CC, Kita K, Gego A, Franetich JF, Mazier D, Hoffman SL, Janse CJ, Sauerwein RW, Khan SM: Two Plasmodium 6-Cys family-related proteins have distinct and critical roles in liver-stage development. FASEB J. 2014, 28: 2158-2170. 10.1096/fj.13-241570.
Saul A, Prescott N, Smith F, Cheng Q, Walliker D: Evidence of cross-contamination among laboratory lines of Plasmodium berghei. Mol Biochem Parasitol. 1997, 84: 143-147. 10.1016/S0166-6851(96)02779-X.
Ramiro RS, Reece SE, Obbard DJ: Molecular evolution and phylogenetics of rodent malaria parasites. BMC Evol Biol. 2012, 12: 219-10.1186/1471-2148-12-219.
Fairlie-Clarke KJ, Allen JE, Read AF, Graham AL: Quantifying variation in the potential for antibody-mediated apparent competition among nine genotypes of the rodent malaria parasite Plasmodium chabaudi. Infect Genet Evol. 2013, 20: 270-275. 10.1016/j.meegid.2013.09.013.
Cheesman S, Tanabe K, Sawai H, O’Mahony E, Carter R: Strain-specific immunity may drive adaptive polymorphism in the merozoite surface protein 1 of the rodent malaria parasite Plasmodium chabaudi. Infect Genet Evol. 2009, 9: 248-255. 10.1016/j.meegid.2008.12.002.
Long GH, Chan BH, Allen JE, Read AF, Graham AL: Experimental manipulation of immune-mediated disease and its fitness costs for rodent malaria parasites. BMC Evol Biol. 2008, 8: 128-10.1186/1471-2148-8-128.
Culleton RL, Inoue M, Reece SE, Cheesman S, Carter R: Strain-specific immunity induced by immunization with pre-erythrocytic stages of Plasmodium chabaudi. Parasite Immunol. 2011, 33: 73-78. 10.1111/j.1365-3024.2010.01251.x.
Gadsby N, Lawrence R, Carter R: A study on pathogenicity and mosquito transmission success in the rodent malaria parasite Plasmodium chabaudi adami. Int J Parasitol. 2009, 39: 347-354. 10.1016/j.ijpara.2008.07.005.
Fidock DA, Nomura T, Talley AK, Cooper RA, Dzekunov SM, Ferdig MT, Ursos LM, Sidhu AB, Naude B, Deitsch KW, Su XZ, Wootton JC, Roepe PD, Wellems TE: Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol Cell. 2000, 6: 861-871. 10.1016/S1097-2765(05)00077-8.
Culleton R, Martinelli A, Hunt P, Carter R: Linkage group selection: rapid gene discovery in malaria parasites. Genome Res. 2005, 15: 92-97. 10.1101/gr.2866205.
Cheesman S, Raza A, Carter R: Mixed strain infections and strain-specific protective immunity in the rodent malaria parasite Plasmodium chabaudi chabaudi in mice. Infect Immun. 2006, 74: 2996-3001. 10.1128/IAI.74.5.2996-3001.2006.
Pollitt LC, Mackinnon MJ, Mideo N, Read AF: Mosquito transmission, growth phenotypes and the virulence of malaria parasites. Malar J. 2013, 12: 440-10.1186/1475-2875-12-440.
Li J, Pattaradilokrat S, Zhu F, Jiang H, Liu S, Hong L, Fu Y, Koo L, Xu W, Pan W, Carlton JM, Kaneko O, Carter R, Wootton JC, Su XZ: Linkage maps from multiple genetic crosses and loci linked to growth-related virulent phenotype in Plasmodium yoelii. Proc Natl Acad Sci U S A. 2011, 108: E374-E382. 10.1073/pnas.1102261108.
Pattaradilokrat S, Culleton RL, Cheesman SJ, Carter R: Gene encoding erythrocyte binding ligand linked to blood stage multiplication rate phenotype in Plasmodium yoelii yoelii. Proc Natl Acad Sci U S A. 2009, 106: 7161-7166. 10.1073/pnas.0811430106.
Frech C, Chen N: Variant surface antigens of malaria parasites: functional and evolutionary insights from comparative gene family classification and analysis. BMC Genomics. 2013, 14: 427-10.1186/1471-2164-14-427.
Clark BJ: The mammalian START domain protein family in lipid transport in health and disease. J Endocrinol. 2012, 212: 257-275. 10.1530/JOE-11-0313.
van Ooij C, Withers-Martinez C, Ringel A, Cockcroft S, Haldar K, Blackman MJ: Identification of a Plasmodium falciparum phospholipid transfer protein. J Biol Chem. 2013, 288: 31971-31983. 10.1074/jbc.M113.474189.
Templeton TJ: The varieties of gene amplification, diversification and hypervariability in the human malaria parasite, Plasmodium falciparum. Mol Biochem Parasitol. 2009, 166: 109-116. 10.1016/j.molbiopara.2009.04.003.
Lopez FJ, Bernabeu M, Fernandez-Becerra C, del Portillo HA: A new computational approach redefines the subtelomeric vir superfamily of Plasmodium vivax. BMC Genomics. 2013, 14: 8-10.1186/1471-2164-14-8.
Fernandez-Becerra C, Yamamoto MM, Vencio RZ, Lacerda M, Rosanas-Urgell A, del Portillo HA: Plasmodium vivax and the importance of the subtelomeric multigene vir superfamily. Trends Parasitol. 2009, 25: 44-51. 10.1016/j.pt.2008.09.012.
UK Home Office: Research and testing using animals. , [https://www.gov.uk/research-and-testing-using-animals]
Pattaradilokrat S, Cheesman SJ, Carter R: Congenicity and genetic polymorphism in cloned lines derived from a single isolate of a rodent malaria parasite. Mol Biochem Parasitol. 2008, 157: 244-247. 10.1016/j.molbiopara.2007.10.011.
Mullikin JC, Ning Z: The phusion assembler. Genome Res. 2003, 13: 81-90. 10.1101/gr.731003.
Zerbino DR, Birney E: Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18: 821-829. 10.1101/gr.074492.107.
Assefa S, Keane TM, Otto TD, Newbold C, Berriman M: ABACAS: algorithm-based automatic contiguation of assembled sequences. Bioinformatics. 2009, 25: 1968-1969. 10.1093/bioinformatics/btp347.
Otto TD, Sanders M, Berriman M, Newbold C: Iterative Correction of Reference Nucleotides (iCORN) using second generation sequencing technology. Bioinformatics. 2010, 26: 1704-1707. 10.1093/bioinformatics/btq269.
Tsai IJ, Otto TD, Berriman M: Improving draft assemblies by iterative mapping and assembly of short reads to eliminate gaps. Genome Biol. 2010, 11: R41-10.1186/gb-2010-11-4-r41.
Otto TD, Dillon GP, Degrave WS, Berriman M: RATT: Rapid Annotation Transfer Tool. Nucleic Acids Res. 2011, 39: e57-10.1093/nar/gkq1268.
Hunt M, Kikuchi T, Sanders M, Newbold C, Berriman M, Otto TD: REAPR: a universal tool for genome assembly evaluation. Genome Biol. 2013, 14: R47-10.1186/gb-2013-14-5-r47.
Swain MT, Tsai IJ, Assefa SA, Newbold C, Berriman M, Otto TD: A post-assembly genome-improvement toolkit (PAGIT) to obtain annotated genomes from contigs. Nat Protoc. 2012, 7: 1260-1284. 10.1038/nprot.2012.068.
Stanke M, Steinkamp R, Waack S, Morgenstern B: AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 2004, 32: W309-W312. 10.1093/nar/gkh379.
Li L, Stoeckert CJ, Roos DS: OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13: 2178-2189. 10.1101/gr.1224503.
Janse CJ, Waters AP: Plasmodium berghei: the application of cultivation and purification techniques to molecular studies of malaria parasites. Parasitol Today. 1995, 11: 138-143. 10.1016/0169-4758(95)80133-2.
Janse CJ, Mons B, Rouwenhorst RJ, Van der Klooster PF, Overdulve JP, Van der Kaay HJ: In vitro formation of ookinetes and functional maturity of Plasmodium berghei gametocytes. Parasitology. 1985, 91: 19-29. 10.1017/S0031182000056481.
Sebastian S, Brochet M, Collins MO, Schwach F, Jones ML, Goulding D, Rayner JC, Choudhary JS, Billker O: A Plasmodium calcium-dependent protein kinase controls zygote development and transmission by translationally activating repressed mRNAs. Cell Host Microbe. 2012, 12: 9-19. 10.1016/j.chom.2012.05.014.
Hoeijmakers WA, Bartfai R, Francoijs KJ, Stunnenberg HG: Linear amplification for deep sequencing. Nat Protoc. 2011, 6: 1026-1036. 10.1038/nprot.2011.345.
Trapnell C, Pachter L, Salzberg SL: TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009, 25: 1105-1111. 10.1093/bioinformatics/btp120.
Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L: Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013, 31: 46-53. 10.1038/nbt.2450.
The R Project for Statistical Computing. [], [www.r-project.org]
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG: Clustal W and Clustal X version 2.0. Bioinformatics. 2007, 23: 2947-2948. 10.1093/bioinformatics/btm404.
Stamatakis A, Ludwig T, Meier H: RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics. 2005, 21: 456-463. 10.1093/bioinformatics/bti191.
Stamatakis A, Hoover P, Rougemont J: A rapid bootstrap algorithm for the RAxML Web servers. Syst Biol. 2008, 57: 758-771. 10.1080/10635150802429642.
Ronquist F, Huelsenbeck JP: MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003, 19: 1572-1574. 10.1093/bioinformatics/btg180.
The European Malaria Reagent Repository. [], [www.malariaresearch.eu]
Acknowledgments
The work was funded by the Wellcome Trust (grant WT 098051). C. Newbold was supported by a Wellcome Trust program grant (082130), B. Franke-Fayard and W.A.M. Hoeijmakers by grants from The Netherlands Organization for Scientific Research (ZonMW TOP Grant No. 9120_6135; NWO Toptalent 021.001.011), T.D. Otto, M. Hunt, H.G. Stunnenberg, and C.J. Janse by a grant from the European Community's Seventh Framework Program (FP7/2007-2013; Grant Agreement No. 242095) and A.P. Water and A.A. Religa were supported by the Wellcome Trust (Ref. 083811/Z/07/Z). Work in the Holder and Langhorne labs is funded by the MRC (U117532067 and U117584248 respectively).
We thank the following individuals: Martine Zilversmit for producing an Augustus training set; Jai Ramesar (LUMC, Leiden) and C. van Overmeir (ITM, Antwerp) for technical support; The European Malaria Reagents Repository [96] as the source for P. chabaudi isolates; Sandra Cheesman for DNA preparation of the P. chabaudi isolates; Richard Carter for helpful discussion and providing information for the P. chabaudi isolates; R. Menard for providing P. berghei NK65 NY; Ph. van den Steen for P. berghei NK65 E; S. J. Boddey and T. Sargeant for assisting in the PEXEL analysis; R. Davies and Q. Lin for help with data release; L. Robertson, D. Harris, K. Segar, A. Babbage, H. Beasley, L. Clark, J. Harley, P. Heath, P. Howden, G. Kerry, S. Pelan, D. Saunders and J.Wood for manual improvement of the P. chabaudi AS assembly; R. Rance, M. Quail and members of DNA Pipelines for sequencing libraries at WTSI; J. Keane, M. Aslett, N de Silva for database support.
We acknowledge Roche (Branford, USA) for the generation of 454 20 kb libraries. The authors have no competing financial interests.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Electronic supplementary material
12915_2014_86_MOESM1_ESM.xlsx
Additional file 1: Novel putative protein coding genes which were absent in the previous genome annotations of P. berghei ANKA and P. c. chabaudi AS. (XLSX 47 KB)
12915_2014_86_MOESM2_ESM.xlsx
Additional file 2: RMP genes with a PEXEL motif as identified by using the updated HMM algorithm ExportPred v2.0. (XLSX 37 KB)
12915_2014_86_MOESM3_ESM.pdf
Additional file 3: A. Chromosomal organization of the expanded fam-d multigene family in the internal region of chromosome 9. The PcAS, PyYM and PbA genomes contain 21, 5 and 1 copy (PBANKA_091920), respectively (shown in blue). B. An ACT (Artemis Comparison Tool) comparison of syntenic centromeric regions (green) of chromosome 7 of PbA and PcAS and chromosome 6 of P. falciparum 3D7, showing size, location and GC-content. Grey bars: forward/reverse DNA strands. The red lines represent sequence similarity (tBLASTx) (related to Table 1). C. Structural organization of several types of full-length pir genes in PbA (birs), PcAS (cirs) and PyYM (yirs). Exons: yellow boxes; with introns: linking lines. The IDs shown represent a single example. (PDF 398 KB)
12915_2014_86_MOESM4_ESM.xlsx
Additional file 4: The different (subtelomeric) multigene families in the genomes of P. berghei ANKA, P. c. chabaudi AS and P. y. yoelii YM: pir; fam-a; fam-b; fam-c; fam-d; ETRAMP; reticulocyte binding protein, putative; lysophospholipase; PCEMA1; haloacid dehalogenase-like hydrolase; `other subtelomeric genes'. (XLSX 153 KB)
12915_2014_86_MOESM5_ESM.xls
Additional file 5: Putative orthologs (OrthoMCL) of RMP and primate malaria proteins: 1) RMP proteins shared with at least one primate malaria protein; 2) RMP specific proteins (no orthologs in primate malarias); 3) primate malaria specific proteins (no orthologs in RMP). (XLS 2 MB)
12915_2014_86_MOESM7_ESM.xlsx
Additional file 7: Information on RMP isolates/lines used in the study for genome sequencing and RNAseq analyses (see also Additional file 6). (XLSX 13 KB)
12915_2014_86_MOESM8_ESM.xls
Additional file 8: Genes with single nucleotide polymorphisms (SNPs) in: 1) different stabilates of P. berghei isolates/lines (NK65NY, NK65 E, SP11 A, SP11 RLL A, K173; compared to the genome of P. berghei ANKA); 2) in P. y. yoelii 17X (compared to the genome of P. y. yoelii YM); and 3) in different isolates of P. c. chabaudi (AJ, CB) and P. c. adami (DS, DK) (compared to the genome of P. c. chabaudi AS). (XLS 3 MB)
12915_2014_86_MOESM9_ESM.xlsx
Additional file 9: RNA-seq analysis: mRNA abundance (FPKM values) of all genes in the different life cycle stages of P. berghei ANKA, P. c. chabaudi AS and P. y. yoelii YM. (XLSX 1 MB)
12915_2014_86_MOESM10_ESM.xlsx
Additional file 10: Differentially expressed genes in 16 hour ookinetes compared to 24 hour ookinetes of P. berghei ANKA and GO-annotation of up- and down-regulated genes. (XLSX 233 KB)
12915_2014_86_MOESM11_ESM.xlsx
Additional file 11: Overview and details of spliced transcripts and alternative splicing (AS) in the RMP genomes based on RNA-seq analyses. (XLSX 79 KB)
12915_2014_86_MOESM15_ESM.xlsx
Additional file 15: RMP pir s in different phylogenetic clades. Classification of pir genes by clade and by species, with information on predicted protein sequence length, base composition and codon usage. (XLSX 121 KB)
12915_2014_86_MOESM16_ESM.pdf
Additional file 16: Expression of PIRs in relation to their phylogenetic relationship. Heat maps of expression (FPKM values >21; normalized by gene) of all PbA pirs in different life cycle stages in association with their location in different clades (L, S) of the phylogenetic tree (black boxes). Ri: ring; Tr: Trophozoite; Sch: Schizont; Gct: Gametocyte; Ook: Ookinetes (16 and 24 hour ookinetes). (PDF 152 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Otto, T.D., Böhme, U., Jackson, A.P. et al. A comprehensive evaluation of rodent malaria parasite genomes and gene expression. BMC Biol 12, 86 (2014). https://doi.org/10.1186/s12915-014-0086-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12915-014-0086-0