
Abstract
Background
Non-alcoholic fatty liver disease (NAFLD) has been well defined as a common chronic liver metabolism disorder. Statins as a first-line therapeutic treatment had some side effects. Here, we found that Fumigaclavine C (FC) was collected from endophytic Aspergillus terreus via the root of Rhizophora stylosa (Rhizophoraceae), had potential anti-adipogenic and hepatoprotective effects both in vitro and in vivo without obvious adverse side effects. However, the mechanisms of the prevention and management of FC for hepatic steatosis are incompletely delineated.
Methods
The pharmacodynamic effects of FC were measured in high-fat diet (HFD)-induced obese mice. Liver index and blood biochemical were examined. Histopathological examination in the liver was performed by hematoxylin & eosin or oil red O. The levels of serum TG, TC, LDL-c, HDL-c, FFA, T-bili, ALT, AST, creatinine, and creatine kinase were estimated via diagnostic assay kits. The levels of hepatic lipid metabolism-related genes were detected via qRT-PCR. The expression levels of hepatic de novo lipogenesis were quantitated with Western blot analysis.
Results
FC-treatment markedly reduced hepatic lipid accumulation in HFD-induced obese mice. FC significantly attenuated the hepatic lipid metabolism and ameliorated liver injury without obvious adverse side effects. Moreover, FC also could dose-dependently modulate the expressions of lipid metabolism-related transcription genes. Mechanically, FC notably suppressed sterol response element binding protein-1c mediated de novo lipogenesis via interfering with the RhoA/ROCK signaling pathway by decreasing the levels of geranylgeranyl diphosphate and farnesyl diphosphate.
Conclusions
These findings suggested that FC could improve hepatic steatosis through inhibiting de novo lipogenesis via modulating the RhoA/ROCK signaling pathway.
Similar content being viewed by others

Background
Non-alcoholic fatty liver disease (NAFLD) has been well defined as a chronic liver metabolism disorder and its prevalence is a serious and increasing clinical problem worldwide, closely related to obesity, diabetes, hyperlipidemia, and hypertension [1]. It contributes to various liver pathologies which range from nonalcoholic simple steatosis, nonalcoholic steatohepatitis (NASH), cirrhosis, to liver cancer [2]. Of these types, simple hepatic steatosis status could be reversed through effective treatment and prevented it from progressing to some more severe stages [3]. Hence, it is an allimportant step in the prevention of hepatic steatosis via ameliorating lipid accumulation and blocking the hepatic lipid metabolism-related genes expressions [4].
Until now, various alternative NAFLD therapeutic strategies including lifestyle intervention and pharmaceutical treatment are either insufficient or have side effects [5, 6]. Although lifestyle intervention is an impactful way to ameliorate NAFLD, prolonged prevention works in half [7, 8]. Currently, some candidates containing natural products and chemical compounds are used for clinical treatment of NAFLD. For instance, statins, HMG-CoA reductase inhibitor, had been used for management of NAFLD-associated hypercholesterolemia [9, 10]. Nevertheless, several safety concerns have been reported for statins. Long-term statins treatment had side effects including liver injury and myopathy [11, 12]. Several articles have reported that statins deplete cholesterol, geranylgeranyl diphosphate (GGPP), and farnesyl diphosphate (FPP) at the beginning of mevalonate pathway [13, 14]. GGPP and FPP are used for the prenylation of proteins at their carboxyl-terminal CAAX motif, like RhoA and Ras [15]. But, various issues remain unresolved elucidating the precise mechanisms of treatment chemical drugs for NAFLD [16, 17]. To overcome above issues, emerging researches have focused on natural products as a selectable effective therapeutic strategy without significant adverse side effects [18, 19].
Teas have long been considered as a dietary supplementation [20]. Rhizophora stylosa (Rhizophoraceae) has been considered as a common edible plant whose roots and leaves were used as an ingredient for Chinese herbal tea [21]. As shown in Fig. 1, fumigaclavine C (FC) which is an indole alkaloid, is collected from endophytic Aspergillus terreus (strain No. FC118) via the root of Rhizophora stylosa (Rhizophoraceae). It has various health beneficial effects, including anti-tumor [22], anti-atherosclerosis [23], anti-inflammation [24,25,26,27], anti-adipogenic [28], immunosuppressive activity [29], and hepatoprotective activity [30]. Our previous paper indicated that FC had potential anti-adipogenic effect in obese animal model [28]. However, the precise molecular mechanisms of the adipogenesis and lipolysis of FC are incompletely delineated. Herein, the aim of this study was to elucidate the effectivity of FC in the prevention and management of NAFLD both in vitro and in vivo without obvious adverse side effects.

The chemical structure of fumigaclavine C
Methods
Materials
FC (≥ 99.5%) was collected from endophytic Aspergillus terreus (strain No. FC118) via the root of Rhizophora stylosa (Rhizophoraceae). The anti-RhoA, anti-prenyl, anti-ROCK, anti-sterol response element binding protein-1c (Srebp-1c), anti-α-tubulin antibodies, and secondary antibody were purchased from Cell Signaling Technology (Danvers, MA, USA). Oleic acid, palmitic acid, and simvastatin (Sim) was purchased from Sigma Aldrich Company (St. Louis, MO, USA). Total cholesterol (TC), Triglyceride (TG), low density lipoprotein-cholesterol (LDL-c), high density lipoprotein-cholesterol (HDL-c), free fatty acid (FFA), total bilirubin (T-bili), alanine transaminase (ALT), aspartate transaminase (AST), creatinine, creatine kinase, and Cell Counting Kit-8 (CCK-8) were measured by diagnostic assay kits from Nanjing Jiancheng Company (NJ, China).
Experimental animals design
All of male C57BL/6 mice were purchased from SJA Laboratory Animal Co., Ltd. (Hunan, China). The four-week-old mice were acclimatized to the experimental facility for one week. The mice were either fed with regular chow (RC, 3.6 kcal/g, 10% fat, 14% protein, and 76% carbohydrates) or high fat diet (HFD, 5.5 kcal/g, 50% fat, 14% protein, and 36% carbohydrates [Shanghai FBSH Biological Pharmaceutical Co., Ltd., Shanghai, China]) for ten weeks to induce NAFLD steatosis model. Experiments with animals were performed following the animal ethics guidelines of the Institutional Animal Ethics Committee. The mice were equally and randomly divided into six groups (ten mice / group): control group (only DMSO), HFD-induced group (only DMSO), HFD-10 mg FC (10 mg/kg of body weight), HFD-20 mg FC, HFD-40 mg FC, and HFD-40 mg Sim. Sim and FC were dissolved into DMSO (final concentration ≤ 5%). FC was administrated via intraperitoneal injection. The injection was performed three times per week for ten weeks. Experimental animals procedures were approved as follow Fig. 2 A [31]. All experimental procedures were approved by the Institutional Animal Care and Use Committee of Liuzhou General Hospital (Protocol No. 1019–3).

FC improves hepatic steatosis in high-fat diet (HFD)-induced obese mice. A The time course of body weight within HFD-induced and FC administration. B The liver weight was examined. C The liver/body weight ratio was calculated. D-F The levels of hepatic TC, TG, and LDL-c were detected. TC, total cholesterol; TG, Triglyceride; LDL-c, low density lipoprotein-cholesterol. Simvastatin (Sim, 40 mg/kg) acted as the positive control group. Each value represents as means ± S.E.M. of triplicate experiments. # P < 0.01 as compared with the control group. * P < 0.05, ** P < 0.01, and *** P < 0.001 as compared with the HFD-induced group
Cell culture and stimulation
AML-12 cells were purchased from American Type Culture Collection (ATCC, Rockville, MD, USA). The cells were cultured in DMEM/F12 medium containing fetal bovine saline (FBS, 10%), penicillin (100 U/mL), and streptomycin (100 μm/mL) at 37 °C with 5% CO2 [32]. Cell viability was evaluated via CCK-8 kit following the manufactures’ instructions. Hepatocytes were stimulated with 500 mM free fatty acids (FFA, oleic acid: palmitic acid = 2:1) for 48 h. Sim and FC were dissolved into DMSO (final concentration ≤ 0.1%). FC was administered with indicated concentrations for 24 h.
Biochemical data measurement
Body weight data were collected twice a week. Hepatocytes/hepatic TC, TG and LDL-c were respectively examined via assay kits following the manufactures’ instructions. The blood samples were collected from orbital venous plexus and centrifuged at 4000 rpm for 15 min at 4 °C. The levels of serum TG, TC, LDL-c, HDL-c, FFA, T-bili, ALT, AST, creatinine, and creatine kinase were respectively measured via diagnostic assay kits following the manufactures’ instructions.
Histopathological analysis
Liver tissues were fixed in 10% formalin saline, embedded in paraffin, and processed following routine histology procedures. Tissue samples (6–7 μm) were stained with hematoxylin & eosin (H & E) or oil red O according to standard protocol by histology, and observed via light microscope (Nikon, Tokyo, Japan).
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was extracted with TRIzol reagent and reversed transcribed with Prime ScriptTM. RT reagent following the manufactures’ protocol (Invitrogen, Carlsbad, CA, USA). Quantitative real-time PCR was conducted using the Quanti Fast SYBR Green PCR Kit (Qiagen, Valencia, CA, USA). The list of PCR primers was shown in Table 1. All data were normalized by β-actin and performed in triplicate.
Western blot analysis
Hepatocytes/hepatic protein extracts were respectively collected according to the manufactures’ protocol (Protein Extraction Kit, Pierce Biotechnology Inc., Nepean, Canada). Protein samples were quantified via using an Enhanced Bicinchoninic Acid Protein Assay Kit (Beyotime, Jiangsu, China). The harvested protein extracts were separated by SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, MA). The PVDF membranes were blocked with 5% non-fat milk and incubated with anti-RhoA, anti-prenyl, anti-ROCK, anti-Srebp-1c, and anti-α-tubulin antibodies at 4°C overnight. The PVDF membranes were washed and exposed to secondary antibodies [33]. Finally, the blots were normalized via β-actin and quantitated with LI-COR Odyssey Analysis software.
Protein prenylation examination
The levels of GGPP and FPP were examined according to previously reported protocol by HPLC–MS/MS in AML-12 cell [34]. To evaluate RhoA prenylation, hepatocyte subcellular fractionation was collected following a Triton X-114 partition method [35]. Briefly, the cells were directly lysed in 2% Triton X-114 on ice for 30 min. To fractionate the lipid-rich cell membrane, 500 μg protein samples were partitioned with 0.1% Triton X-114 at 37°C for 10 min. Subcellular phases were immunoprecipitated with anti-RhoA and anti-prenyl, then with protein A/G coupled to agarose beads. After a thorough wash, cell samples were measured to immunoblotting analysis.
Statistical analysis
Results were represented as means ± S.E.M. or a percentage of triplicate tests. All data were performed via one-way ANOVA followed by the Newman-Keuls test. All statistical analyses were performed via SPSS 21.0 (IBM Co., Armonk, NY, USA). Differences of P < 0.05 (*) were considered statistical significant. P < 0.01 (# or **) and P < 0.001 (***) were considered statistically very significant.
Results
FC improves liver steatosis in HFD-induced obese mice
Five-week-old C57BL/6 mice were fed with HFD diet for ten weeks the lipid data like the body weight (Fig. 2A), the liver weight (Fig. 2B), the liver/body weight ratio (Fig. 2C), hepatic TC (Fig. 2D), hepatic TG (Fig. 2E), and hepatic LDL-c (Fig. 2F) were significantly elevated. Thereafter the mice were treated with FC via intraperitoneal injection three times per week for ten weeks. As shown in Fig. 2A, significant differences in body weight and body weight gain were found between the control group and HFD-induced group (P < 0.01). Moreover, treatment with FC significantly reduced the body weight gain of the HFD-induced groups (Fig. 2B) (P < 0.05). The liver /body weight ratios of the HFD-induced group and the FC-treatment groups were significantly higher than that of the control group (Fig. 2C) (P < 0.05). The hepatic TC, TG, and LDL-c levels were also notably reduced in FC-treatment groups (Fig. 2D-F) (P < 0.05). The livers of HFD-induced mice revealed obvious lipid accumulation. Many vacuoles lipid droplets were examined by H & E staining (Fig. 3A, arrows). Great deals of oil red positive areas were detected by Oil-Red Staining (Fig. 3B, arrows). Nevertheless, FC could alleviate lipid deposition. Effect of HFD-20 mg FC could be comparable to positive reference HFD-40 mg Sim. These results demonstrated that FC improved liver steatosis in HFD-induced mice.

FC ameliorates the hepatic steatosis in HFD-induced obese mice. Histopathological analysis was exhibited via hematoxylin and eosin (H&E) staining A, and Oil-Red Stating B. HFD-induced liver steatosis obese mice showed abundant vacuoles lipid droplets in H & E staining (A, arrows) and oil red positive area in Oil-Red Staining (B, arrows). Simvastatin (Sim, 40 mg/kg) acted as the positive control group
FC ameliorates liver injury without significant adverse side effects
Since acute liver injury and myopathy were the most adverse side effects of statins administration reported previously [11, 12]. Thus, we measured some serum lipid profiles. As shown in Fig. 4A-E, the serum TG, TC, LDL-c, HDL-c, and FFA levels were all significantly elevated in HFD-induced group (P < 0.01). However, FC could dose-dependently attenuate these hepatic lipid metabolisms (P < 0.05). FC-treatment which reduced the levels of serum T-bili, ALT, and AST could markedly ameliorate liver injury (Fig. 4F-H) (P < 0.05). Meanwhile, prolonged statins administration had significant adverse side effects. The creatinine and creatine kinase levels were elevated in HFD-40 mg Sim group (P < 0.001). As shown in Fig. 4I-J, there was no difference of two parameters in FC-treatment groups. These data indicated that FC ameliorated liver injury without significant adverse side effects in HFD-induced mice.

FC ameliorates liver injury without significant adverse side effects in HFD-induced obese mice. A-E The levels of serum TG A, TC B, LDL-c C, HDL-c D, and FFA E were measured. F–H Liver injury was evaluated via serum T-bili F, ALT G, and AST H levels. Adverse side effects of FC were examined via serum creatinine I and creatine kinase J. TG, Triglyceride; TC, total cholesterol; LDL-c, low density lipoprotein-cholesterol; HDL-c, high density lipoprotein-cholesterol; FFA, free fatty acid; T-bili, total bilirubin; ALT, alanine transaminase; AST, aspartate transaminase. Sim (40 mg/kg) acted as the positive control group. Each value represents as means ± S.E.M. of triplicate experiments. # P < 0.01 as compared with the control group. * P < 0.05, ** P < 0.01, and *** P < 0.001 as compared with the HFD-induced group
FC alleviates liver steatosis via attenuating de novo lipogenesis
Substantial evidences have revealed that the level of hepatic TG depended on de novo lipogenesis, β-oxidation, FFA uptake, and very low density lipoprotein (VLDL) export [36, 37]. To evaluate which the most possible pathway involved in FC-treatment, we determined the hepatic lipid metabolism-related genes expressions involved in these four stages. Interestingly, FC could markedly and dose-dependently inhibit the de novo lipogenesis related mRNA expressions such as sterol response element binding protein-1c (Srebp-1c), peroxisome proliferator activated receptor-γ (Ppar-γ), fatty acid synthase (Fas), and Acetyl-CoA (Acc) (Fig. 5A) (P < 0.05). Furthermore, FC reduced the β-oxidation related mRNA expressions like carnitine palmitoyltransferase 1 (Cpt1), peroxisome proliferator activated receptor-α (Ppar-α), and medium chain acyl dehydrogenase (Mcad) (Fig. 5B) (P < 0.05). Moreover, FC attenuated the FFA uptake related mRNA expressions including fatty acid translocase (Fat/CD36), fatty acid binding protein 1 (Fabp1), and fatty acid transport protein 1 (Fatp1) (Fig. 5C) (P < 0.05). In addition, FC decreased the VLDL related mRNA expressions like Apo lipoprotein B (ApoB), Apo lipoprotein E (ApoE), and microsomal triglyceride transfer protein (Mttp) (Fig. 5D) (P < 0.05). As shown in Fig. 5E, FC-treatment could reverse gradually hepatic FFA content of HFD-induced mice (P < 0.05). These results concluded that FC could dose-dependently modulate the expressions of lipid metabolism-related transcription genes via attenuating hepatic de novo lipogenesis and FFA uptake.

Effects of FC-treatment on the hepatic lipid metabolism-related genes expressions in HFD-induced obese mice. Hepatic lipid metabolism-related genes expressions, such as DNL A, β-oxidation B, FFA uptake C and VLDL export D were examined. E The level of hepatic FFA was measured. DNL, de novo lipogenesis; FFA, free fatty acid; VLDL, very low density lipoprotein; Srebp-1c, sterol response element binding protein-1c; Ppar, peroxisome proliferator activated receptor; Fas, fatty acid synthase; Acc, Acetyl-CoA; Cpt1, carnitine palmitoyltransferase 1; Mcad, medium chain acyl dehydrogenase; CD36, fatty acid translocase; Fabp1, fatty acid binding protein 1; Fatp1, fatty acid transport protein 1; ApoB, Apo lipoprotein B; ApoE, Apo lipoprotein E; Mttp, microsomal triglyceride transfer protein. Sim (40 mg/kg) acted as the positive control group. Each value represents as means ± S.E.M. of triplicate experiments. # P < 0.01 as compared with the control group. * P < 0.05, ** P < 0.01, and *** P < 0.001 as compared with the HFD-induced group
FC inhibits Srebp-1c mediated de novo lipogenesis through modulating of the RhoA/ROCK signaling pathway
Some previous researches have reported that de novo lipogenesis was controlled via activating the expressions of Srebp-1c [38, 39]. To investigate the mechanism of which FC could modulate Srebp-1c-related pathways, we further evaluated Srebp-1c-related kinases. As shown in Fig. 6A, the tested concentrations of FC showed no obvious cytotoxicity in AML-12 cells. FC could remarkably and dose-dependently reduce the TG level in AML-12 cells (Fig. 6B) (P < 0.05). Moreover, the levels of GGPP and FPP were decreased after FC-treatment in AML-12 cells (Fig. 6C) (P < 0.01). Furthermore, FC notably suppressed the maturation of Srebp-1c. The RhoA prenylation and the expression of ROCK, which is a downstream effector of RhoA, were also attenuated by FC exposure (Fig. 6D-E) (P < 0.05). Together these data suggested that FC inhibited Srebp-1c mediated de novo lipogenesis via interfering with the RhoA/ROCK signaling pathway by decreasing the levels of GGPP and FPP.

FC suppresses Srebp-1c mediated hepatic DNL by modulating the RhoA/ROCK signaling pathway in FFA-induced AML-12 cell. A FC-treatment on AML-12 cell viability. B The level of hepatocellular TG was examined. C The levels of FPP and GGPP were measured by HPLC–MS/MS. D, E Western blotting technique was used to evaluate the expressions of prenyl-RhoA, ROCK, and Srebp-1c. FFA, free fatty acid; TG, Triglyceride; FPP, farnesyl diphosphonate; GGPP, geranylgeranyl diphosphonate; Srebp-1c, sterol response element binding protein-1c. Sim (20 μM) acted as the positive control group. Each value represents as means ± S.E.M. of triplicate experiments. # P < 0.01 as compared with the control group. * P < 0.05, ** P < 0.01, and *** P < 0.001 as compared with the FFA-induced group
Discussion
Currently, various alternative NAFLD therapeutic strategies including lifestyle intervention and pharmaceutical treatment are either insufficient or have side effects [5, 6]. Emerging researches have focused on natural products as a selectable effective therapeutic strategy without significant adverse side effects [18, 19]. In the present study, we investigated the potential NAFLD therapeutic effect of FC. Our data demonstrated that FC could ameliorate hepatic steatosis and alter GGPP and FPP levels both in vitro and in vivo without obvious adverse side effects. The mechanisms were involved in inhibiting de novo lipogenesis, altering the levels of GGPP and FPP, and modulating the RhoA/ROCK signaling pathway.
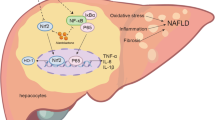
Substantial evidences have revealed that the level of hepatic TG depended on de novo lipogenesis, β-oxidation, FFA uptake, and VLDL export [36, 37]. Thus, we tried to elucidate which the possible pathway FC took part in attenuating hepatic lipid accumulation. First of all, FC could markedly and dose-dependently inhibit the mRNA expressions of de novo lipogenesis, particularly in HFD-40 mg FC group. As FC-treatment notably suppressed the levels of hepatic TG and FFA, it is reasonable to unravel that the hepatic TG reduction cause by attenuating de novo lipogenesis. Secondly, de novo lipogenesis contributed to twenty-six percent of TG deposition, FFA uptake accounted for fifty-nine percent in NAFLD [36]. Some studies have reported that normal subjects were one-third of rates of de novo fatty acid synthesis compared with NAFLD subjects [37]. The livers of HFD-induced mice revealed obvious lipid accumulation [40]. Nevertheless, FC could alleviate lipid deposition [41, 42]. Hence, we next assessed whether FC reduced hepatic lipid accumulation via interfering with de novo lipogenesis. Thirdly, some previous researches have reported that de novo lipogenesis was controlled via activating the expressions of Srebp-1c [38, 39]. To investigate the mechanism of which FC could modulate Srebp-1c-related pathways, we further evaluated Srebp-1c-related kinases. Srebp-1c was suggested to be cleaved to be maturated under the RhoA/ROCK signaling pathway [13]. Especially, Rho need to undergo prenylation to be activated [14]. In this study, the levels of GGPP and FPP were decreased after FC-treatment in AML-12 cells. This might be due to the reduction of the maturation of Srebp-1c via blocking the RhoA prenylation and the expression of ROCK (Fig. 7). Together these data suggested that FC inhibited Srebp-1c mediated de novo lipogenesis via interfering with the RhoA/ROCK signaling pathway by decreasing the levels of GGPP and FPP.

Scheme of the molecular mechanisms of the hepatoprotective effects of FC
Until now, great administrations for NAFLD have been always limited [16, 17]. Statins, HMG-CoA reductase inhibitor, had been usually used for management of NAFLD-associated hypercholesterolemia [9, 10]. However, long-term statins treatment had side effects including hepatotoxicity and myopathy [11, 12], due to depletion of cholesterol, GGPP, and FPP at the beginning of mevalonate pathway [13, 14]. In this study, some results demonstrated that FC could not only ameliorate liver steatosis, but also do not worsen, as well as improve obesity-induced liver injury in HFD -induced obese mice. Possible adverse side effects of FC were not reported, such as myalgia, fatigue, hepatotoxicity, and fever [21,22,23,24,25,26,27,28,29,30]. Interestingly, FC has scarcely obvious adverse side effects. There was no difference in the levels of serum ALT, AST, T-bili, creatinine, and creatine kinase in FC-treatment groups. These data indicated that FC ameliorated liver injury without significant adverse side effects in HFD-induced mice.
Furthermore, there are still many limitations in this study. Besides RhoA-prenylation, other small G proteins (such as Rab and Rac1) and G-γ subunit of GTPase proteins also need to go through prenylation and subsequent membrane-association to be activated [13,14,15]. Hence, it is very important to further research the potentiality of FC on other G-γ subunit of GTPase proteins and small G proteins. This research only provides a potential way to ameliorate liver steatosis with FC-treatment, while the precise molecular mechanisms remain to be elucidated.
Conclusion
This study elucidated that FC could ameliorate hepatic steatosis and alter GGPP and FPP levels both in vitro and in vivo without obvious adverse side effects (Fig. 7). The mechanisms were involved in inhibiting de novo lipogenesis, altering the levels of GGPP and FPP, and modulating the RhoA/ROCK signaling pathway.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- NAFLD:
-
Non-alcoholic fatty liver disease
- NASH:
-
Nonalcoholic steatohepatitis
- FC:
-
Fumigaclavine C
- HFD:
-
High-fat diet
- RC:
-
Regular chow
- GGPP:
-
Geranylgeranyl diphosphate
- FPP:
-
Farnesyl diphosphate
- Sim:
-
Simvastatin
- Srebp-1c:
-
Sterol response element binding protein-1c
- TC:
-
Total cholesterol
- TG:
-
Triglyceride
- LDL-c:
-
Low density lipoprotein-cholesterol
- HDL-c:
-
High density lipoprotein-cholesterol
- FFA:
-
Free fatty acid
- T-bili:
-
Total bilirubin
- ALT:
-
Alanine transaminase
- AST:
-
Aspartate transaminase
- CCK-8:
-
Cell Counting Kit-8
- DMSO:
-
Dimethyl Sulphoxide
- H & E:
-
Hematoxylin & eosin
- PVDF:
-
Polyvinylidene difluoride
- DNL:
-
De novo lipogenesis
- VLDL:
-
Very low density lipoprotein
- Ppar:
-
Peroxisome proliferator activated receptor
- Fas:
-
Fatty acid synthase
- Acc:
-
Acetyl-CoA
- Cpt1:
-
Carnitine palmitoyltransferase 1
- Mcad:
-
Medium chain acyl dehydrogenase
- CD36:
-
Fatty acid translocase
- Fabp1:
-
Fatty acid binding protein 1
- Fatp1:
-
Fatty acid transport protein 1
- ApoB:
-
Apo lipoprotein B
- ApoE:
-
Apo lipoprotein E
- Mttp:
-
Microsomal triglyceride transfer protein
References
Gao Z, Song GY, Ren LP, Ma HJ, Ma BQ, Chen SC. β-catenin mediates the effect of GLP-1 receptor agonist on ameliorating hepatic steatosis induced by high fructose diet. Eur J Histochem. 2020;64:225–33.
Tao WW, Wu J, Xie BX, Zhao YY, Shen N, Jiang S, Wang XX, Xu N, Jiang C, Chen S, Gao X, Xue B, Li CJ. Lipid-induced muscle Insulin resistance is mediated by GGPPS via modulation of the RhoA/Rho kinase signaling pathway. J Biol Chem. 2015;33:20086–97.
Rahman MS, Kang I, Lee Y, Habib MA, Choi BJ, Kang JS, Park DS, Kim YS. Bifidobacterium longum subsp infantis YB0411. inhibits adipogenesis in 3T3-L1 Pre-adipocytes and reduces high-fat-diet-induced obesity in mice. J Agric Food Chem. 2021;69:6032–42.
Tang QL, Jiang S, Jia WJ, Shen D, Qiu YD, Zhao Y, Xue B, Li CJ. Zoledronic acid, an FPPS inhibitor, ameliorates liver steatosis through inhibiting hepatic de novo lipogenesis. Eur J Pharmacol. 2017;814:169–77.
Lee MR, Kim JE, Park JW, Kang MJ, Choi HJ, Bae SJ, Choi YW, Kim KM, Hong JT, Hwang DY. Fermented mulberry (Morus alba) leaves suppress high fat diet-induced hepatic steatosis through amelioration of the inflammatory response and autophagy pathway. BMC Complementary Med Ther. 2020;20:283.
Yao LY, Sun JY, Liang Y, Feng T, Wang HT, Sun M, Yu WG. Volatile fingerprints of Torreya grandis hydrosols under different downstream processes using HS-GC–IMS and the enhanced stability and bioactivity of hydrosols by high pressure homogenization. Food Control. 2022;139:109058.
Cui HP, Lin YX, Xie L, Zhao J. Urantide decreases hepatic steatosis in rats with experimental atherosclerosis via the MAPK/Erk/JNK pathway. Mol Med Rep. 2021;23:284.
Ai S, Qin Y, Hong YX, Liu LY, Yu WG. Removal of C3–C4 diols in ethylene glycol via selective dehydration reactions over Beta zeolite with acidity tailored. J Catal. 2022;413:870–9.
Li WJ, Ren YP, Meng TY, Yang W, Zhang W. miR-129-5p attenuates hypoxia-induced apoptosis in rat H9c2 cardiomyocytes by activating autophagy. J Gene Med. 2020;22:e3200.
Feng D, Zou J, Su DF, Mai HY, Zhang SS, Li PY, Zheng XM. Curcumin prevents high-fat diet-induced hepatic steatosis in ApoE-/- mice by improving intestinal barrier function and reducing endotoxin and liver TLR4/NF-κB inflammation. Nutr Metab. 2019;16:79.
Zhou C, Zhen MM, Yu ML, Li X, Yu T, Liu JC, Jia W, Liu S, Li L, Li J, Sun ZH, Zhao ZP, Wang XY, Zhang XY, Wang CR, Bai CL. Gadofullerene inhibits the degradation of apolipoprotein B100 and boosts triglyceride transport for reversing hepatic steatosis. Sci Adv. 2020;6:e1586.
Yang Z, Li P, Shang Q, Wang Y, He J, Ge S, Jia R, Fan X. CRISPR-mediated BMP9 ablation promotes liver steatosis via the down-regulation of PPARα expression. Sci Adv. 2020;6:e5022.
Tsai CC, Chen YJ, Yu HR, Huang LT, Tain YL, Lin IC, Sheen JM, Wang PW, Tiao MM. Long term N-acetylcysteine administration rescues liver steatosis via endoplasmic reticulum stress with unfolded protein response in mice. Lipids Health Dis. 2020;19:105.
Xu H, Chen GF, Ma YS, Zhang HW, Zhou Y, Liu GH, Chen DY, Ping J, Liu YH, Mou X, Fu D. Hepatic proteomic changes and Sirt1/AMPK signaling activation by oxymatrine treatment in rats with non-alcoholic steatosis. Front Pharmacol. 2020;11:216.
Okuyama T, Shirakawa J, Tajima K, Ino Y, Vethe H, Togashi Y, Kyohara M, Inoue R, Miyashita D, Li JH, Goto N, Ichikawa T, Yamasaki S, Ohnuma H, Takayanagi R, Kimura Y, Hirano H, Terauchi Y. Linagliptin ameliorates hepatic steatosis via non-canonical mechanisms in mice treated with a dual inhibitor of insulin receptor and IGF-1 receptor. Int J Mol Sci. 2020;21:7815.
Li L, Yang L, Yang F, Zhao XI, Xue SJ, Gong FH. Ginkgo biloba extract 50 (GBE50) ameliorates insulin resistance, hepatic steatosis and liver injury in high fat diet-fed mice. J Inflammation Res. 2021;14:1959–71.
Ai S, Qin Y, Liu LH, Yu WG. Ion-exchange modification of zeolite for the controllable catalytic transformation of diols and selective conversion of 1,2-butanediol to butanal. Compr Mater Chem Phys. 2022;292:126836.
Nakade Y, Kitano R, Yamauchi T, Kimoto S, Sakamoto K, Inoue T, Kobayashi Y, Ohashi T, Sumida Y, Ito K, Yoneda M. Effect of adrenergic agonists on high-fat diet-induced hepatic steatosis in mice. Int J Mol Sci. 2020;21:9392.
Wollman A, Daniel T, Rosenzweig T. Sarcopoterium spinosum inhibited the development of non-alcoholic steatosis and steatohepatitis in mice. Nutrients. 2019;11:3044.
Wang S, Huang Y, Xu H, Zhu Q, Lu H, Zhang M, Hao S, Fang C, Zhang D, Wu X, Wang X, Sheng J. Oxidized tea polyphenols prevent lipid accumulation in liver and visceral white adipose tissue in rats. Eur J Nutr. 2017;56:2037–48.
Wang Y, Zhu HW, Tam NFY. Polyphenols, tannins and antioxidant activities of eight truemangrove plant species in South China. Plant Soil. 2014;374:549–63.
Guo WJ, Hu SS, Elgehama A, Shao F, Ren R, Liu W, Zhang WJ, Wang XL, Tan RX, Xu Q, Sun Y, Jiao RH. Fumigaclavine C ameliorates dextran sulfate sodium-induced murine experimental colitis via NLRP3 inflammasome inhibition. J Pharmacol Sci. 2015;129:101–6.
Zhao Y, Liu JY, Wang J, Wang L, Yin H, Tan RX, Xu Q. Fumigaclavine C improves concanavalin A-induced liver injury in mice mainly via inhibiting TNF-α production and lymphocyte adhesion to extracellular matrices. J Pharm Pharmacol. 2004;56:775–82.
Tan Y, Wu XX, Sun J, Guo WJ, Gong FY, Shao FL, Tan T, Cao Y, Zheng BF, Gu YH, Sun Y, Xu Q. A fumigaclavine C isostere alleviates Th1-mediated experimental colitis via competing with IFN-γ for binding to IFN-γ receptor 1. Biochem Pharmacol. 2017;123:63–72.
Yu WG, Qian J, Lu YH. Hepatoprotective Effects 2′,4′-dihydroxy-6′- methoxy-3′,5′-dimethylchalcone on CCl4-induced acute liver injury in mice. J Agric Food Chem. 2011;59:12821–9.
Yu WG, He H, Qian J, Lu YH. Dual role of 2′,4′-dihydroxy-6′- methoxy-3′,5′-dimethylchalcone in inhibiting high-mobility group box 1 secretion and blocking its Pro-inflammatory activity in hepatic inflammation. J Agric Food Chem. 2014;62:11949–56.
Yu WG, He H, Yao JY, Zhu YX, Lu YH. Dimethyl cardamonin exhibits anti-inflammatory effects via interfering with the PI3K-PDK1-PKCα signaling pathway. Biomol Ther. 2015;23:549–56.
Yu WG, He Y, Chen YF, Gao XY, Ning WE, Liu CY, Tang TF, Liu Q, Huang XC. Fumigaclavine C attenuates adipogenesis in 3T3-L1 adipocytes and ameliorates lipid accumulation in high-fat diet-induced obese mice. Korean J Physiol Pharmacol. 2019;23:161–9.
Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Investig. 2005;115:1343–51.
Yu WG, Pan ZH, Zhu YX, An FL, Lu YH. Fumigaclavine C exhibits anti-inflammatory effects by suppressing high mobility group box protein 1 relocation and release. Eur J Pharmacol. 2017;812:234–42.
Ye Q, Liu Y, Zhang GJ, Deng HJ, Wang XJ, Tuo L, Chen C, Pan XM, Wu K, Fan JG, Pan Q, Wang K, Huang AL, Tang N. Deficiency of gluconeogenic enzyme PCK1 promotes metabolic-associated fatty liver disease through PI3K/AKT/PDGF axis activation in male mice. Nat Commun. 2023;14:1402.
Mukerjee N, Das A, Jawarkar RD, Maitra S, Das P, Castrosanto MA, Paul S, Samad A, Zaki MEA, Al-Hussain SA, Masand VH, Hasan MM, Bukhari SNA, Perveen A, Alghamdi BS, Alexiou A, Kamal MA, Dey A, Malik S, Bakal RL, Abuzenadah AM, Ghosh A, Ashraf GM. Repurposing food molecules as a potential BACE1 inhibitor for Alzheimer’s disease. Front Aging Neurosci. 2022;14:878276.
An SH, Vo TTL, Son T, Choi H, Kim J, Lee J, Kim BH, Choe M, Ha E, Surh YJ, Kim KW, Seo JH. SAMHD1-induced endosomal FAK signaling promotes human renal clear cell carcinoma metastasis by activating Rac1-mediated lamellipodia protrusion. Exp Mol Med. 2023;55:779–93.
Wang XX, Ying P, Diao F, Wang Q, Ye D, Jiang C, Shen N, Xu N, Chen WB, Lai SS, Jiang S, Miao XL, Feng J, Tao WW, Zhao NW, Yao B, Xu ZP, Sun HX, Li JM, Sha JH, Huang XX, Shi QH, Tang H, Gao X, Li CJ. Altered protein prenylation in Sertoli cells is associated with adult infertility resulting from childhood mumps infection. J Exp Med. 2013;210:1559–74.
Goalstone ML, Leitner JW, Golovchenko I, Stjernholm MR, Cormont M, Marchand-Brustel YL, Draznin B. Insulin promotes phosphorylation and activation of geranylgeranyltransferase II. J Biol Chem. 1999;29:2880–4.
Du RH, Li EG, Cao Y, Song YC, Tan RX. Fumigaclavine C inhibits tumor necrosis factor α production via suppression of toll-like receptor 4 and nuclear factor κB activation in macrophages. Life Sci. 2011;89:235–40.
Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. 2014;146:726–35.
Horton JD, Shimomura I. Sterol regulatory element-binding proteins: activators of cholesterol and fatty acid biosynthesis. Curr Opin Lipidol. 1999;10:143–50.
Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Investig. 2002;109:1125–31.
Brown GT, Kleiner DE. Histopathology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Metabolism. 2016;65:1080–6.
Khan S, Choudhary S, Kumar A, Tripathi AM, Alok A, Adhikari JS, Rizvi MA, Chaudhury NK. Evaluation of sesamol-induced histopathological, biochemical, haematological and genomic alteration after acute oral toxicity in female C57BL/6 mice. Toxicol Rep. 2016;3:880–94.
Elizabeth M, Brunt MD. Histological assessment of nonalcoholic fatty liver disease in adults and children. Clin Liver Dis. 2012;4:108–11.
Acknowledgements
Not applicable.
Funding
This study was supported by the Guangxi Scientific Base and Talent Special Project (AD19110052 and AD22080013), National Natural Science Foundation of China (32060205 and 22268011), the Innovation Project of Guangxi Graduate Education (YCSW2021326), the Open Fund of Key Laboratory for Processing of Sugar Resources of Guangxi Higher Education Institutes (GXTZY201903), and the Special Fund for Guangxi University of Science and Technology (No.19Z05).
Author information
Authors and Affiliations
Contributions
W.G.Y. and S.A. participated in every step of the trial, including design of study, acquisition of data, analysis of data, and drafting the manuscript. Y.X.G., Z.Y.Z., and X.F.L. participated in acquisition of data. Y.Y. was in charge of the procurement of materials.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All experimental animals’ procedures were approved by the Institutional Animal Care and Use Committee of Liuzhou General Hospital (Protocol No. 1019–3). The ARRIVE guidelines were adhered to throughout this study. All methods were performed in accordance with the relevant guidelines and regulations.
Consent for publication
Not applicable.
Competing interests
All authors declared that have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yu, W., Gao, Y., Zhao, Z. et al. Fumigaclavine C ameliorates liver steatosis by attenuating hepatic de novo lipogenesis via modulation of the RhoA/ROCK signaling pathway. BMC Complement Med Ther 23, 288 (2023). https://doi.org/10.1186/s12906-023-04110-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12906-023-04110-9