
Abstract
Background
Pearl millet (PM), i.e., Pennisetum glaucum, is widely grown in Africa and known for its anti-oxidant and anti-hyperlipidemic properties.
Methods
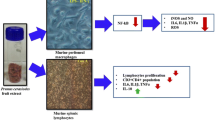
The P. glaucumgrains were obtained from the region of Ouled Aïssa (South of Algeria). We assessed the effects of phenolic compounds and lipids, extracted from seeds of P. glaucum, on rat lymphocyte proliferation, activated by phorbol 12-myristate 13-acetate and ionomycin. In order to explore signaling pathway, triggered by these compounds, we assessed interleukin-2 (IL-2) mRNA expression and extracellular signal-regulated kinase-1/2 (ERK1/ERK2) phosphorylation. Finally, we determined increases in free intracellular Ca2+ concentrations, [Ca2+]i, by employing Fura-2/AM in rat lymphocytes.
Results
The composition of P. glaucum grains in polyphenols was estimated to be 1660 µg gallic acid equivalents (GAE)/g. Lipids represented 4.5 %, and more than 72% of the fatty acids belonged to unsaturated family. Our investigation showed that both lipid and phenolic compounds inhibited mitogen-induced T-cell proliferation. Compared with phenolic compounds, lipids exerted weaker effects on ERK-1/ERK2 phosphorylation and Ca2+ signaling in mitogen-activated T-cells.
Conclusion
We conclude that the immunomodulatory effects of P. glaucum could be contributed by its phenolic and lipid contents.
Similar content being viewed by others


Background
Current recommendations from international health and nutritional organizations, like Food and Drug Administration (FDA), include an increase in the consumption of high-bran cereals because of their potential benefits on human health [1]. Millets are cereals that have been cultivated for more than 3500 years in all Sahelian Africa and tropical countries of Western Africa. Indeed, millets are extremely resistant to dryness and well adapted to manure-poor soil [2]. Millet refers to a number of different species, and all of them are small-grained and annual cereal grasses [3].
Millet grains have been shown to exert beneficial effects in health and disease [4–6]. Lee et al. [4] reported that foxtail and proso millet decreased plasma triglycerides in hyperlipidemic rats. Shobana et al. [5] reported that feeding a diet containing 20 % finger millet decreased hyperglycemia and its associated complications in streptozotocin-induced diabetic rats. In clinical studies, finger millet exerted anti-hyperglycemic effects in diabetic patients [6]. The beneficial effects of pearl millet (PM) have not been well studied except a few available reports that have shown anti-oxidant activity because of its high contents in polyphenols [7]. Luteolin, a flavone present in millets, has been reported to exert antioxidant, anti-inflammatory and cancer-preventive properties [8, 9]. Van Rensburg [10] and Chen et al. [11] reported that populations consuming cereals including millet had lower incidences of esophageal cancer compared to those consuming wheat or maize. Nani et al. [12] have shown that streptozotocine (STZ)-induced diabetic Wistar rats fed with pearl millet-enriched diet underwent a significant curtailment in glycaemia and an improvement of body weight. Regarding its chemical composition, pearl millet has been attributed to having several health promoting abilities: anemia, constipation, cancer, and diabetes [13].
As far as immune system is concerned, cereal consumption has been reported to exert immune-modulating activities. Recent studies have shown that cereals contain a wide range of phenolic compounds [14–17]. The immunomodulatory effects of polyphenols have drawn considerable attention in recent years [18–20]. González et al. [21] have reported that flavonoids and related polyphenolic compounds possess anti-inflammatory activity. Furthermore, the mechanisms of action of other polyphenols, e.g., resveratrol, curcumin, genistein and epigallocatechin, in the modulation of immune system and the secretion of pro-inflammatory mediators have been reviewed [22]. In addition to dietary polyphenols, a great attention has been paid to dietary lipids which are able to modulate inflammatory status, depending on their fatty acid content and composition [23–25]. In vitro and in vivo studies have shown that fatty acids modulate a number of lymphocyte functions [25], including proliferation [26], cytokine release [27], and mitogenic signaling [28].
Pearl millet (PM), i.e., Pennisetum glaucum, is the most widely grown species in Africa [29], and it constitutes the daily basic food for 50 million inhabitants of the Sahel [2]. PM contains several compounds including lipids and polyphenols that could modulate immune system. To our knowledge, no study concerning the effects of PM polyphenolic or lipidic fractions on the modulation of immune system is available. We, therefore, investigated the effect of phenolic compounds and lipids, extracted from PM, on T-cell activation. Since an increase in free intracellular Ca2+ concentration, [Ca2+]i and mitogen-activated protein kinase (MAPK) phosphorylation, are the part of early events of T-cell activation, it was thought worthwhile to elucidate the effect of PM lipids and phenolic extracts on Ca2+ and MAPK signaling in T-cells.
Methods
Materials
Grains of pearl millet, Pennisetum glaucum, obtained from the region of Ouled Aïssa (174 km in the North of Adrar city and 70 km to the North-West of Timimoun, Algeria), were used in this study. Wistar rats were obtained from Janvier Elevage (Le Genest-st-isle, France). RPMI 1640 medium and L-glutamine were purchased from Lonza Verviers SPRL (Verviers, Belgium). Fura-2 AM was procured from Life Technologies (France). Anti-Phospho-p44/42 mitogen-activated protein kinase (MAPK, Erk1/2) and anti-p44/42 MAPK (Erk1/2) were obtained from Cell Signaling (France). All other chemicals were purchased from Sigma (USA).
The general guidelines for the care and use of laboratory animals, recommended by the council of European Economic Communities, were followed. The experimental protocol was approved by the Regional Ethical Committee (Dijon).
Extraction and determination of phenolic compounds
The plant was recognized by a botanist (Pr Benabadji Nouri, Université Aboubekr Belkaïd, Tlemcen) of the Herbarium Center of the Faculty of Pharmacy (Tlemcen) which contained the voucher specimen (PM 1681). PM phenolic extracts were obtained according to the method of Liyana-Pathirana and Shahidi [30] with slight modifications. Briefly, 2 g of PM grain powder were extracted two times (2 h for each extraction) with 40 ml of methanol–acetone–water (7:7:6, v/v/v) at room temperature (25 ± 2 °C) with constant stirring. The mixtures were centrifuged (20 min at 4000 g) and supernatants were collected and subjected to extraction with an equal volume of hexane to eliminate lipids.
Total phenolic contents in plant extracts were determined by Folin-Ciocalteu method [31] as described by Miliauskas et al. [32]. Briefly, 0.5 ml polyphenol extract was reacted with 2.5 ml of Folin-Ciocalteu reagent (0.2 mol/l) for 4 min, then 2 ml saturated sodium carbonate solution (75 g/l) was added into the reaction mixture. After 2 h incubation at room temperature, the absorbance at 760 nm was determined. The content of phenolic compounds was determined with reference to standard curve determined with gallic acid. The content of phenolic compounds was expressed as μg gallic acid equivalents (GAE)/g dry matter (μg GAE/g).
Phenolic extract (20 μl) was analyzed by HPLC (Model Agilent Technologies 1260, Germany) with reverse phase Zorbax Eclipse XDB-C18 column (4.6 × 100 mm) and a diode array UV-detector (operating at 280 nm). The gradient mobile phase was composed of two solvents: A and B. Solvent A was methanol and solvent B was 0.1 % formic acid (v/v). Phenolic acid separation was achieved using a 35 min linear solvent gradient at a flow rate of 0.4 ml/min, as follows: 0 min 90 % B, 5 min 80 % B, 10 min 70 % B, 15 min 50 % B, 20 min 30 % B, 25 min 10 % B, 30 min 50 % B, 35 min 90 % B. PM phenolic compounds were identified with reference to retention time of authentic standards and quantified on the basis of their peak area. Standard phenolic compounds used were: p-coumaric, chlorogenic, ferulic, gallic, trans-hydroxycinnamic, syringic acid, ellagic acids, quercetin and apigenin.
Lipid extraction
Lipids were extracted according to the method of Hara and Radin [33]. In order to avoid oxidation, all solvents contained 0.01 % butylated hydroxytoluene (BHT). Briefly, 1 g of millet grain powder plus 20 μg of internal standard (C19:0) were blended with 8 ml isopropanol and heated at 80 °C for 5 min. After cooling to room temperature, the blended grain powder was crushed in 12 ml hexane. The mixture was briefly centrifuged, and then the upper phase was collected. For complete recovery, the pellet was re-extracted with 9 ml hexane and 2 ml isopropanol, and then the extract was combined with the upper phase of the previous step. To remove non-lipid fraction, the extract was partitioned into an upper hexane phase by the addition of aqueous sodium sulphate 6.5 % (0.5/1 : v/v). The upper phase, lipid extract, was transferred into a new tube, dried under a stream of nitrogen and stored at −20 °C until fatty acid analysis by gas–liquid chromatography (GLC).
Preparation of fatty acid methyl esters (FAMEs)
In brief, 0.1 ml of hexane, containing lipid extract, was transferred into screw cap tubes and dried under nitrogen, then 1 ml of methanolic NaOH (0.5 N) was added into tubes, and heated at 80 °C during 20 min. After cooling at 4 °C, 2 ml of boron trifluoride-methanol solution (BF3) were added, and the methylation was performed at 80 °C for 20 min. After cooling-down in ice, 2 ml of NaCl (35 %) plus 2 ml of hexane were added into the tubes. After vigorous agitation and centrifugation (1200 g/5 min), the upper phase containing fatty acid methyl esters was transferred into new tubes, and analyzed by gas − liquid chromatography (GLC).
Gas liquid chromatography (GLC)
GLC was performed in a Packard Model 417 gas–liquid chromatograph, equipped with a flame ionization detector and a 30-m capillary gas column coated with carbowax 20 M. The analysis conditions were as follows: oven temperature was 85 °C/1 min, increased to 150 °C at 30 °C/min, then increased at 4 °C/min to 210 °C. Helium was used as carrier gas, with a flow rate of 0.4 ml/min. Analysis of fatty acid peaks was achieved with reference to the internal standards (Nu-Chek-Prep, Elysian, MN) by using DELSI ENICA 31 (Delsi Nermag, Rungis, France). The fatty acid levels were expressed as g/100 g of total fatty acids.
Isolation and preparation of splenic T-cells
Fresh splenocytes were harvested from Wistar rat spleens under aseptic conditions. The removed spleens were immediately transferred to the petri dishes, containing RPMI-1640 complete medium (RPMI 1640 medium supplemented with 10 % foetal calf serum, 2 mM L-glutamine, 100 U/ml of penicillin, and 100 μg/ml of streptomycin and 25 mM HEPES) . Spleens were teased apart using a wire gauge. After lysis of red blood cells, with Red Cell Lysing Buffer (Sigma, USA) and centrifugation (200 × g, 5 min), the pellet was resuspended in RPMI-1640 complete medium, and placed into a sterile petri dish for 1 h at 37 °C, in a humidified chamber containing 95 % air and 5 % CO2, to remove the macrophages by adherence. T lymphocytes were isolated by panning [34]. In brief, the unadhered cells were decanted, centrifuged (200 × g, 5 min) and transferred to the petri dishes that were previously coated with anti-rat IgG (5 μg/ml) overnight at 4 °C. Hence, selective depletion of B lymphocytes was accomplished because they adhered to the substratum of the petri dishes. After an incubation of 1 h at 4 °C, the T-lymphocyte–rich supernatant was decanted and centrifuged (200 × g, 5 min) twice with RPMI-1640 complete medium. This technique provided us with an enriched (99 %) T-cell population as verified by cyotofluorimetry (not shown). Cell numbers were determined by hemocytometer.
T-cell proliferation assay
The proliferation of splenic T-cells in response to PM lipids and polyphenols was assessed according to the method of Bonin and Khan [35] with slight modification. Splenic T-cells were resuspended in RPMI complete medium and plated into 96-well microplates at the concentration of 5 × 105 cells/well. Lipid and polyphenol extracts obtained from pearl millet, were solubilized in ethanol, and added to cells, final ethanol volume was below 0.1 % (v/v), 1 h before their activation with PMA (50 nM) and ionomycin (500 nM). After 48 h of treatment, T-cell proliferation was measured by Cayman’s WST-8 cell proliferation assay kit (Cayman Chemical, USA). The stimulation index (SI) was calculated as follows: SI = optical density (450 nm) of stimulated cells/optical density (450 nm) of unstimulated cells × 100.
Cell preparation for western blot analysis
Splenic T-cells were serum starved for 6 h in RPMI 1640 medium without serum. Then, splenic T- cells (5 × 106/ml) were pre-incubated for 5 min with either PGL or PGPC or vehicle before stimulation with PMA (200 nM) for an additional 30 min, according to Nel et al. [36]. Incubation was stopped by centrifugation, and cell pellets were washed twice with PBS and resuspended in 50 μl of lysis buffer (HEPES, 20 mM, pH 7.3; EDTA, 1 mM; EGTA, 1 mM; NaCl, 0.15 mM; Triton X-100, 1 %; glycerol, 10 %; phenylmethylsulfonyl fluoride, 1 mM; sodium orthovanadate, 2 mM; antiprotease cocktail, 2 μl in 1 ml of buffer). After centrifugation (2500 g for 1 min), the protein in the supernatant was quantified with the bicinchoninic acid (BCA) assay (Thermo Fisher Scientific, France) and either used immediately for Western blot detection or stored at −80 °C.
Western blot detection of phosphorylated MAP kinases
Denatured proteins (60 μg) were separated by SDS-PAGE (10 %) and transferred onto polyvinylidine difluoride membranes, and immunodetection was performed by using rabbit antibodies raised against phosphorylated or non-phosphorylated P44/P42 MAPK. Primary antibodies were detected with a horse radish peroxidase conjugated mouse anti-rabbit antibody and visualised using an ECL Kit (Merck Millipore) on Bio-Rad ChemiDoc XRS+ system. Densitometric analysis was performed on Bio-Rad Image Lab Software (version 4.1).
RNA isolation and real time quantitative PCR
Cells were cultured as described above in the presence of either PGL or PGPC extracts and stimulated with anti-CD3 antibodies for 2 h [37, 38]. Total RNA was extracted using TRIzol Reagent and underwent DNase treatment using the RNase-free DNase Set (Life Technologies). 500 ng of total RNA was reverse transcribed with Super script II H-reverse transcriptase (Life Technologies) using oligo (dT) according to the manufacturer’s instructions. Real time PCR was carried out on the iCycler iQ real time detection system and amplification was undertaken by using SYBR® Green PCR Master Mix (Life Technologies) as described elsewhere [39]. Oligonucleotide primers were as follow: beta-actin forward: 5′-ATGATA TCGCCGCGCTCGTCGTC-3′, beta-actin reverse 5′-AGGTCCCGGCCAGCCAGGTCCAG-3′; IL-2 forward 5′ CACTAATTCTTGCACTTGTCAC-3′, IL-2 reverse 5′- CTTCTTGGGCATGTAAAACT-3′. Relative quantification of IL 2 mRNA was determined by ΔΔCt method as follows: ΔCt = Ct of IL-2 - Ct of beta actin. ΔΔCt = ΔCt of treated cells - ΔCt control cells. Relative quantity (RQ) was calculated as follows: RQ = (1 + E)-(ΔΔCt) .
Measurement of free intracellular Ca2+concentrations; [Ca2+]i
Splenic T-cells (2 × 106 cells/ml) were washed with phosphate-buffered saline, pH 7.4, and then incubated with Fura-2/AM (1 μM) for 60 min at 37 °C in loading buffer containing: 110 mM, NaCl; 5.5 mM, KCl; 25 mM, NaHCO3; 0.8 mM, MgCl2; 0.4 mM, KH2PO4; 0.33 mM, Na2HPO4; 20 mM, HEPES; 1.2 mM, CaCl2, and the pH was adjusted to 7.4. After loading, the cells were washed three times (720 g × 10 min) and remained suspended in the identical medium. The fluorescence intensities were measured in the ratio mode in the PTI spectrofluorometer at 340 and 380 nm (excitation filters) and 510 nm (emission filters). The cells were continuously stirred throughout the experiment. The intracellular concentrations of free Ca2+, [Ca2+]i, were calculated by using the following equation: [Ca2+]i = Kd × (R-Rmin)/(Fmax-F)(Sf2/Sb2). A value of 224 nM for Kd was added into the calculations. Rmax value was obtained by the addition of ionomycin (5 μM) and Rmin value was obtained by the addition of MnCl2 (2 mM), Triton X-100 (0.1 %) and EGTA (24 mM).
For experiments conducted in the absence of external calcium (0 % Ca2+), CaCl2 was replaced by 1 mM EGTA in the buffer [40]. All test molecules were added in small volumes with no interruption in recordings.
Statistical analyses
Results are shown as mean ± SD (standard error deviation) for a given number of experiments (n). Data were analysed by using Statistica (4.1 version, Statsoft, Paris, France). The significance of differences between mean values was determined by one-way ANOVA, followed by Fisher’s least-significant-difference (LSD) test. Differences with p < 0.05 were considered to be significant.
Results
Phenolic acid and lipid composition of PM grains
The total content of phenolic compounds in PM grains was estimated to be 1660 μg GAE/g. Table 1 shows that p-coumaric acid represents 81 %, and ferulic acid represents 12 % of the total phenolic compounds.
Total lipids were estimated to be 4.5 % (Table 2). Alpha-linoleic acid (18:2n-6) was the most abundant fatty acid (44.95 %), and oleic acid (18:1 n-9) was the second most abundant fatty acid (24.88 %). The proportion of unsaturated fatty acids was estimated to be 72 % (Table 2).
PGL and PGPC decrease T-cell proliferation
Figure 1a shows the effects of increasing concentration of PGL on T-splenocyte proliferation either in the presence or absence of mitogens (PMA + Iono). We observed that PGL until 20 μg/ml concentration exerted no significant effect on basal splenic T-cells proliferation. However, PGPC at 20 μg/ml decreased basal T-cells proliferation. We also observed that both PGL and PGPC curtailed, in a dose-dependent manner, T-cell proliferation induced by PMA + Iono. PGPC exerted more inhibitory effect on T-cell proliferation than PGL (Fig. 1b).

Effect of PGL and PGPC on T cell proliferation. The cells (5 × 105 cells/ml) were stimulated with different concentrations of the extracts as described in Methods. Cell number were determined with a hemocytometer. Inserts show the stimulation index (SI) of T-cell proliferation in response to PGL (left panel) and PGPC (right panel). Data represent means ± SD (n = 6). p < 0.01 as compared to cells without PGL or PGPC, *represents p < 0.001 as compared to PMA + Iono-stimulated T-cells. NS = insignificant differences. p values were obtained by one-way ANOVA, followed by Fisher’s LSD test
PGL and PGPC diminish PMA-induced ERK1/ERK2 activation
Figure 2 shows that both PGL and PGPC dose dependently diminished PMA-induced ERK1/2 phosphorylation in splenic T-cells. PGPC completely blocked MAP kinase phosphorylation even at a low concentration (10 μg/ml) (Fig. 2b), whereas the effects of PGL was weaker than that of PGPC. (Fig. 2a).

Effects of PGL and PGPC on PMA + Iono-stimulated ERK1/ERK2 phosphorylation in splenic T-cells. Data were quantified by densitometry and expressed as phosphorylated /non phosphorylated ERK1/ERK2 ratio. Splenic T-cells (5 × 106 cells/ml), before determination of MAP kinase phosphorylation, were incubated for 6 h in RPMI 1640 medium without serum, and treated with increasing concentrations (0 to 40 μg/ml) of PGL in (a) and PGPC in (b). After 5 min of incubation, cells were stimulated with PMA (50 nM) and Iono (500 nM) for another 30 min at 37 °C. Cells were lysed and phosphorylated MAP kinases were detected performed as described in Materials and Methods. Results are expressed as arbitrary units in bar graphs. *Represents P < 0.001 as compared to PMA + Iono-stimulated T-cells in the absence of PGL (a) and PGPC (b). NS = insignificant differences. p values were obtained by one-way ANOVA, followed by Fisher’s LSD test
PGL and PGPC decrease IL-2 mRNA expression
Our results show that PGL and PGPC exerted no effect on basal expression of IL-2 mRNA in splenic T-cells. However, both PGL and PGPC extracts diminished, in a dose-dependent manner, PMA + Iono-induced IL-2 mRNA expression (Fig. 3). As observed for T-cell proliferation and ERK1/2 phosphorylation, PGPC exerted more important inhibitory effect than PGL on IL-2 mRNA expression.

PGL and PGPC modulate IL-2 mRNA expression. Splenic T-cells (5 × 105 cells/ml) were incubated with increasing concentrations (0 to 40 μg/ml) of PGL (a) and PGPC (b), and stimulated with anti-CD3 antibodies for 2 h. Each value represents the mean of three determinations. * Represents P < 0.001 as compared to PMA + Iono-stimulated T-cells. NS = insignificant differences. The p values were obtained by one-way ANOVA, followed by Fisher’s LSD test
PGL and PGPC induce increases in [Ca 2+]i in splenic T-cells
Figure 4 shows that both PGL and PGPC evoked a dose-dependent increase in [Ca2+]i in splenic T-cells; however, the increase in [Ca2+]i triggered by PGPC was significantly higher than that triggered by PGL (Fig. 4). In order to assess the origin of Ca2+ mobilized by lipids and phenolic compounds, we conducted experiments in the absence (0 % Ca2+) and presence (100 % Ca2+) of Ca2+ in the extracellular medium. Figure 5 shows that the PGPC and PGL induced a weak and discrete increase in [Ca2+]i in 0 % Ca2+ medium as compared to that induced in 100 % Ca2+ medium.

Ca2+ signaling modulation by PGL and PGPC in splenic T-cells. Cells (2 × 106/ml) were loaded with the fluorescent probe, Fura-2/AM, as described in Methods. The arrow head indicates the time when PGL or PGPC were added into the cuvette without interruptions in recordings. Figures show the single traces of observations which were reproduced independently (n = 6). Inserts show the increase in [Ca2+]i evoked by the increasing concentrations (10 to 40 μg/ml) of PGL (a) and PGPC (b). *Represents P < 0.001 as compared to control (untreated cells). NS = insignificant differences. The p values were obtained by one-way ANOVA, followed by Fisher’s LSD test

Ca2+ signaling modulation by PGL (a) and PGPC (b) in splenic T-cells in 0 % Ca2+-buffer and 100 % Ca2+-buffer. PGL and PGPC-evoked increases in [Ca2+]i are curtailed in 0 % Ca2+-buffer in T-cells. Cells (2 × 106/ml) were loaded with the fluorescent probe, Fura-2/AM, as described in Methods. *Represents P < 0.001 as compared to [Ca2+]i increases in 0 % Ca2+-buffer. The p values were obtained by one-way ANOVA, followed by Fisher’s LSD test
As the polyphenolic and lipidic extracts were able to induce increases in [Ca2+]i even in the absence of external calcium, it was thought worthwhile to examine the nature of intracellular stores involved in this rise in [Ca2+]i. We used thapsigargin, known to induce increase in [Ca2+]i by inhibiting the endoplasmic reticulum Ca2+-ATPase [41, 42]. Figure 6 illustrates that thapsigargin alone triggered a calcium peak, and addition of PGL or PGPC after thapsigargin or vice versa evoked additive effects on the increases in [Ca2+]i in these cells.

Effects of thapsigargin (TG) on PGL and PGPC-induced rise in [Ca2+]i in lymphocytes that were tretaed as follows : TG (a), PGL followed by TG (b), TG followed by PGL (c), TG followed by PGPC (d, and PGPC followed by TG (e). Cells (2 × 106/ml) were loaded with the fluorescent probe, Fura-2/AM, as described in Methods. The arrow head indicates the time when 20 μg/ml of PGL, PGPC, or TG (5 μM) were added into the cuvette. The figure shows the single traces of observations reproduced independently (n = 6)
Discussion
Millet grains, widely consumed in many areas of Asia, Africa and Latin America, have been shown to exert several beneficial effects in health and disease [5]. Among millets species, pearl millet (PM) has been the least studied. In this study, we examined the effects of polyphenols and lipids, extracted from PM, on T-cell proliferation. We investigated the involvement of calcium and MAP kinase signaling in this process.
Most of the millet species contain phenolic compounds, which are detected in the pericarp, testa, aleurone layer, and endosperm [43]. In our study, the total polyphenol content of the PM extract was estimated to be 1660 μg GAE/g, in agreement with other studies in which the values ranged from 1387 to 2580 μg GAE/g [44, 45]. Similarly to the finding of Shahidi and Chandrasekara [46], we observed that PM grains contained principally p-coumaric acid and ferulic acid. In addition, apigenin, a flavonoid, was detected in PM grains [46].
It is well established that, except finger millet, millet species have higher lipid contents, ranging from 3.5 % to 5.2 %, than other cereals [46]. In our investigation, total lipids were estimated to be 4.5 % in PM grains. Indeed, Ragaee et al. [44] found that PM had the highest content of lipids (4.2 %) compared to wheat flours and other cereal whole grains. The high content of lipids in PM grains might be due to the presence of embryo in which lipids are concentrated. Daniel et al. [47] had reported that PM oil yielded three fatty acids as major components. Hence, alpha-linoleic acid amounted to be 45.6 % followed by oleic acid (28.5 %) and palmitic acid (20.6 %), whereas linolenic and stearic acids were the minor fatty acids. In our samples, we obtained a high amount of both linolenic and stearic acid (3.03 % and 5.11 %, respectively).
T-lymphocytes represent a fundamental component of the adaptive immune response. The lymphocyte transformation assay is an important tool to measure, in vitro, mitogen-induced lymphocyte proliferation [48, 49]. Following T-cell receptor (TCR) engagement, one of the early events in T-cell activation is the phosphorylation of tyrosine kinases and the generation of inositol 1,4,5-triphosphate (IP3), leading to the release and influx of Ca2+, and the rise in cytoplasmic Ca2+ concentration [50]. The rise in [Ca2+]i activates via calcineurin induces IL-2 gene expression [51]. To our knowledge, the present report is the first study assessing the immunomodulatory effects of PM polyphenols and lipids. PM extracts were rich in apigenin, p-coumaric acid and other phenolic acids. Apigenin has been shown to inhibit T-cell proliferation [52], without exerting any toxic effect [53]. Interestingly, p-coumaric acid has been reported to exert anti-cancer [54], anti-mutagenic [55], and anti-inflammatory activities [56]. PGPC strongly inhibited T-cell proliferation and IL-2 mRNA expression. Other investigators have also reported that plant polyphenols inhibited proliferation and IL-2 production in human lymphocytes [39, 57, 58]. Gao et al. [59] reported that resveratrol, a stilbene, inhibited the proliferation and IL-2 and interferon (IFN)-γ production by splenic lymphocytes. Kaempferol, a flavonoid, was able to reduce IFN-γ and IL-2 production by murine T-cells [60]. Curcumin, that gives rise mainly to ferulic acid and vanillin, also inhibited IL-2-induced T-proliferation of splenic cells [61].
PGL also, to a lesser extent than PGPC, inhibited T-cell proliferation. In PGL, n-3:n-6 ratio was estimated to be 1:14 which is very close to the recommended ratio (1:10), reported by Ma et al. [62]. The inhibitory effect of lipid extract of PM may be attributed to linoleic acid, an n-6 fatty acid. Linoleic acid (18: 2n-6) which represents 44.95 % of total fatty acids in PGL, could be involved in T-cell immunosuppression. Indeed, Liu et al. [63] had reported that linoleic acid inhibited IL-2 mRNA expression and, consequently, lymphocyte proliferation. Another study has shown that linoleic acid was a potent inducer of cell death in human peripheral blood lymphocytes. The mechanism of action of linoleic acid on cell apoptosis involved alterations in mitochondrial transmembrane potential and ROS production [64]. Similarly, linolenic acid (18: 2n-3) could be involved in T-cell immunosuppression. Indeed, Denys et al. [65] have shown that n-3 fatty acids inhibited mitogen-induced nuclear translocation of NF-κB and IL-2 mRNA expression in Jurkat T-cells. The inhibitory effect of PGPC and PGL on T-cell proliferation could be mediated by their capacity to reduce IL-2 mRNA expression. In fact, the transition of T-cells via S phase of cell cycle is associated to the expression of IL-2 mRNA. The newly synthesized IL-2 acts in an autocrine manner in order to assure the T-cell cycle progression. Furthermore, inhibition of IL-2 production is associated with cell cycle arrest [66].
Mitogen-activated protein (MAP) kinases including the extracellular signal-regulated kinase-1/2 (ERK1/ERK2) have been shown to play a critical role in the events leading to increased IL-2 production in mammalian cells [36, 67]. Both PGL and PGPC dose-dependently diminished PMA-induced ERK1/ERK2 phosphorylation in splenic T-cells; however, the inhibitory effect of PGPC was more pronounced. Our results agree with the observations of Neuhaus et al. [68] who had demonstrated that the phosphorylation of ERK1/ERK2 was inhibited by epigallocatechin-3 gallate (EGCG). Similarly, the treatment of ECV304 cells with ferulic acid, a major phenolic acid in pearl millet, inhibited both cell proliferation and ERK1/ERK2 phosphorylation [69]. Besides, the PM lipid contents might be responsible for the inhibition of MAPK phosphorylation as reported previously [65].
We examined the actions of PGL and PGPC on the increases in [Ca2+]i in T-cells. In the presence of 100 % Ca2+, PGPC and PGL induced high increases in [Ca2+]i, suggesting that Ca2+ influx plays a major role in the increase in [Ca2+]i evoked by theses extracts. The kinetic study of PGL and PGPC-induced Ca2+ mobilization showed that these compounds produced a sustained increase in [Ca2+]i. We also noted that PGPC induced stronger and more important increases in Ca2+ (around 6-fold), compared to PGL. These sustained [Ca2+]i increases are correlated with the immunosuppressive effects of PM extracts, as reported for the prickly pear phenolic compounds [40].
To ascertain the nature of the intracellular Ca2+ pool mobilized by lipids and phenolic compounds, thapsigargin, an inhibitor of Ca2+-ATPase of endoplasmic reticulum [41], was employed. The addition of thapsigargin during the PGL or PGPC-induced Ca2+ peak, and vice versa, suggesting that PGL and PGPC did not seem to act on Ca2+-ATPase.
Conclusion
We can state that both PM polyphenols and lipids exhibited an immunosuppressive effects. The PM polyphenols seem to be more active than lipids. Two molecular mechanisms seem to be involved in the immunosuppressive activity of PM extracts: i) the sustained increases in intracellular free Ca2+ concentration and ii) the inhibition of IL-2 mRNA expression and MAP kinase phosphorylation. Our results argue for the use of millet diet as dietary supplements for treatment of diseases associated with a sustained activation of the immune system such as autoimmune diseases.
Abbreviations
- [Ca2+]i:
-
Free intracellular calcium concentration
- EGCG:
-
Epigallocatechin-3 gallate
- ER:
-
Endoplasmic reticulum
- ERK:
-
Extracellular signal-regulated kinase
- FAMEs:
-
Fatty acid methyl esters
- GAE:
-
Gallic acid equivalents
- IFN:
-
Interferon
- IL-2:
-
Interleukin-2
- LA:
-
Linolenic acid
- MAPK:
-
Mitogen-activated protein kinase
- n-3 PUFA:
-
Polyunsaturated fatty acids of the n-3 family
- n-6 PUFA:
-
Polyunsaturated fatty acids of the n-6 family. PBS, Phosphate-buffered saline, pH 7.4
- PGL:
-
Pennisetum glaucum lipids
- PGPC:
-
Pennisetum glaucum phenolic compounds
- PKC:
-
Protein kinase C
- PM:
-
Pearl millet
- TCR:
-
T-cell receptor
- TG:
-
Thapsigargin
References
US Food and Drug Administration. Food Labeling: Health Claims; Soluble Fiber from Certain Foods and Coronary Heart Disease. Federal Register 63 FR 8103, 1998. https://www.federalregister.gov/articles/1998/02/18/98-4074/food-labeling-health-claims-soluble-fiber-from-certain-foods-and-coronary-heart-disease.
Vigouroux Y. Le mil, aliment du futur au Sahel. IRD. 2009. Fiche n°325.
Bender DA, Bender AE. Benders’ Dictionary of Nutrition and Food Technology. 7th ed. Abington: Woodhead Publishing; 1999.
Lee SH, Chung IM, Cha YS, Park Y. Millet consumption decreased serum concentration of triglyceride and C-reactive protein but not oxidative status in hyperlipidemic rats. Nutr Res. 2010;30:290–6.
Shobana S, Harsha MR, Platel K, Srinivasan K, Malleshi NG. Amelioration of hyperglycaemia and its associated complications by finger millet (Eleusine coracana L.) seed coat matter in streptozotocin-induced diabetic rats. Br J Nutr. 2010;104:1787–95.
Shukla K, Srivastava S. Evaluation of finger millet incorporated noodles for nutritive value and glycemic index. J Food Sci Technol. 2014;51:527–34.
Chandrasekara A, Shahidi F. Content of insoluble bound phenolics in millets and their contribution to antioxidant capacity. J Agric Food Chem. 2010;58:6706–14.
Duke JA. Biological Active Phytochemicals and Their Activities. Boca Raton: CRC Press, Inc.; 1992.
Watanabe M. Antioxidative phenolic compounds from Japanese barnyard millet (Echinochloa utilis) grains. J Agric Food Chem. 1999;47:4500–5.
Van Rensburg SJ. Epidemiological and dietary evidence for a specific nutritional predisposition to esophageal cancer. J Natl Cancer Inst. 1981;67:243–51.
Chen F, Cole P, Mi Z, Xing LY. Corn and wheat-flour consumption and mortality from esophageal cancer in Shanxi, China. Int J Cancer. 1993;53:902–6.
Nani A, Belarbi M, Soualem Z, Ghanemi FZ, Borsali N, Amamou F. Study of the impact of millet (Pennisetum glaucum) on the glucidic metabolism in diabetic rats. Ann Biol Res. 2011;2:21–3.
Nambiar VS, Dhaduk JJ, Sareen N, Shahu T, Desai R. Potential Functional Implications of Pearl millet (Pennisetum glaucum) in Health and Disease. J Appl Pharm Sci. 2011;01:62–7.
Adom KK, Sorrells ME, Liu RH. Phytochemical profiles and antioxidant activity of wheat varieties. J Agric Food Chem. 2003;51:7825–34.
Andreasen MF, Christensen LP, Meyer AS, Hansen A. Content of phenolic acids and ferulic acid dehydrodimers in 17 rye (Secale cereale L.) varieties. J Agric Food Chem. 2000;48:2837–42.
Lloyd BJ, Siebenmorgen TJ, Beers KW. Effects of commercial processing on antioxidants in rice bran. Cereal Chem. 2000;77:551–5.
Maillard M, Berset C. Evolution of antioxidant activity during kilning: role of insoluble bound phenolic acids of barley and malt. J Agric Food Chem. 1995;43:1789–93.
Middleton E. Effect of plant flavonoids on immune and inflammatory cell function. Adv Exp Med Biol. 1998;439:175–82.
Serrano A, Palacios C, Roy G, Cespon C, Villar M, Nocito M, et al. Derivatives of gallic acid induce apoptosis in tumoral cell lines and inhibit lymphocyte proliferation. Arch Biochem Biophys. 1998;350:49–54.
Miles EA, Zoubouli P, Calder PC. Effects of polyphenols on human Th1 and Th2 cytokine production. Clin Nutr. 2005;24:780–4.
González R, Ballester I, López-Posadas R, Suárez MD, Zarzuelo A, Martínez-Augustin O, et al. Effects of flavonoids and other polyphenols on inflammation. Crit Rev Food Sci Nutr. 2011;51:331–62.
Ghiringhelli F, Rebe C, Hichami A, Delmas D. Immunomodulation and anti-inflammatory roles of polyphenols as anticancer agents. Anticancer Agents Med Chem. 2012;12:852–73.
Tappia PS, Grimble RF. Complex modulation of cytokine induction by endotoxin and tumour necrosis factor from peritoneal macrophages of rats by diets containing fats of different saturated, monounsaturated and polyunsaturated fatty acid composition. Clin Sci. 1994;87:173–8.
Wallace FA, Neely SJ, Miles EA, Calder PC. Dietary fats affect macrophage-mediated cytotoxicity towards tumour cells. Immunol Cell Biol. 2000;78:40–8.
Kelley DS. Modulation of human immune and inflammatory responses by dietary fatty acids. Nutrition. 2001;17:669–73.
Cavaglieri CR, Nishiyama A, Fernandes LC, Curi R, Miles EA, Calder PC. Differential effects of short-chain fatty acids on proliferation and production of pro- and anti-inflammatory cytokines by cultured lymphocytes. Life Sci. 2003;73:1683–90.
Kurita-Ochiai T, Seto S, Ochiai K. Role of cell–cell communication in inhibiting butyric acid-induced T-cell apoptosis Infections. Infect Immun. 2004;72:5947–54.
Yusufi AN, Cheng J, Thompson MA, Walker HJ, Gray CE, Warner GM, et al. Differential effects of low-dose docosahexaenoic acid and eicosapentaenoic acid on the regulation of mitogenic signaling pathways in mesangial cells. J Lab Clin Med. 2003;141:318–29.
Lestienne I, Buisson M, Lullien-Pellerin V. Losses of nutrients and anti-nutritional factors during abrasive decortication of two pearl millet cultivars (Pennisetum glaucum). Food Chem. 2007;100:1316–23.
Liyana-Pathirana CM, Shahidi F. Importance of insoluble-bound phenolics to antioxidant properties of wheat. J Agric Food Chem. 2006;54:1256–64.
Folin O, Ciocalteu V. On tyrosine and tryptophane determinations in proteins. J Biol Chem. 1927;27:627–50.
Miliauskas G, Venskutonis PR, van Beek TA. Screening of radical scavenging activity of some medicinal and aromatic plant extracts. Food Chem. 2004;85:231–7.
Hara A, Radin NS. Lipid extraction of tissues with a low-toxicity solvent. Anal Biochem. 1978;90:420–6.
Triboulot C, Hichami A, Denys A, Khan NA. Dietary (n-3) Polyunsaturated Fatty Acids Exert Antihypertensive Effects by Modulating Calcium Signaling in T Cells of Rats. J Nutr. 2001;131:2364–9.
Bonin A, Khan NA. Regulation of calcium signaling by docosahexaenoic acid in human T-cells: implication of CRAC channels. J Lipid Res. 2000;41:277–84.
Nel AE, Hanekom C, Rheeder A, Williams K, Pollack S, Katz R, et al. Stimulation of MAP-2 kinase activity in T lymphocytes by anti-CD3 or anti-Ti monoclonal antibody is partially dependent on protein kinase C. J Immunol. 1990;144:2683–9.
Yessoufou A, Plé A, Moutairou K, Hichami A, Khan NA. Docosahexaenoic acid reduces suppressive and migratory functions of CD4 + CD25+ regulatory T-cells. J Lipid Res. 2009;50:2377–88.
Sojka DK, Bruniquel D, Schwartz RH, Singh NJ. IL-2 secretion by CD4+ T cells in vivo is rapid, transient, and influenced by TCR-specific competition. J Immunol. 2004;172:6136–43.
Abdoul-Azize S, Bendahmane M, Hichami A, Dramane G, Simonin AM, Benammar C, et al. Effects of Zizyphus lotus L. (Desf.) polyphenols on Jurkat cell signaling and proliferation. Int Immunopharmacol. 2013;15:364–71.
Aires V, Adote S, Hichami A, Moutairou K, Boustani EE, Khan NA. Modulation of intracellular calcium concentrations and T cell activation by prickly pear polyphenols. Mol Cell Biochem. 2004;260:103–10.
Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proc Natl Acad Sci. 1990;87:2466–70.
Pan Z, Zhao X, Brotto M. Fluorescence-based Measurement of Store-operated Calcium Entry in Live Cells: from Cultured Cancer Cell to Skeletal Muscle Fiber. J Vis Exp. 2012;10:3415–791.
Dykes L, Rooney LW. Sorghum and millet phenols and antioxidants. J Cereal Sci. 2006;44:236–51.
Ragaee S, El-Sayed M, Abdel-Aal MN. Antioxydant activity and nutrient composition of selected cereals for food use. Food Chem. 2006;98:32–8.
Hithamani G, Srinivasan K. Effect of domestic processing on the polyphenol content and bioaccessibility in finger millet (Eleusine coracana) and pearl millet (Pennisetum glaucum). Food Chem. 2014;164:55–62.
Shahidi F, Chandrasekara A. Millet grain phenolics and their role in disease risk reduction and health promotion: A review. J Funct Foods. 2013;5:570–81.
Daniel M, Denni M, Chauhan D. Polyphenols, phospholipids and fixed oil composition of pearl millet [Pennisetum glaucum (L.) R. Br.]. IJPLS. 2012;3:2098–102.
Baba Hamed Y, Medjdoub A, Mostefa Kara B, Merzouk H, Villemin D, Narce M. 5,6-Dihydro-2H-pyranones and 5,6-dihydro-2H-pyridones and their derivatives modulate in vitro human T lymphocyte function. Mol Cell Biochem. 2012;360:23–33.
Christiansen J, Farm G, Eid-Forest R, Anderson C, Cederbrant K, Hultman P. Interferon-gamma secreted from peripheral blood mononuclear cells as a possible diagnostic marker for allergic contact dermatitis to gold. Contact Dermatitis. 2006;55:101–12.
Donnadieu E, Bismuth G, Trautmann A. X Calcium fluxes in T-lymphocytes. J Biol Chem. 1992;267:25864–72.
Negulescu PA, Shastri N, Cahalan MD. Intracellular calcium dependence of gene expression in single T lymphocytes. Proc Natl Acad Sci. 1994;91:2873–7.
YiN Y, Gong FY, Wu XX, Sun Y, Li YH, Chen T, et al. Anti-inflammatory and immunosuppressive effect of flavones isolated from Artemisia vestita. J Ethnopharmacol. 2008;120:1–6.
Liang ZD, Zeng YY, Huang XY, He F. Effect of Apigenin on proliferation, cell cycle and apoptosis of mouse T cells in vitro. Chinese J Cell Mol Immunol. 2008;24:337–40.
Hudson EA, Dinh PA, Kokubun T, Simmonds MS, Gescher A. Characterization of potentially chemopreventive phenols in extracts of brown rice that inhibit the growth of human breast and colon cancer cells. Cancer Epidemiol Biomarkers Prev. 2000;9:1163–70.
Ferguson LR, Lim IF, Pearson AE, Ralph J, Harris PJ. Bacterial antimutagenesis by hydroxycinnamic acids from plant cell walls. Mutat Res. 2003;542:49–58.
Luceri C, Guglielmi F, Lodovici M, Giannini L, Messerini L, Dolara P. Plant phenolic 4-coumaric acid protects against intestinal inflammation in rats. Scand J Gastroenterol. 2004;39:1128–33.
Atluru D, Jackson TM, Atluru S. Genistein, a selective protein tyrosine kinase inhibitor, inhibits interleukin-2 and leukotriene B4 production from human mononuclear cells. Clin Immunol Immunopathol. 1991;59:379–87.
Devi MA, Das NP. In vitro effects of natural plant polyphenols on the proliferation of normal and abnormal human lymphocytes and their secretions of interleukin-2. Cancer Lett. 1993;69:191–6.
Gao X, Xu YX, Janakiraman N, Chapman RA, Gautman SC. Immunomodulatory activity of resveratrol: suppression of lymphocyte proliferation, development of cellmediated cytotoxicity, and cytokine production. Biochem Pharmacol. 2001;62:1299–308.
Okamoto I, Iwaki K, Koya-Miyata S, Tanimoto T, Kohno K, Ikeda M, et al. The flavonoid kaempferol suppresses the graftversus- host reaction by inhibiting type 1 cytokine production and CD8+ T cell engraftment. Clin Immunol. 2002;103:132–44.
Wang YJ, Pan MH, Cheng AL, Lin LI, Ho YS, Hsieh CY, et al. Stability of curcumin in buffer solutions and characterization of its degradation products. J Pharm Biomed Anal. 1997;15:1867–76.
Ma X, Jiang Z, Lai C. Significance for Human Health of Increasing n-3PUFA Content in Pork. Crit Rev Food Sci Nutr. 2015. doi:10.1080/10408398.2013.850059.
Liu Y, Gong L, Li D, Feng Z, Zhao L, Dong T. Effects of fish oil on lymphocyte proliferation, cytokine production and intracellular signaling in weanling pigs. Arch Tierernahr. 2003;57:151–65.
Cury-Boaventura MF, Gorjão R, de Lima TM, Newsholme P, Curi R. Comparative toxicity of oleic and linoleic acid on human lymphocytes. Life Sci. 2006;78:1448–56.
Denys A, Hichami A, Khan NA. n-3 PUFAs modulate T-cell activation via protein kinase C-α and -ε and the NF-κB signaling pathway. J Lipid Res. 2005;46:752–8.
Crabtree GR. Generic signals and specific outcomes: Signaling through Ca2+, calcineurin, and NF-AT. Cell. 1999;96:611–4.
Cantrell D. T cell antigen receptor signal transduction pathways. Annu Rev Immunol. 1996;14:259–74.
Neuhaus T, Pabst S, Stier S, Weber AA, Schrör K, Sachinidis A, et al. Inhibition of the vascular-endothelial growth factor-induced intracellular signaling and mitogenesis of human endothelial cells by epigallocatechin-3 gallate. Eur J Pharmacol. 2004;483:223–7.
Hou Y, Yang J, Zhao G, Yuan YJ. Ferulic acid inhibits endothelial cell proliferation through NO down-regulating ERK1/2 pathway. Cell Biochem. 2004;93:1203–9.
Acknowledgements
The study was supported, in part, by a bilateral Franco-Algerian collaborative project “Tassili” (grant number 30850QG.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
The authors’ contributions are as follows: NAK, AH and MB designed research (project conception, development of overall research plan); AN conducted research; AN wrote the MS; NAK, AH and FG supervised in vitro studies; JWK-M did make technical part of polyphenol analysis. S A-A helped the in vitro experiments. CB did the statistical analysis. All authors have read and approved the final content of the manuscript.
Aziz Hichami and Naim Akhtar Khan contributed equally to this work.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Nani, A., Belarbi, M., Ksouri-Megdiche, W. et al. Effects of polyphenols and lipids from Pennisetum glaucum grains on T-cell activation: modulation of Ca2+ and ERK1/ERK2 signaling. BMC Complement Altern Med 15, 426 (2015). https://doi.org/10.1186/s12906-015-0946-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12906-015-0946-3