
Abstract
Background
Inflammatory myofibroblastic tumour (IMT) is a mesenchymal neoplasm of intermediate biological potential that may affect a wide range of anatomic sites but has a particular predilection for the lung and intra-abdominal soft tissues.
Case presentation
We report here an exceptional case of inflammatory myofibroblastic tumor arising in the lacrimal gland and presenting as an orbital mass in a 24-year-old male.
Conclusion
This report aims to discuss the importance of histopathological and immunohistochemical findings in arriving at the diagnosis, which helps dictate the management, treatment and prognosis of the patient.
Similar content being viewed by others


Background
Inflammatory myofibroblastic tumor (IMT) is a very rare mesenchymal tumour that can arise in various anatomic locations [1, 2]. IMT is characterized by a proliferation of myofibroblastic cells admixed with inflammatory elements. Approximately half of IMTs have a rearrangement of the anaplastic lymphoma kinase (ALK). Moreover, this entity is quite curable if completely and appropriately excised [1,2,3]. We present here an exceptional case of orbital IMT occuring in the lacrimal gland.
Case presentation
A 24-year-old male, was referred to our medical institution with a 4 months history of progressive and indolent mass in the super-external area of the left eye with no notion of trauma or insect bite.
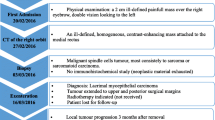
On clinical examination, there was a soft tissue swelling at the superolateral angle of the left orbit (Fig. 1). The patient also had left upper eyelid ptosis with no associated diplopia, proptosis or other clinical symptoms. Pupils were round and reactive. Visual acuity was 20/20 in each eye. Visual fields and intraocular pressures were normal.

Picture showing the patient with swelling at the superolateral angle of the left orbit
Computerized tomography (CT) scan revealed a well circumscribed and homogeneous left lacrimal gland tumor without bone erosion, measuring 22 mm × 21 mm (Fig. 2).

Computed Tomography scan showing a well circumscribed and homogeneous left supraconal tumor
The patient underwent a complete surgical excision of the lesion within its smooth pseudocapsule and without fragmentation (Fig. 3).

Gross picture of the excised mass
Histopathology of the excised mass showed a tumor composed of haphazardly arranged spindle-shaped and oval cells in a predominantly fibrous background, interspersed with prominent dense bands of collagen that clamped residual glands (Figs. 4 and 5). Mitosis and necrosis were absent. The stroma was infiltrated by numerous inflammatory cells composed of lymphocytes, plasma cells and rare eosinophils (Fig. 6). These histopathological features were suggestive of IMT.

Low magnification showing a lesion with fascicular architecure in a predominantly fibrous background (haematoxylin & eosin stain, × 50)

Higher magnification showing the residual glands that are encircled by the proliferation (haematoxylin & eosin stain, × 200)

Higher magnification showing a stroma infiltrated by numerous inflammatory cells composed of lymphocytes, plasma cells and eosinophils (haematoxylin & eosin stain, ×400)
The immunohistochemical analysis showed that the spindle-shaped cells to be diffusely positive for smooth muscle actin, vimentin and ALK with a low proliferative index (Ki-67 = 10%) (Fig. 7).

Immunohistochemical staining revealed the expression of smooth muscle actin (a) and ALK (b) by the neoplastic cells. Keratin highlights the residual glands (c). Ki-67 immunostaining showed a low proliferation index (d)
These cells were negative for pancytokeratin AE1/AE3 (positive in residual glands), S100 protein, desmin, CD34.
Co-relating histopathological aspect and immunophenotype profile, a final diagnosis of inflammatory myofibroblastic tumor (IMT) of the lacrimal gland was established.
The patient is currently under regular follow-up with no evidence of recurrence till date (followed for approximately 7 months)
Discussion
Inflammatory myofibroblastic tumor (IMT) is defined as a benign tumor composed of myofibroblastic spindle cells over an inflammatory background. This lesion has long been reported under many synonyms, including plasma cell granuloma, plasma cell pseudotumour, inflammatory myofibrohistiocytic proliferation, myxoid hamartoma and inflammatory pseudotumour.
This rare neoplasm affects predominantly children and young adults but can also be seen in elderly, with no sex and race predilection [1,2,3].
IMT occurs most commonly in the lungs, mesentery, pelvis, retroperitoneum, and abdominal soft tissue. Its occurrence in the lacrimal gland is very exceptional [1, 2, 4].
This tumor has historically been reported by many synonyms; such as inflammatory pseudotumor, xanthogranuloma, plasma cell granuloma and plasma cell pseudotumor. This confusion in nomenclature was because of confusion concerning its pathogenesis that whether it is a reactive process or a true neoplasm. Inflammatory reactions to surgery, trauma, irritation and infection are some of triggers for its development. In the 2013 WHO classification, IMTs are recognized as a clonal neoplasm that are considered to have intermediate malignancy that rarely metastasizes. They are characterized by rearrangements involving the ALK (anaplastic lymphoma kinase) gene locus on 2p23 in 50% of cases, leading to constitutive activation of the tyrosine kinase [3, 5, 6].
The mode of presentation of IMT varies. They usually present as a local swelling. Although, systemic symptoms, such as fever, malaise, weight loss and night sweats, are also observed [2, 7].
On computed tomography imaging, IMTs appear as a mildly enhancing soft tissue mass. In general, the radiological features of IMT are variable and non-specific [2, 8].
Grossly, the tumor is well-circumscribed, lobulated or multinodular and may be firm or fleshy with a tan-white cut surface [3, 9, 10].
Characteristic histological findings include the presence of stellate/spindle shaped myofibroblasts and inflammatory spindle cells. The spindled to stellate cells are ranged haphazardly in short or storiform fascicles and exhibit plump, oval nuclei, with no appreciable atypia or hyperchromasia. Cytoplasm is generally sparse or pale eosinophilic in these cells.
Mitotic activity is often low, and atypical mitotic forms are very rare. The stroma shows prominent vasculature and admixed inflammatory cells, consisting chiefly of lymphocytes, plasma cells and occasional neutrophils and eosinophils. Presence of foamy histiocytes has also been reported. Several histological patterns have been described in IMT: loose or myxoid stroma with prominent vascularity; compact spindle cells and densely collagenous with fewer spindle cells and inflammatory cells [1, 10,11,12]. Histology of our case showed the pattern of densely collagenous stroma with compact spindle cells.
Immunohistochemical study is generally realized to affirm and support the myofibroblastic phenotype of the tumor spindle cells, which consistently express vimentin (99%) and smooth muscle actin (92%). Desmin and other myogenic markers are less frequently expressed and may be focal. Cytokeratins have been identified in about one-third of cases. IMTs are typically negative to myoglobin and S-100 protein [1, 10, 12,13,14].
In our case also the tumour was strongly positive for vimentin and smooth muscle actin. It was negative for desmin and cytokeratin.
In IMTs, ALK overexpression is typically detectable by immunohistochemistry, which demonstrates cytoplasmic reactivity and is also detectable by FISH [5, 15]. In our case, indeed, tumor cells exhibited cytoplasmic staining with ALK.
The differential diagnosis of IMTs is extensive and broad, comprising nodular fasciitis, smooth muscle and myofibroblastic sarcomas, idiopathic orbitaliInflammation (IOI), inflammatory fibrosarcoma, pseudotumor resulting from mycobacterial infection, gastrointestinal stromal tumours (GIST), fibrous histiocytoma and fibromatosis amongst others [7]. The ALK status has a role in a number of this differential diagnoses. In fact, the majority of this various neoplasms are ALK-negative [16].
Surgery is the mainstay of treatment for IMTs and complete surgical resection, when feasible, is typically curative [2, 11, 12, 17, 18]. Although local Recurrence or progression is site dependent, seen in 25% of extra-pulmonary locations [18, 19]. Several cases have been treated with chemotherapy, Radiotherapy, and/or corticosteroids. Systemic or malignant transformation of IMT is rare and has only been reported in a few cases [2, 11, 17,18,19,20,21,22].
The recognition of the molecular basis of IMT has led to the increasing use of new biological therapies using inhibitors of the kinase domain of ALK protein with spectacular success and impressive response rate, especially in patients with ALK-positive IMTs [15, 16, 18, 20,21,22].
Conclusion
Inflammatory myofibroblastic tumour is a very rare spindle cell neoplasm of intermediate biological potential that may arise in a wide range of anatomic sites. Lacrimal gland represents a distinct and an exceptional location. Differential diagnoses of IMT are varied and immunohistochemistry can play a central role in resolving diagnostic issues. Complete surgical resection is the gold standard.
Abbreviations
- ALK:
-
Anaplastic lymphoma kinase
- CT:
-
Computerized tomography
- IMT:
-
Inflammatory myofibroblastic tumour
References
Gleason BC, Hornick JL. Inflammatory myofibroblastic tumours: where are we now? J Clin Pathol. 2008;61:428–37.
Coffin CM, Watterson J, Priest JR, Dehner LP. Extrapulmonary inflammatory myofibroblastic tumor (inflammatory pseudotumor). A clinicopathologic and immunohistochemical study of 84 cases. Am J Surg Pathol. 1995;19:859–72.
Fletcher CD, Mertens F, Bridge JA. WHO classification of Tumours of soft tissue and bone. International Agency for Research on Cancer. 4th ed. Geneva: WHO Press; 2013.
Tawfik HA, Raslan AO. Infantile inflammatory myofibroblastic tumor of the orbit with apical bone involvement. Ophthal Plast Reconstr Surg. 2013;29:e44–6.
Lawrence B, Perez-Atayde A, Hibbard MK, Rubin BP, Dal Cin P, Pinkus JL, et al. TPM3-ALK and TPM4-ALK oncogenes in inflammatory myofibroblastic tumours. Am J Pathol. 2000;157:377–84.
Patnana M, Sevrukov AB, Elsayes KM, et al. Inflammatory pseudotumor: the great mimicker. Am J Roentgenol. 2012;198:W217–27.
Coffin CM, Hornick JL, Fletcher CD. Inflammatory myofibroblastic tumor: comparison of clinicopathologic, histologic and immunohistochemical features including ALK expression in atypical and aggressive cases. Am J Surg Pathol. 2007;31:509–20.
Park SB, Lee JH, Weon YC. Imaging findings of head and neck inflammatory pseudotumor. AJR Am J Roentgenol. 2009;193:1180–6.
Dutta V, Manoj MG, Malik A, Kumar P. ALK negative inflammatory myofibroblastic tumor of the orbit: a masquerading entity. Indian J Ophthalmol. 2014;62(5):627–9.
McDermott M. Myofibroblastic inflammatory tumour. Semin Diagn Pathol. 2016;33(6):358–66.
Coffin CM, Humphrey PA, Dehner LP. Extrapulmonary inflammatory myofibroblastic tumor: a clinical and pathological survey. Semin Diagn Pathol. 1998;15(2):85–101.
Pettinato G, Manivel JC, De Rosa N, Dehner LP. Inflammatory myofibroblastic tumor (plasma cell granuloma). Clinicopathologic study of 20 cases with immunohistochemical and ultrastructural observations. Am J Clin Pathol. 1990;94(5):538–46.
Hussong JW, Brown M, Perkins SL, Dehner LP, Coffin CM. Comparison of DNA ploidy, histologic, and immunohistochemical findings with clinical outcome in inflammatory myofibroblastic tumors. Mod Pathol. 1999;12(3):279–86.
Cessna MH, Zhou H, Sanger WG, et al. Expression of ALK1 and p80 in inflammatory myofibroblastic tumor and its mesenchymal mimics: a study of 135 cases. Mod Pathol. 2002;15(9):931–8.
Cools J, Wlodarska I, Somers R, et al. Identification of novel fusion partners of ALK, the anaplastic lymphoma kinase, in anaplastic large-cell lymphoma and inflammatory myofibroblastic tumor. Genes Chromosom Cancer. 2002;34(4):354–62.
Sukov WR, Cheville JC, Carson AW, Shearer BM, Piatigorsky EJ, Grogg KL, Sebo TJ, Sinnwell JP, Ketterling RP. Utility of ALK-1 protein expression and ALK rearrangements in distinguishing inflammatory myofibroblastic tumor from malignant spindle cell lesions of the urinary bladder. Mod Pathol. 2007;20:592–603.
Souid AK, Ziemba MC, Dubansky AS, et al. Inflammatory myofibroblastic tumor in children. Cancer. 1993;172(6):2042–8.
Karnak I, Senocak ME, Ciftci AO, et al. Inflammatory myofibroblastic tumor in children: diagnosis and treatment. J Pediatr Surg. 2001;36(6):908–12.
Donner LR, Trompler RA, White RR. Progression of inflammatory myofibroblastic tumor of soft tissue into sarcoma after several recurrences. Hum Pathol. 1996;27(10):1095–8.
Butrynski JE, D’Adamo DR, Hornick JL, et al. Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N Engl J Med. 2010;363:1727–33.
Polito E, Pichierri P, Loffredo A, Moramarco A, Occhini R. Inflammatory myofibroblastic tumor of the orbit. Ophthalmologica. 2007;221(5):353–5.
Sa HS, Ji JY, Suh YL, et al. Inflammatory myofibroblastic tumor of the orbit presenting as a subconjunctival mass. Ophthal Plast Reconstr Surg. 2005;21(3):211–5.
Acknowledgments
Not applicable.
Funding
Not applicable.
Availability of data and materials
The datasets used during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
AB, MA, FEA, IR and MREO analyzed and interpreted the patient data, drafted the manuscript and made the figures. AB, MA, MT and HC performed the histological examination and proposed the study. AB, MA, AAB and MO made substantial contributions to conception and design, and revised the manuscript. All authors of this paper have read and given final approval of the version to be published.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor of this journal.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Boudhas, A., Allaoui, M., El Asri, F. et al. Inflammatory myofibroblastic tumor of the lacrimal gland: case report of an exceptional location. BMC Clin Pathol 17, 12 (2017). https://doi.org/10.1186/s12907-017-0050-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12907-017-0050-3