
Abstract
Background
Cancer is one of the leading causes of death worldwide. Recently, it was shown that many natural extracts have positive effects against cancer, compared with chemotherapy or recent hormonal treatments. A. annua is an annual medicinal herb used in the traditional Chinese medicine. It has also been shown to inhibit the proliferation of various cancer cell lines.
Methods
Multi-level modes of action of A. annua constituents in cancer therapy were investigated using an integrated approach of network pharmacology, molecular docking, dynamic simulations and in-vitro cytotoxicity testing on both healthy and cancer cells.
Results
Network pharmacology-based analysis showed that the hit Artemisia annua constituents related to cancer targets were 3-(2-methylpropanoyl)-4-cadinene-3,11-diol, artemisinin G, O-(2-propenal) coniferaldehyde, (2-glyceryl)-O-coniferaldehyde and arteamisinin III, whereas the main cancer allied targets were NFKB1, MAP2K1 and AR. Sixty-eight significant signaling KEGG pathways with p < 0.01 were recognized, the most enriched of which were prostate cancer, breast cancer, melanoma and pancreatic cancer. Thirty-five biological processes were mainly regulated by cancer, involving cellular response to mechanical stimulus, positive regulation of gene expression and transcription. Molecular docking analysis of the top hit compounds against the most enriched target proteins showed that 3-(2-methylpropanoyl)-4-cadinene-3,11-diol and O-(2-propenal) coniferaldehyde exhibited the most stabilized interactions. Molecular dynamics simulations were performed to explain the stability of these two compounds in their protein-ligand complexes. Finally, confirmation of the potential anticancer activity was attained by in-vitro cytotoxicity testing of the extract on human prostate (PC-3), breast (MDA-MB-231), pancreatic (PANC-1) and melanoma (A375) cancerous cell lines.
Conclusion
This study presents deeper insights into A. annua molecular mechanisms of action in cancer for the first time using an integrated approaches verifying the herb’s value.
Similar content being viewed by others


Introduction
Cancer is an essential health problem and one of the most leading causes of death worldwide [1]. Carcinogenesis is a multistage process that initiates with genetic alteration due to the activation of oncogenes and the inactivation of tumor suppressor genes resulting in a loss of control of cell proliferation [2]. The alteration of normal cells into malignant ones is regulated by several steps, each may serve as a target for anticancer agents [3].
Prevention of cancer is one of the most crucial medicinal issues in the past few years [4]. Recently, many natural products have been studied for their chemo-preventive effects. It has been shown that many extracts from natural products have positive effects against cancer, compared with chemotherapy or recent hormonal treatments due to their availability, lower side effects and cost effectiveness [5]. Hence, they form approximately 60% of all anti-cancer drugs and have captivated scientific attention as promising anti-cancer therapies. The assessment of traditional phytomedicines for cytotoxic activity may contribute to the innovation of new chemotherapeutic regimens either alone or combined with the existing chemotherapeutic drugs so that to combat various cancer types [6]. Cytotoxicity of these phytomedicines arises from their ability to suppress certain tumor cell characteristics, such as increased metabolism, elevated concentration of iron and transferrin, and susceptibility to Reactive Oxygen Species (ROS) [7].
Artemisia annua (family Asteraceae) also known as ‘Sweet wormwood’ (English) or ‘Qinghao’ (Chinese), is an annual medicinal herb naturally grown in China, with a long history in treating various diseases in the traditional Chinese medicine including fever, amoebiasis, cancer, schistosomiasis, HIV and Leishmaniasis [8]. This plant is known for its aromatic scent, bitter taste, and aromatic pale yellow blossoms [9]. Beside its promising anti-inflammatory, antimalarial and immunosuppressive activities, it has also been shown to inhibit the proliferation of various cancer cell lines. Sesquiterpene lactones as artemisinin were identified as important active principles of Artemisia annua extract, however, available evidence assumed that artemisinin might not be the most active anticancer ingredient of this medicinal plant. Accordingly, Artemisia annua may serve as a source for new anti-cancer components [10] as it is enriched with phenolic compounds with high antioxidant power classified into mainly five major groups; coumarin, flavones, flavonols and phenolic acids [9]. Up to our knowledge, few studies have explained the potential role of these metabolites and their synergistic effects in the treatment of cancer, thus, the molecular mechanisms for the anti-cancer activities of Artemisia annua still need elucidation.
Owing to the multi-chemical components of the plant extracts, their multi-pharmacological effects as well as their multi-action targets in the treatment of different diseases, it is challenging for traditional methods to demonstrate the different mechanisms behind their pharmacological activities in an adequate manner [11, 12]. Yet, a recently emerging approach named network pharmacology had combined bioinformatics and pharmacology organically thus can clarify and explain the multi-mechanisms of action of these plants via emphasizing the relationship between targets, components and diseases [13, 14]. Accordingly, this approach was capable of affording new ideas for research and development of herbal medicine [15].
In this study, we aimed to overview the material basis and the detailed mechanism of Artemisia annua in the treatment of different cancer types and highlight the specific type of cancer cells that are mostly inhibited for their proliferation using network pharmacology analysis. The interaction between the most enriched compounds and bio-macromolecule targets was illustrated by molecular docking to estimate the binding patterns and affinity. However, network pharmacology and molecular docking are just in-silico techniques that provide a simulation model but cannot reflect realistic complexity, accordingly, in-vitro cytotoxicity experiments against the top hit cancer cells – retrieved from network pharmacology- were conducted to verify and provide complementary information. Ultimately, the study in hand could provide a new perceptiveness and guidance for the development of Artemisia annua in cancer treatment.
Results and discussion
Network pharmacology analysis
From the dictionary of natural products database, 80 Artemisia annua compounds were identified while, their predicted targets were collected from the Swiss Target Prediction platform, generating a total of 340 targets. 487 cancer-related target genes were yielded from the GeneCards (Table S1), and 29 intersecting druggable target genes of the corresponding active ingredients of Artemisia annua were identified by mapping the compounds’ genes to the cancer-associated genes (Fig. 1).

Venn diagram of Artemisia annua compounds and cancer targets
It is worth mentioning that 25 compounds out of 80 Artemisia annua metabolites showed interactions with cancer-related targets. Based on the compounds’ predicted interaction probabilities retrieved from Swiss Target Prediction database, they contributed differently to the pharmacological profile of the plant where, 4-Cadinene-3,11-diol; (1α,3xi,6α,7β,10α)-form, 3-(2-Methylpropanoyl) accounted for 16% of the total interactions followed by artemisinin G, O-(2-Propenal)coniferaldehyde and O-(2-Glyceryl) coniferaldehyde contributing with 13%, 12% and 11% respectively of the total interactions (Fig. 2). This revealed that these compounds might be the core metabolites for the anti-cancer activity of Artemisia annua.

The distributions % of Artemisia annua L. constituents’ interactions with 29 intersecting cancer-related target genes
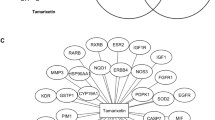
As far as it can be noted, this plant appeared to exhibit its therapeutic effect on cancer by regulating these 29 intersecting target genes, among them, Nuclear factor NF-kappa-B p105 subunit (NFKB1), mitogen-activated protein kinase kinase 1 (MAP2K1), Androgen Receptor (AR) and p53-binding protein (MDM2) had the highest sum of interaction probability scores as reflected by their percentages of interactions (22%, 12%, 6% and 5%, respectively) (Fig. 3; Table 1). This indicated their valuable contribution as therapeutic cancer targets on which Artemisia annua could act. Cancer is regulated by NFKB1 gene as indicated by the notion that genetic variants of NFKB1 might increase the susceptibility of liver [16] and gastric cancer [17]. Moreover, targeting NFKB1/RELA interaction could mitigate Ets1-mediated metastatic breast cancer [18]. Not only NFKB1, MAP2K1 is one of the target proteins in cancer pathogenesis where, a previous study proved that miRNAs repressed the carcinogenesis of the human non-small cell lung cancer (NSCLC) by inhibiting MAP2K1 expression of MAP2K1 via direct targeting its 3’UTR [19]. Furthermore, MAP2K1 mutation lead to poor response to EGFR inhibition and increase pathogenesis of colorectal cancer [20] and thyroid gland cancer [21]. AR inhibitors are regarded as promising agents in cancer treatment as they decreased IL-6 production by breast cancer cells (MB-231 and MB-231BR) so inhibit their proliferation [22]. In addition, inhibition of AR diminished the synthesis of androgens in prostate cancer [23]. MDM2 protein also takes a significant part in the pathology of cancer in which, MDM2 inhibitors reactivated p53 and treated some types of cancer as Malignant pleural mesothelioma (MPM) [24]. Furthermore, targeting over-expressed MDM2 interrupted membrane integrity by pore formation, triggered membrane destabilization and rapid cancer cell necrosis [25].

The distributions % of target genes interactions with Artemisia annua L. constituents
From STRING analysis, the overlapping genes were closely associated in the protein-protein interaction diagram (Fig. 4, Table S2).

Protein-Protein interaction diagram of Artemisia annua
Based on the aforementioned 29 intersecting target genes, 68 significant signaling KEGG pathways with p < 0.01 were generated, from which, the most enriched 17 cancer – related pathways were picked up for further analysis including Prostate cancer, breast cancer, melanoma and pancreatic cancer. As can be observed (Table 2), prostate cancer was recognized as the crucial key pathway in cancer with lowest p value (4.06E-12) and highest observed gene count [10].
Cytoscape software was used to build “component-target’’ and ‘’ gene-pathway” networks (Fig. 5a and b). As can be observed, Artemisia annua related multi-component, multi-target, and multi-pathway mechanisms for cancer treatment. Figure (5a) showed interactions between components and target genes. The network included 55 nodes (sorted as 25 components and 29 genes) and 310 edges. It also suggested that not only the single component acted on multiple targets but also the same target was hit by different components. Similarly, KEGG pathway annotation revealed that the 29 potential target genes were involved in 18 significantly cancer - correlated pathways (Fig. 5b).

(a) Compounds-targets network. (b) Genes-pathway network. The nodes represent targets (green circles), compounds (red hexagons) and pathways (green hexagons). The edges were regarded as the association between the nodes. Enrichment of nodes are indicated by different color intensities
KEGG pathway and gene ontology (GO) enrichment analysis
To investigate the biological functions and pathway of the key targets of Artemisia annua, molecular functions (MF), biological process (BP), cellular components (CC) and KEGG were performed through the functional annotation tool of DAVID. As shown in Fig. (6a), 35 BPs were mainly regulated by cancer involving cellular response to mechanical stimulus, positive regulation of gene expression and positive regulation of transcription, DNA-templated. It was recently reported that a cytotoxic effect can be achieved when excessive levels of exogenous mechanically forced stimulation that used deep tissue-penetrating and focusable energy sources (e.g., ultrasound and magnetic fields) were directed to a cell [26]. Furthermore, regulation of the YAP and TAZ (YAP/TAZ) proteins controlled the cellular response to different mechanical stimuli causing its death [27]. Previous literature reviews confirmed the contribution of AML1, Sp1, and p300 in cancer through promoting E-cadherin expression in breast cancer cells thus, reducing cell motility [28]. Anqi Ge et al. conferred the possible antitumor activity of some intrinsic metabolites in herbal drugs such as baicalin by regulating DNA-templated RNA transcription in MCF-7 cancer cells which may afford new a therapeutic target in treating breast cancer [29]. Another study expressed the potential role of KIF1A in the positive regulation of DNA-templated RNA transcription via cell adhesion molecules (CAMs) and primary immunodeficiency. This mechanism suggested its contribution as one of the biomarkers causing poor prognosis in ovarian cancer [30].
Among the observed 22 CC terms, macromolecular complex, nucleus and cytoplasm were the most prominent cancer- related components. Enrichment results also revealed 31 molecular function items, including enzyme binding, transcription coactivator binding and RNA polymerase II transcription factor activity.

Enrichment analysis results of cancer-related targets in Artemisia annua. (A) BP (orange), CC (green) and MF (blue) results in GO annotation analysis. Y-axis stands for gene count (columns) and Log P values (red line), while the x-axis represents the genes in the GO annotation results. (B) Pathway enrichment bar chart where enriched pathways of the targets are represented by y-axis, and the x-axis stands for KEGG (orange) and BIOCARTA (green) pathways. Gene count is presented in columns and probabilities are denoted in a red line
GO enrichment pathways analysis retrieved 50 pathways (Fig. 6b) from which, 17 BIOCARTA pathways were obtained where pathways in cancer, Chemical carcinogenesis, Prostate cancer and Proteoglycans in cancer were strikingly related to cancer.
Molecular docking analysis of top-hit compounds in the active sites of the most enriched cancer-associated target genes
Prior to docking the top hit compounds into the most enriched target genes, a validation process was performed to determine the binding site and evaluate the ability of the docking program to reproduce the orientation of the native co-crystallized ligand. This was attained by extraction and re-docking of the co-crystallized ligands into their respective enzyme’s active site. Results revealed that the used docking software (Glide) was accurate in reproducing the native co-crystallized orientation of the redocked ligands with RMSD values ranging from 0.1 to 0.4 Å (Table 3, Figure S1).
The Glide module of the Schrodinger suite software was then used for calculating the docking XP G scores of A. annua hit compounds 4-cadinene-3,11-diol; (1α,3xi,6α,7β,10α)-form, 3-(2-methylpropanoyl), artemisinin G, O-(2-propenal) coniferaldehyde, (2-glyceryl)-O-coniferaldehyde and arteamisinin III against the active sites of the most enriched cancer-associated target genes NFKB1, MAP2K1, AR, MDM2, CDK2 and PTGS2. From Table 1, it can be observed that 4-cadinene-3,11-diol; 3-(2-methylpropanoyl) had the lowest XP G score against androgen receptor, P53-binding protein and cyclooxygenase-2. Whereas (2-glyceryl)-O-coniferaldehyde exhibited the most stabilized interaction with nuclear factor NF-kappa-B, mitogen-activated protein kinase and cyclin-dependent kinase 2.
The 2D and 3D interaction diagrams of 4-cadinene-3,11-diol; 3-(2-methylpropanoyl) in the active site of androgen receptor (PBD ID 2PIW) (Fig. 7A) revealed the formation of a hydrogen bond between ketone group and Thr877. Polar interactions were also observed with Gln711 and Asn705 in addition to hydrophobic interactions with Leu701, Leu707, Trp741, Met742, Val746, Met749, Met780, Met787 and Phe876.

2D and 3D interaction diagrams of 4-Cadinene-3,11-diol; 3-(2-methylpropanoyl) in the active site of (A) androgen receptor (PBD ID 2PIW), (B) P53-binding protein (PDB ID 4IPF) and (C) cyclooxygenase-2 (PDB ID 3LN1).
Meanwhile, the interaction of 4-cadinene-3,11-diol; 3-(2-methylpropanoyl) with P53-binding protein (PDB ID 4IPF) involved charged positive interaction with Lys47. In addition to a polar interaction with His92. There were also hydrophobic interactions with Leu50, Tyr51, Leu53, Ile57, Val71, Phe87, Val89, Ile95, Tyr96 and Ile99 (Fig. 7B).
Furthermore, 4-cadinene-3,11-diol; 3-(2-methylpropanoyl) interacted cyclooxygenase-2 (PDB ID 3LN1) using one hydrogen bonds between ketonic group and Thr877. In addition to polar interactions with Asn705 and Gln711 and hydrophobic interaction with Leu701, Leu704, Leu707, Trp741, Met742, Val746, Phe764, Met780, Met787, Leu873, Phe876, Leu880 and Met895. There was also a charged positive interaction with Arg752 (Fig. 7C).
Whereas the stabilization of (2-glyceryl)-O-coniferaldehyde interaction with nuclear factor NF-kappa-B (PDB ID 1LV2) was through the formation of two hydrogen bonds between hydroxyl group in the glyceryl moiety and Glu210 and Gln304. There were also polar interactions with Gln304 and hydrophobic interactions with Ile135, Met142, Leu180, Leu194, Ile211, Val214, Ala215, Val218, Met301, Ile305, Phe307, Val308, Val314 and Val316. In addition to negatively charged interaction with Glu210 (Fig. 8A).

2D and 3D interaction diagrams of O-(2-glyceryl) coniferaldehyde in the active site of (A) nuclear factor NF-kappa-B (PDB ID 1LV2), (B) mitogen-activated protein kinase 1 (PDB ID 4AN9) and (C) Cyclin-dependent kinase 2 (PDB ID 1B39)
On the other hand, the interaction of (2-glyceryl)-O-coniferaldehyde with mitogen-activated protein kinase 1 (PDB ID 4AN9) involved three hydrogen bonds between aldehydic ketone and Met146 residue, hydroxyl group in the glyceryl moiety and Ser194 and between methoxyl oxygen and Lys97. There were also hydrophobic interactions with Leu74, Ala76, Val82, Ala95, Met143, Met146, Leu197 and Cys207 residues and polar interaction with Asn78, Hie145, Ser150, Asn195 and Ser194. In addition to a charged negative interaction with Glu144 and Asp208 and charged positive ones with Lys97 and Lys 192(Fig. 8B). While the interaction of (2-glyceryl)-O-coniferaldehyde with cyclin-dependent kinase 2 (PDB ID 1B39) was stabilized via the formation of a hydrogen bond between aldehydic ketone group and Lys89 and between hydroxyl group in the glyceryl moiety and Gln131. In addition to the formation of coordination reaction with Mg382 and hydrophobic interactions with Ile10, Val18, Ala31, Phe82, Leu83, Leu134 and Ala144. There were also a charged positive interaction with Lys 33 and Lys89 and negative ones with Glu12, Asp86 and Asp145. Moreover, polar interactions with Hie84, Gln85, Gln131 and Asn132 were also observed (Fig. 8C).
Molecular dynamics simulations stability analyses of hit protein-ligand complexes
Molecular dynamics simulations are a molecular modeling technique often used to in silico [31, 32] explain the stability of protein-ligand complexes [33]. In this study, the compound 4-cadinene-3,11-diol; 3-(2-methylpropanoyl) with target proteins androgen receptor, P53-binding and cyclooxygenase-2, and target proteins of compound (2-glyceryl)-O-coniferaldehyde with nuclear factor NF-kappa-B, mitogen-activated protein kinase 1 and cyclin-dependent kinase 2 protein-ligand complexes created by molecular docking were analyzed by molecular dynamics simulation of 100 ns duration. RMSD measurements were used to quantify the changes and movements of compounds in protein active pockets per frame [34]. The RMSD value between 4-cadinene-3,11-diol; 3-(2-methylpropanoyl) and the androgen receptor was below 0.3 nm up to 80 ns and averaged 0.21 as shown in (Fig. 9A). 4-Cadinene-3,11-diol; 3-(2-methylpropanoyl) and P53-binding complex RMSD peaked below 0.7 nm up to 90 ns and then at 1 nm as shown in (Fig. 9B) and averaged 0.67. As given in (Fig. 9C), 4-Cadinene-3,11-diol; 3-(2-methylpropanoyl) at the cyclooxygenase-2 active site generally gave a value below 0.2 nm, with an average RMSD of 0.13 nm. The RMSD of the second compound, (2-glyceryl)-O-coniferaldehyde, was measured below 0.4 nm with a mean of 0.32, as shown in (Fig. 9D), according to the nuclear factor NF-kappa-B. (2-glyceryl)-O-coniferaldehyde and mitogen-activated protein kinase 1 gave RMSD value below 0.2 nm and an average of 0.11 nm as shown in (Fig. 9E). With the other target protein cyclin-dependent kinase 2, the first 25 ns fluctuated up to 0.4 nm, then elevated with mean RMSD value of 0.54 as shown in (Fig. 9F).

RMSD analysis numerically showing the stability of compounds 4-Cadinene-3,11-diol; 3-(2-methylpropanoyl) and O-(2-glyceryl) coniferaldehyde relative to proteins from molecular dynamics’ simulations. (A-B-C) RMSD of 4-Cadinene-3,11-diol; 3-(2-methylpropanoyl) with androgen receptor, P53-binding and cyclooxygenase-2. (D-E-F) RMSD of O-(2-glyceryl)-coniferaldehyde with nuclear factor NF-kappa-B, mitogen-activated protein kinase 1 and cyclin-dependent kinase 2 for 100 ns, respectively
RMSF analysis, which calculates the mean square deviation of each amino acid residue from its initial position, provides an indication of the degree of flexibility or rigidity in distinct regions of a protein. This methodology was utilized to ascertain the average flexibility of the target proteins in this study. In the initial phase of the investigation, the interaction between the compound 4-Cadinene-3,11-diol; 3-(2-methylpropanoyl) and the target proteins, namely the androgen receptor, P53-binding protein, and cyclooxygenase-2, was analyzed (Fig. 10A-B-C). The data indicated that the average RMSF value for cyclooxygenase-2 was 0.104 nm, which was notably higher than that of the other target proteins. This suggests that cyclooxygenase-2 displayed a higher degree of average flexibility in comparison to the androgen receptor and P53-binding protein. The average RMSF values for the androgen receptor and P53-binding protein were computed to be 0.069 and 0.075 nm, respectively. These findings denote that these proteins showed a comparable degree of average flexibility, with a marginally higher value noted in the P53-binding protein. In the subsequent phase of the investigation, the interactions between the compound O-(2-glyceryl) coniferaldehyde and a different set of target proteins, namely nuclear factor NF-kappa-B, mitogen-activated protein kinase 1, and cyclin-dependent kinase 2, were explored (Fig. 10D-E-F). It was found that the average RMSF value for cyclin-dependent kinase 2, at 0.103 nm, was the highest among these target proteins, suggesting that cyclin-dependent kinase 2 exhibited a greater degree of average flexibility compared to nuclear factor NF-kappa-B and mitogen-activated protein kinase 1. The average RMSF values for nuclear factor NF-kappa-B and mitogen-activated protein kinase 1 were computed to be 0.086 and 0.100 nm, respectively. This implies that these proteins showed similar degrees of average flexibility, albeit with a slightly higher average flexibility discerned in the mitogen-activated protein kinase 1.

RMS fluctuation analyzes for flexibility and mobility in protein structures. (A-B-C) RMSF of 4-Cadinene-3,11-diol; 3-(2-methylpropanoyl) with androgen receptor, P53-binding and cyclooxygenase-2. (D-E-F) RMSF of O-(2-glyceryl)-coniferaldehyde with nuclear factor NF-kappa-B, mitogen-activated protein kinase 1 and cyclin-dependent kinase 2 for 100 ns, respectively
The binding poses of the protein-ligand complexes at the end of 100 ns were analyzed to examine interaction changes [35]. As shown in (Fig. 11A), 4-cadinene-3,11-diol; 3-(2-methylpropanoyl) and the androgen receptor molecular docking pose was stabilized by hydrogen bonding with Thr877(4.05 Å), and Gln711(4.15 Å) and Met745 (3.47 Å), and continued hydrophobic interactions with Leu701, Asn705, Gly708, Trp741, Met742, Val746, Met749, Met780, Met787 and Phe876. Menwhile, 4-cadinene-3,11-diol;3-(2-methylpropanoyl) and P53-binding protein complex, as shown in (Fig. 11B), was stabilized by hydrophobic interactions with Leu50, Thr51, Met58, Gly54, Ile57, Leu62, Thr63, Gln68, His69, Ile70, Val71, Val89, His92 and Ile95. The hydroogen bond between the hydroxyl group of 4-cadinene-3,11-diol; 3-(2-methylpropanoyl) and Thr341 of cyclooxygenase-2 remained stable, and hydrophobic interactions with Val102, Ser339, Phe367, Met508 and Ser516 has been preserved as shown in (Fig. 11C).

Protein-ligan interactions at the end of 100 ns molecular dynamics simulation. (A-B-C) Androgen receptor, P53-binding and cyclooxygenase-2 with 4-cadinene-3,11-diol; 3-(2-methylpropanoyl) and (D-E-F) Nuclear factor NF-kappa-B, mitogen-activated protein kinase 1, and binding poses of cyclin-dependent kinase 2 with (2-glyceryl)-O-coniferaldehyde, respectively
Regarding the other compound (2-glyceryl)-O-coniferaldehyde and the target nuclear factor NF-kappa-B, the hydrogen bond with Glu210 and Gln304 remained stable at the 100th ns, as shown in (Fig. 11D), and hydrophobic interactions with Ala215, Met301, Ile305, Phe307, Val308 continued. Metal-acceptor interaction between (2-glyceryl)-O-coniferaldehyde and mitogen-activated protein kinase 1 with Mg++ (2.12 Å and 2.10 Å), hydrogen bond with Ser194 (3.97 Å) and Met146 (4.24 Å) interactions are preserved as shown in (Fig. 11E). As given (Fig. 11F), the metal-acceptor interaction with Mg++ (1.99 Å and 2.13 Å) and the hydrogen bond interaction with Gln131 remained stable between the other target cyclin-dependent kinase 2 and (2-glyceryl)-O-coniferaldehyde, as in the molecular docking pose. Finally, to examine the stability states of these 6 protein-ligand complexes, molecular dynamic animation videos were created from 250 frames recorded for 100 ns and are given in Supporting Information (Video S1-6) [36]. According to post-molecular dynamics simulation trajectory analyses, the compounds 4-cadinene-3,11-diol; 3-(2-methylpropanoyl) and (2-glyceryl)-O-coniferaldehyde have the potential to form stable interactions with selected target proteins.
Assessment of the in-vitro cytotoxicity activity of A.annua extract
Cytotoxicity of A.annua on healthy (Vero E6) cells
The effect of A.annua extract on the viability of Vero E6 cells was evaluated using MTT assay. It showed 50% cytotoxicity (CC50) at concentration 38.25 μg/mL (i.e. more than 30 μg/mL) indicating their safety on these cells [37] (Fig. 12a).
In vitro anticancer activity of A.annua extract on human prostate (PC-3), breast (MDA-MB-231), pancreatic (PANC-1) and melanoma (A375) cancer cell lines
Artemisia extract was subjected to in vitro anticancer activity testing on human prostate (PC-3), breast (MDA-MB-231), pancreatic (PANC-1) and melanoma (A375) cancer cell lines. The results shown in (Fig. 12b-e) indicated variable potency of artemisia against the studied cancer types, where it showed high potency against human prostate (PC-3) cell lines with an IC50 value of 30.13 μg/mL, followed by breast (MDA-MB-231), pancreatic (PANC-1) and melanoma (A375) cancer cell lines scoring IC50 values 86.21, 64.40 and 34.89 μg/mL respectively. These values were moderately competing to those of doxorubicin (IC50 values 6.4, 4.1, 43 and 0.5 μg/mL, respectively). Results also showed concordance with the literature survey where it was suggested that artesunate – a semi-synthetic artemisinin derivative - suppressed tumor growth of prostatic cancer cells (PC-3) via modulating AR-DNMT3b pathway [38]. In the same context, artemisinin exhibited anti-proliferative effects in prostate cancer cells through transcriptional down-regulation of CDK4 expression by disruption of Sp1 interactions with the CDK4 promoter [39]. Other study confirmed that Artemisia annua extract exerted a concentration-dependent effect on breast (MCF-7) cancer cells [40], however, its isolated flavonol Chrysosplenol d activated ERK1/2, and induced autophagy in MDA-MB-231 (breast cancer) cells [41]. The total polyphenolic fraction inhibited the proliferation of the same cell line either through suppression of vascular cell adhesion molecule-1 [42] or stem Cell Phenotype, β-Catenin, and MMP-9 [6]. Significant inhibition of melanoma and breast cancer cells by some other constituents including, artemisinin, scopoletin, 1,8-Cineole, arteanuine B, artemisitene was also reported [43].

Dose response curve of the cytotoxic activity of A.annua extract on healthy cells (Vero E6) (a), human prostate (PC-3) (b), breast (MDA-MB-231) (c), pancreatic (PANC-1) (d) and melanoma (A375) (e) cell lines
KEGG pathway analysis of human prostate (ID: hsa05215) (Figure S2), breast (ID: hsa05224) (Figure S3), melanoma (ID: hsa05212) (Figure S4) and pancreatic cancer (ID: hsa05218) (Figure S5) illustrated the potential targets and pathways of A. annua chemical constituents where the orange ovals indicated the targets where the molecules interacted and the red rectangles indicated the targeted pathways [44,45,46].
Materials and methods
Network pharmacology analysis
Collection of Artemisia annua compounds and corresponding targets
Dictionary of natural products database (http://dnp.chemnetbase.com/, CRC press) is a platform containing many compounds from herbal medicine [47]. It was used to collect the potential active eighty components of Artemisia annua (table S3). Protein targets interacting with those potential active compounds were anticipated by importing the molecular structure files into Swiss Target Prediction (http://www.swisstargetprediction.ch/) databases. Probability score was used to evaluate the interaction strength of the compound to each target.
Sorting cancer-related target genes
By searching for “cancer” in the GeneCards database (https://www.genecards.org), targets related to cancer were identified and a disease-target database was constructed.
Screening common drug and disease targets
The online Venny 2.1.0 program (https://bioinfogp.cnb.csic.es/tools/venny/) was employed to identify intersecting Artemisia annua and cancer targets, through which, Venn diagrams were drafted.
Pathway enrichment analysis and protein-protein interaction (PPI) network
Kyoto Encyclopedia of Genes and Genomes (KEGG) and protein-protein interaction (PPI) data were compiled and retrieved from String (https://string-db.org/), using the species “homo sapiens”. A minimum required interaction score to the highest confidence level (0.900) was set aiming to generate protein-protein interaction information.
Construction of the component-target-pathway network
Two networks namely, compound-gene-network and Gene-pathway-network were constructed via Cytoscape 3.7.2 software, displaying the active components, target genes, and pathways as nodes and the inter-node relationships as edges.
KEGG pathway and gene ontology (GO) enrichment analysis
GO enrichment analysis is an important computational tool used to provide a logical framework of gene function [48]. It can effectively recognize the biological processes, cellular components and molecular functions related to a certain ailment and obtain more profound information about a list of genes [49]. KEGG pathway analysis and GO Enrichment analysis was conducted using functional annotation tool in Annotation, Visualization, and Integrated Discovery (DAVID) (https://david.ncifcrf.gov/) where ‘’ homo sapiens’’ species were selected. Prior to enrichment, genes names were converted into corresponding UniProt accession numbers. GO terms possessing p-values < 0.05 were considered statistically significant.
Molecular docking analysis
The Protein Data Bank (PDB) was used for retrieving the crystal structure of the most enriched six targets revealed from network pharmacology analysis, which are Nuclear factor NF-kappa-B p105 subunit (1LV2), Mitogen-activated protein kinase 1 (4AN9), Androgen receptor (2PIW), P53-binding protein (4IPF), Cyclin-dependent kinase 2 (1B39) and Cyclooxygenase-2 (3LN1). The selection of crystal structure of each protein was relied on best resolution available. The 3D structures of top-five compounds reveled from network pharmacology were imported as SDF into the LigPrep module of Maestro 10.2 molecular modeling (Schrodinger®) software package to obtain low energy structures of compounds. Ionization at pH 7 was performed to produce all possible states. High-resolution crystal structures of protein targets were retrieved from Protein Data Bank (PDB). Protein preparation was accomplished using protein preparation module of Maestro where they were preprocessed by assigning bond orders and hydrogens in addition to removing all the water molecules beyond 5 °A from the active site. Assignment of H-bonds was performed via PROPKA at PH = 7, then energy minimization using OPLS 3 force field was performed till the relative mean standard deviation (RMSD) of the minimized structure compared to the crystal structure was above 0.30 °A [50]. Receptor grid generation module using boxes enclosing the centroids of co-crystallized ligands were set as the grids. Molecular docking analysis was achieved using Glide docking program of Maestro molecular modeling package implementing extra-precision (XP-Glide) mode. The binding modes of the compounds with targets were visualized using Maestro interface.
Molecular dynamics simulations
The stability of the hit protein-ligand complexes was tested by molecular dynamics simulations performed with Gromacs v2021.2 [51]. System preparations such as solvation and neutralization of protein-ligand complexes obtained by molecular docking for molecular dynamics simulation and creation of topology files were done with the Solution Builder tool in the CHARMM-GUI server. Amber FF99SB [52, 53] was preferred for topology files. A temperature of 300 K and a pressure of 1 atm was equilibrated by the Nose-Hoover thermostat and Parrinello-Rahman barostat methods for 0.3 ns, and 100 ns molecular dynamics simulation was performed to 2 fs under PBC. Post molecular dynamic trajectory analysis with root mean square deviation (RMSD) and root mean square fluctuation (RMSF) gmx rms script, analysis and visualization of protein-ligand binding poses at 100 ns with [54] UCSF ChimeraX v1.4, and creation of animation videos with PyMOL Molecular Graphics System v2.4.1 were performed.
In vitro anticancer activity screening of A. annua
Preparation of A. annua extract
Aerial parts of Artemisia annua (1.5 kg) were collected from a cultivated area in Cairo. The plant material was identified by Mrs. Teresa Labib, Taxonomist at the El-Orman Botanical Garden, Giza. Our study complies with relevant institutional, national, and international guidelines and legislation. A voucher sample was kept at the Pharmacognosy Department, Faculty of pharmacy, Alexandria University. The plant was carefully air-dried to obtain 500 g that were extracted with 70% ethanol (10 g/100 ml) twice at room temperature (27.50 C) by sonication for two hours. The extract was filtered and concentrated to dryness using the rotary evaporator at 40oC under vacuum then kept at 4oC before use.
Cytotoxicity of A. annua extract on healthy (Vero E6) cells
The dilution of the tested extract was done with Dulbecco’s Modified Eagle’s Medium (DMEM). The stock solutions of the tested extract were prepared in 10% DMSO in double distilled H2O. MTT method with minor modifications was used to evaluate the cytotoxic effects in Vero E6 cells (Green monkeys kidney cells) [55, 56]. In 96 well plates, Vero E6 cells were seeded (100 μl/well at a density of 3 × 105 cells/ml) followed by their incubation for 24 h at 37 oC in 5% CO2. After 24 h, treatment of cells with various concentrations of the tested extract in triplicates was done followed by their incubation for another 24 h. After that, the supernatant layer was discarded, and washing of cell monolayers with sterile PBS 3 times was carried out. To each well, MTT solution (20 μl of 5 mg/ml stock solution) was added followed by incubation at 37 °C for 4 h then medium aspiration. The formed formazan crystals were dissolved with 200 μl of acidified isopropanol (0.04 M HCl in absolute isopropanol, 0.073 ml HCl in 50 ml isopropanol). Finally, a multi-well plate reader was used to measure the absorbance of formazan solutions at λmax 540 nm using 620 nm as a reference wavelength. The percentage of cell cytotoxicity 50 compared to the untreated cells was calculated with the following equation:
A0 is the absorbance of cells without treatment.
A is the absorbance of cells with treatment.
The concentration which exhibited 50% cytotoxicity (CC50) was determined by plotting % cytotoxicity versus sample concentration.
In vitro sulforhodamine B (SRB) cytotoxicity assay
Human prostate (PC-3), breast (MDA-MB-231), pancreatic (PANC-1) and melanoma (A375) cancer cells were separately preserved in Dulbecco’s Modified Eagle Media (DMEM) supplied with 100 units per mL of penicillin, 100 mg/mL of streptomycin, and 10% of heat-inactivated fetal bovine serum in humidified, 5% (v/v) CO2 atmosphere at 37 °C. Sulforhodamine B (SRB) assay was applied for assessment of cell viability [57, 58] Aliquots of 100 mL cell suspension (5 × 103 cells) from each cell line were separately placed in 96-well plates and incubated for 24 h in complete media. Then treatment of the cells with another aliquot of 100 mL media containing A.annua at 0.001, 0.01, 0.1, 1, 10, 100 mg/mL concentrations was carried out. After 72 h exposure to drug, cells were fixed by exchanging media with 150 mL of 10% trichloroacetic acid (TCA) and then the cells were incubated at 4 °C for 1 h. The TCA solution was removed, and distilled water was used for washing the cells 5 times. Aliquots of 70 mL SRB solution (0.4% w/v) were added and incubated in a dark place at room temperature for 10 min. Washing the plates 3 times with 1% acetic acid was carried out, then the plates were allowed to air-dry overnight. After that, protein-bound SRB stain was dissolved using 150 mL of TRIS (10 mM); finally, the measurement of the absorbance was performed at 540 nm by the use of a BMG LABTECH® –FLUO star Omega microplate reader (Ortenberg, Germany). The whole experiment has been conducted in triplicate for each tested concentration to attain reproducibility. Doxorubicin was used as a reference anti-cancer drug.
Conclusion
Owing to the multi-chemical components of the plant extracts, their multi-pharmacological effects as well as their multi-action targets in the treatment of different diseases, network pharmacology had combined bioinformatics and pharmacology organically to clarify and explain the multi-mechanisms of action of these plants via emphasizing the relationship between targets, components and diseases. In this study, the network pharmacology-based analysis of A. annua showed that the hit phytoconstituents related to cancer targets were 3-(2-methylpropanoyl)-4-cadinene-3,11-diol, artemisinin G, O-(2-propenal) coniferaldehyde, (2-glyceryl)-O-coniferaldehyde and arteamisinin III, whereas the main cancer allied targets were NFKB1, MAP2K1, AR, MDM2, CDK2 and PTGS2. Sixty-eight significant signaling KEGG pathways were recognized where the most enriched ones were prostate cancer, breast cancer, melanoma and pancreatic cancer. Among hit compounds, molecular docking and dynamic simulation studies revealed that 4-cadinene-3,11-diol; 3-(2-methylpropanoyl) had the lowest XP G score against androgen receptor, P53-binding protein and cyclooxygenase-2. Whereas (2-glyceryl)-O-coniferaldehyde exhibited the most stabilized interaction with nuclear factor NF-kappa-B, mitogen-activated protein kinase and cyclin-dependent kinase 2. This study represents a thorough explanation of the proposed mechanism of action of A. annua phytoconstituents in cancer and suggests this natural product as a potential source for cancer prevention or treatment. making it worthy of clinical application and promotion. However, only part of the specific mechanism of action has been clinically verified and further extensive in vivo and clinical studies are required to confirm the anti-cancer potential of the concluded top hit natural compounds.
Data Availability
All data generated or analyzed during this study are included in this article (and its supplementary information files).
References
Sitarek P, Merecz-Sadowska A, Śliwiński T, Zajdel R, Kowalczyk T. An in vitro evaluation of the molecular mechanisms of action of medical plants from the Lamiaceae family as effective sources of active compounds against human cancer cell lines. Cancers (Basel). 2020;12:2957.
Ali S, et al. Amomum subulatum: a treasure trove of anti-cancer compounds targeting TP53 protein using in vitro and in silico techniques. Front Chem. 2023;11:1174363
Ahmad HM, et al. Characterization of fenugreek and its natural compounds targeting AKT-1 protein in cancer: Pharmacophore, virtual screening, and MD simulation techniques. J King Saud Univ - Sci. 2022;34:102186.
Umar HI, et al. Discovery of novel HSP27 inhibitors as prospective anti-cancer agents utilizing computer-assisted therapeutic discovery approaches. Cells. 2022;11:2412.
Taghizadeh Rabe SZ, Mahmoudi M, Ahi A, Emami SA. Antiproliferative effects of extracts from Iranian Artemisia species on cancer cell lines. Pharm Biol. 2011;49:962–9.
Ko YS, et al. Polyphenols extracted from Artemisia annua L. exhibit anti-cancer effects on radio-resistant MDA-MB-231 human breast cancer cells by suppressing stem cell phenotype, β-Catenin, and MMP-9. Molecules. 2020;25:1916.
Konstat-Korzenny E, Ascencio-Aragón JA, Niezen-Lugo S, Vázquez-López R. Artemisinin and its synthetic derivatives as a possible therapy for cancer. Med Sci. 2018;6:19.
van Loggenberg S, et al. Evaluating in vitro cytotoxic effects of Artemisia afra and Artemisia annua infusions against selected lung cancer cell lines. South Afr J Bot. 2022;150:404–11.
Alesaeidi S, Miraj SA. Systematic review of anti-malarial Properties, Immunosuppressive properties, anti-inflammatory properties, and anti-cancer properties of Artemisia Annua. electron physician. 2016;8:3150–5.
Lang SJ, et al. Antitumor activity of an Artemisia annua herbal preparation and identification of active ingredients. Phytomedicine. 2019;62:152962.
Rafique Q, et al. Reviewing methods of deep learning for diagnosing COVID-19, its variants and synergistic medicine combinations. Comput Biol Med. 2023;163:107191.
Tang Y, et al. Network pharmacology-based predictions of active components and pharmacological mechanisms of Artemisia annua L. for the treatment of the novel Corona virus disease 2019 (COVID-19). BMC Complement Med Ther. 2022;22:56.
Ali MK, et al. Exploring the multifunctional roles of quantum dots for unlocking the future of biology and medicine. Environ Res. 2023;232:116290.
Mazhar T, et al. The role of machine learning and deep learning approaches for the detection of skin cancer. Healthcare. 2023;11(3):415.
Zhang S, Mo Z, Zhang S, Li XA. Network pharmacology approach to reveal the underlying mechanisms of Artemisia annua on the treatment of hepatocellular carcinoma. Evidence-Based complement. Altern. Med 2021:8947304.
Gao J, Xu HL, Gao S, Zhang W, Tan YT, Rothman N, et al. Genetic polymorphism of NFKB1 and NFKBIAgenes and liver cancer risk: a nested case–control study in Shanghai, China. BMJ Open. 2014;4(2):e004427.
Li D, Wu C, Cai Y, Liu B. Association of NFKB1 and NFKBIA gene polymorphisms with susceptibility of gastric cancer. Tumor Biol. 2017;39:1010428317717107.
Kim G-C, et al. Upregulation of Ets1 expression by NFATc2 and NFKB1/RELA promotes breast cancer cell invasiveness. Oncogenesis. 2018;7:1–15.
You J, et al. MiR-449a suppresses cell invasion by inhibiting MAP2K1 in non-small cell lung cancer. Am J Cancer Res. 2015;5:2730–44.
Chuang J, et al. MAP2K1 mutations in Advanced colorectal cancer predict poor response to anti-EGFR therapy and to vertical targeting of MAPK pathway. Clin Colorectal Cancer. 2021;20:72–8.
Bu R, et al. Recurrent somatic MAP2K1 mutations in papillary thyroid cancer and colorectal cancer. Front Oncol. 2021;11:670.
Madden KS, Szpunar MJ, Brown EB. β-Adrenergic receptors (β-AR) regulate VEGF and IL-6 production by divergent pathways in high β-AR-expressing breast cancer cell lines. Breast Cancer Res Treat. 2011;130:747–58.
Knudsen KE, Penning TM. Partners in crime: deregulation of AR activity and androgen synthesis in prostate cancer. Trends Endocrinol Metab. 2010;21:315–24.
Urso L, Calabrese F, Favaretto A, Conte P, Pasello G. Critical review about MDM2 in cancer: possible role in malignant mesothelioma and implications for treatment. Crit Rev Oncol Hematol. 2016;97:220–30.
Shaikh MF, et al. Emerging role of MDM2 as target for anti-cancer therapy: a review. Ann Clin Lab Sci. 2016;46:627–34.
An J, et al. Mechanical stimuli-driven cancer therapeutics. Chem Soc Rev. 2023;52:30–46.
Dasgupta I, McCollum D. Control of cellular responses to mechanical cues through YAP/TAZ regulation. J Biol Chem. 2019;294:17693–706.
Liu Y-N, et al. Regulatory mechanisms controlling human E-cadherin gene expression. Oncogene. 2005;24:8277–90.
Ge A, Liu L, Deng X, Luo J, Xu Y. Exploring the mechanism of baicalin intervention in breast cancer based on MicroRNA microarrays and bioinformatics strategies. Evidence-Based complement. Altern. Med 2021:7624415.
Lu X, et al. Bioinformatics analysis of KIF1A expression and gene regulation network in ovarian carcinoma. Int J Gen Med. 2021:3707–17.
Celik I, Tallei TE. A computational comparative analysis of the binding mechanism of molnupiravir’s active metabolite to RNA-dependent RNA polymerase of wild‐type and Delta subvariant AY. 4 of SARS‐CoV‐2. J Cell Biochem. 2022;123:807–18.
Khan MA, et al. Anti-quorum sensing, antibiofilm, and antibacterial activities of extracts of Centella asiatica L. leaves, and in vitro derived leaves-calli through tissue culture: a potential for biofouling-prevention. Biofouling. 2022;38:715–28.
Tabti K, et al. Profiling the structural determinants of pyrrolidine derivative as gelatinases (MMP-2 and MMP-9) inhibitors using in silico approaches. Comput Biol Chem. 2023;104:107855.
Paşayeva L, Fatullayev H, Celik I, Unal G, Bozkurt NM, Tugay O, Abdellattif MH. Evaluation of the chemical composition, antioxidant and antidiabetic activity of Rhaponticoides iconiensis flowers: effects on key enzymes linked to type 2 diabetes in vitro, in silico and on alloxan-induced diabetic rats in vivo. Antioxidants. 2022;11:2284.
Yadav R, et al. Identification and in-vitro analysis of potential proteasome inhibitors targeting PSMβ5 for multiple myeloma. Biomed Pharmacother. 2023;157:113963.
Bender O, et al. Discovery of oxindole-based FLT3 inhibitors as a promising therapeutic lead for acute myeloid leukemia carrying the oncogenic ITD mutation. Arch Pharm (Weinheim). 2022;356(2):e2200407.
Darwish RS, et al. Efficacy-directed discrimination of the essential oils of three Juniperus species based on their in-vitro antimicrobial and anti-inflammatory activities. Ethnopharmacology. 2020;259:1–12.
Wang Z, et al. Artesunate suppresses the growth of prostatic cancer cells through inhibiting androgen receptor. Biol Pharm Bull. 2017;40:479–85.
Willoughby JAS, et al. Artemisinin blocks prostate cancer growth and cell cycle progression by disrupting Sp1 interactions with the cyclin-dependent kinase-4 (CDK4) promoter and inhibiting CDK4 gene expression. J Biol Chem. 2009;284:2203–13.
Worku N, et al. Evaluation of the in vitro efficacy of Artemisia annua, Rumex abyssinicus, and Catha edulis Forsk extracts in cancer and Trypanosoma brucei cells. Int Sch Res Not. 2013;2013.
Lang SJ, et al. Chrysosplenol d, a flavonol from Artemisia annua, induces ERK1/2-Mediated apoptosis in triple negative human breast cancer cells. Int J Mol Sci. 2020;21(11):4090.
Ko YS, et al. Polyphenols from Artemisia annua L inhibit adhesion and EMT of highly metastatic breast cancer cells MDA-MB‐231. Phyther Res. 2016;30:1180–8.
Efferth T, Herrmann F, Tahrani A, Wink M. Cytotoxic activity of secondary metabolites derived from Artemisia annua L. towards cancer cells in comparison to its designated active constituent artemisinin. Phytomedicine. 2011;18:959–69.
Kanehisa M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019;28:1947–51.
Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30.
Kanehisa M, Furumichi M, Sato Y, Kawashima M, Ishiguro-Watanabe. M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023;51:D587–92.
Kibble M, et al. Network pharmacology applications to map the unexplored target space and therapeutic potential of natural products. Nat Prod Rep. 2015;32:1249–66.
Qiu Z-K, et al. A network pharmacology study with molecular docking to investigate the possibility of licorice against posttraumatic stress disorder. Metab Brain Dis. 2021;36:1763–77.
Chen G, et al. A Network Pharmacology Approach to Uncover the Potential Mechanism of Yinchensini decoction. Evidence-Based complement. Altern. Med 2018:2178610.
Ibrahim RS, et al. Biologically guided isolation and ADMET profile of new factor Xa inhibitors from Glycyrrhiza glabra roots using in vitro and in silico approaches. RSC Adv. 2021;11:9995–10001.
Abraham MJ, et al. High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1. 2015;GROMACS:19–25.
Jo S, Kim T, Iyer VG, Im W. CHARMM-GUI: a web‐based graphical user interface for CHARMM. J Comput Chem. 2008;29:1859–65.
Lee J, et al. CHARMM-GUI supports the Amber force fields. J Chem Phys. 2020;153:35103.
Pettersen EF, et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 2021;30:70–82.
Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63.
Darwish RS, et al. Chemical profiling and unraveling of anti-COVID-19 biomarkers of red sage (Lantana camara L.) cultivars using UPLC-MS/MS coupled to chemometric analysis, in vitro study and molecular docking. J Ethnopharmacol. 2022;291:115038.
Skehan P, et al. New colorimetric cytotoxicity assay for anticancer-drug screening. JNCI J Natl Cancer Inst. 1990;82:1107–12.
Ibrahim RS, El-Banna AA. Network pharmacology-based analysis for unraveling potential cancer-related molecular targets of egyptian propolis phytoconstituents accompanied with molecular docking and in vitro studies. RSC Adv. 2021;11:11610–26.
Acknowledgements
The numerical calculations reported in this paper were partially performed at TUBITAK ULAKBIM, High Performance and Grid Computing Center (TRUBA resources).
Funding
The authors declare that this study was self-funded.
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
H.D. and R.S.I.: data acquisition, analysis and interpretation, drafting the work conceptualization of work, experimental design and revision of the manuscript. I.C.: conducting and writing the molecular dynamic simulation.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The aerial parts of A. annua (Family Asteraceae) were collected from a cultivated area in Giza in May 2022 with license approval from the Faculty of Pharmacy, Cairo University, Egypt according to relevant guidelines and regulations. The plant material was kindly identifed by Mrs. Teresa Labib, Taxonomist at the El-Orman Botanical Garden, Giza, Egypt. A voucher sample was deposited in the Herbarium of the Faculty of pharmacy, Alexandria University, Alexandria, Egypt.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Material 1: Figure S1
: 3D interaction diagrams between the top six target protein crystal structures with top hit ligands together with the co-crystallized ligands for molecular docking validation.
Supplementary Material 2: Figure S2
: KEGG pathway analysis of human prostate cancer (ID: hsa05215) illustrating the potential targets and pathways of A. annua chemical constituents where the orange ovals indicated the targets where the molecules interacted and the red rectangles indicated the targeted pathways.
Supplementary Material 3: Figure S3
: KEGG pathway analysis of human breast cancer (ID: hsa05224) illustrating the potential targets and pathways of A. annua chemical constituents where the orange ovals indicated the targets where the molecules interacted and the red rectangles indicated the targeted pathways.
Supplementary Material 4: Figure S4
: KEGG pathway analysis of human melanoma (ID: hsa05212) illustrating the potential targets and pathways of A. annua chemical constituents where the orange ovals indicated the targets where the molecules interacted and the red rectangles indicated the targeted pathways.
Supplementary Material 5: Figure S5
: KEGG pathway analysis of human pancreatic cancer (ID: hsa05218) illustrating the potential targets and pathways of A. annua chemical constituents where the orange ovals indicated the targets where the molecules interacted and the red rectangles indicated the targeted pathways.
Supplementary Material 6: Video S1
: MD Simulation animation of 250 snapshots between 0 and 100 ns of androgen receptor with 4-cadinene-3,11-diol; 3-(2-methylpropanoyl) (PDB ID: 2PIW). Video S2. MD Simulation animation of 250 snapshots between 0 and 100 ns of P53-binding protein with 4-cadinene-3,11-diol; 3-(2-methylpropanoyl) (PDB ID: 4IPF). Video S3. MD Simulation animation of 250 snapshots between 0 and 100 ns of cyclooxygenase-2 with 4-cadinene-3,11-diol; 3-(2-methylpropanoyl) (PDB ID: 3NL1). Video S4. MD Simulation animation of 250 snapshots between 0 and 100 ns of NF-kappa-B with O-(2-Glyceryl)-coniferaldehyde (PDB ID: 1LV2). Video S5. MD Simulation animation of 250 snapshots between 0 and 100 ns of mitogen-activated protein kinase 1 with O-(2-Glyceryl)-coniferaldehyde (PDB ID: 4AN9). Video S6. MD Simulation animation of 250 snapshots between 0 and 100 ns of cyclin-dependent kinase 2 with O-(2-Glyceryl)-coniferaldehyde (PDB ID: 1B39).
Supplementary Material 7: Table S1
: Cancer-related target genes retrieved from Genecards database.
Supplementary Material 8: Table S2
: Protein-Protein interactions retrieved from String database.
Supplementary Material 9: Table S3
: Database of A. annua active constituents and their SMILES.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Dawood, H., Celik, I. & Ibrahim, R.S. Computational biology and in vitro studies for anticipating cancer-related molecular targets of sweet wormwood (Artemisia annua). BMC Complement Med Ther 23, 312 (2023). https://doi.org/10.1186/s12906-023-04135-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12906-023-04135-0