
Abstract
Background
Hip osteoarthritis (OA) is a leading cause of musculoskeletal pain. Exercise is a core recommended treatment. Despite some clinical guidelines also recommending weight loss for hip OA, there is no evidence from randomised controlled trials (RCT) to substantiate these recommendations. This superiority, 2-group, parallel RCT will compare a combined diet and exercise program to an exercise only program, over 6 months.
Methods
One hundred people with symptomatic and radiographic hip OA will be recruited from the community. Following baseline assessment, participants will be randomly allocated to either, i) diet and exercise or; ii) exercise only. Participants in the diet and exercise group will have six consultations with a dietitian and five consultations with a physiotherapist via videoconferencing over 6 months. The exercise only group will have five consultations with a physiotherapist via videoconferencing over 6 months. The exercise program for both groups will include prescription of strengthening exercise and a physical activity plan, advice about OA management and additional educational resources. The diet intervention includes prescription of a ketogenic very low-calorie diet with meal replacements and educational resources to support weight loss and healthy eating. Primary outcome is self-reported hip pain via an 11-point numeric rating scale (0 = ‘no pain’ and 10 = ‘worst pain possible’) at 6 months. Secondary outcomes include self-reported body weight (at 0, 6 and 12 months) and body mass index (at 0, 6 and 12 months), visceral fat (measured using dual energy x-ray absorptiometry at 0 and 6 months), pain, physical function, quality of life (all measured using subscales of the Hip Osteoarthritis Outcome Scale at 0, 6 and 12 months), and change in pain and physical activity (measured using 7-point global rating of change Likert scale at 6 and 12 months). Additional measures include adherence, adverse events and cost-effectiveness.
Discussion
This study will determine whether a diet intervention in addition to exercise provides greater hip pain-relief, compared to exercise alone. Findings will assist clinicians in providing evidence-based advice regarding the effect of a dietary intervention on hip OA pain.
Trial registration
ClinicalTrials.gov . Identifier: NCT04825483. Registered 31st March 2021.
Similar content being viewed by others


Background
Hip OA is a major public health problem and symptomatic hip OA affects one in four adults over their lifetime [1]. There is no cure for OA and joint replacement is reserved for end-stage disease. Of concern is the life-time risk of hip replacement for hip OA is as high as 1 in 7 for women and 1 in 10 for men [2]. In 2018, 77% of the total hip joint replacements performed in Australia were for people with hip OA with comorbid overweight/obesity [3]. Effective treatments to reduce hip OA pain are urgently needed.
Clinical guidelines [4,5,6,7,8] recommend exercise as the core treatment for hip OA symptoms. Systematic reviews and meta-analyses of clinical trials evaluating land-based exercise report small-to-moderate benefits for hip OA pain [9, 10]. Muscle weakness is common in hip OA [11] and the majority of evidence for exercise is strength-related [9]. In addition to strengthening exercise, regular physical activity is important as people with OA are at increased of risk of death due to cardiovascular disease [12]. Indeed, many people with hip OA do not meet physical activity recommendations [13]. Exercise may indeed delay hip joint replacement for some people with hip OA [14]. However, up to 85% of people with hip OA have overweight or obesity [15] and exercise therapy in isolation may be insufficient to reduce pain.
Recommendations for weight loss to manage hip OA symptoms are inconsistent across clinical guidelines, due to the absence of clinical trials in hip OA evaluating weight loss. Clinical guidelines [4, 6,7,8] including the 2019 American College of Rheumatology guidelines [8], recommend weight loss for hip OA based on evidence from clinical trials in knee OA. However, the 2019 Osteoarthritis Research Society International guidelines for hip OA management do not recommend weight loss for hip OA [5] due to the absence of clinical trials in hip OA evaluating weight loss. A reduction in body weight by at least 5% is recommended for improvement in clinical and mechanistic outcomes, however this is based on evidence in people with knee OA [8]. Irrefutable health benefits are associated with weight loss for those with overweight or obesity [16]. However, our systematic review and meta-analysis of clinical trials found that a combination of diet and exercise reduced knee OA pain compared to control, but not for diet only interventions compared to control [17]. Notably, the control was considered either an active (non-diet treatment) or no treatment (including placebo or waiting list) [17]. It remains uncertain whether weight loss in addition to exercise and regular physical activity is more benefical for hip OA pain compared to exercise alone.
Among the myriad of weight loss strategies, the ketogenic very low calorie diet (VLCD) leads to greater short-term weight loss compared to low-fat diets [18] and low calorie diets [19]. This program is typically characterised by an intensive weight loss phase followed by a transition and maintenance phase. A systematic review and meta-analysis of ketogenic VLCDs found an average 14% reduction in body weight loss in studies with a ketogenic phase up to 12 weeks with loss in body weight stable up to 2 years follow-up [19]. Additionally, a VLCD was associated with improvements in factors related to cardiovascular disease such as blood pressure, total cholesterol and triglycerides. Importantly, in combination with exercise, using nutritionally complete meal replacement products is considered safe in adults aged up to 80 years or older [20]. A VLCD is perceived as acceptable to people with knee OA [21], and our pilot study in hip OA (n = 18) demonstrated feasibility of a ketogenic VLCD and exercise intervention delivered remotely [22]. We observed an average of 10% [95% CI 8 to 12%] reduction in body weight at 3 months and no adverse events related to the diet program were reported [22].
The primary aim of this randomised controlled trial (RCT) is to determine if a 6-month diet and exercise program results in significantly greater improvements in hip pain compared to an exercise only program at 6 months. We hypothesise that diet and exercise will reduce pain more than exercise alone.
The secondary aims of this study are to test the hypothesis that: i) a diet and exercise program will improve hip pain more than exercise alone at 12-months; ii) a diet and exercise program will improve other clinical outcomes (physical function, body composition, quality of life; physical activity) at 6 months and 12 months.
Methods
Design and management
The ECHO trial is a superiority, 2-group, parallel RCT. The trial will be based in Melbourne, Australia. The University of Melbourne Human Research Ethics Committee approved the study (HREC 201516) and the trial was prospectively registered with ClinicalTrials.gov (NCT04825483). The trial is sponsored by the University of Melbourne. The trial was designed according to the Consolidated Standards of Reporting Trials statement [23] and is reported according to the Standard Protocol Items: Recommendations for Interventional Trials statement [24]. The current study protocol (version 2) is available online as Additional File 1 and any protocol amendments will be detailed in the trial registration following ethics approval. All participants will provide written informed consent. Due to the expected low risk of harm based on our pilot study [22], we have not planned a data safety monitoring committee. There is also no planned interim analysis or stopping guidelines.
Participants
We will recruit 100 participants with hip OA from metropolitan Melbourne and Sydney through advertisement, print/radio/social media and our research volunteer database. Hip OA will be classified according to the American College of Rheumatology classification criteria (pain in the groin or hip region on most days of the past month and femoral or acetabular osteophytes and joint space narrowing (superior, axial and/or medial) on x-ray) [25]. Participants will be included if they:
-
i)
are aged 50 years or older;
-
ii)
report a history of hip pain ≥3 months;
-
iii)
report an average pain score of at least 4 on an 11-point numeric rating scale (anchored at 0 = no pain, 10 = worst pain imaginable) over the previous week;
-
iv)
report pain in the groin or hip region on most days of the past month [25];
-
v)
have femoral or acetabular osteophytes and joint space narrowing (superior, axial and/or medial) on x-ray) [25];
-
vi)
have access to a device with internet connection;
-
vii)
have a BMI > 27 kg/m2; based on the Royal Australian College of General Practitioners guideline for obesity management, which indicates usage of a very low energy diet to induce rapid weight loss for those with BMI > 30 kg/m2 or those with BMI > 27 kg/m2 who also suffer comorbidities such as OA [26];
-
viii)
are willing and able to give informed consent and participate fully in the interventions and assessment procedures;
-
ix)
have the ability to weigh themselves (e.g. access to scales);
-
x)
pass the Exercise and Sports Science Australia stage 1 adult pre-exercise screening system [27] or obtain general practitioner clearance for participation in the study.
Participants will be excluded if they:
-
i)
weigh > 150 kg (due to the added complexities of additional nutritional requirements for individuals above this weight);
-
ii)
are unable to speak and read English;
-
iii)
are on a waiting list for/planning back/lower limb surgery or bariatric surgery in the next 12 months;
-
iv)
have had previous arthroplasty on affected hip;
-
v)
have had recent hip surgery on affected hip (past 6 months);
-
vi)
have self-reported inflammatory arthritis (e.g. rheumatoid arthritis);
-
vii)
lost > 2 kg weight over the previous 3 months (to ensure that participants were not already engaging in weight change behaviour)
-
viii)
are already actively trying to lose weight by any of the following mechanisms:
-
a.
using meal replacements for weight loss;
-
b.
being a member of a commercial weight loss program (e.g. Weight Watchers);
-
c.
receiving support from any health care professional for weight loss (i.e. participating in ongoing consultations about weight loss with a health care professional);
-
d.
using any drugs prescribed to aid in weight loss;
-
e.
using structured meal programs for weight loss such as ‘Lite n’ Easy’;
-
ix)
are unable to undertake a ketogenic VLCD without close medical supervision including self-reported:
-
a.
diagnosis of Type 1 diabetes;
-
b.
Type 2 diabetes requiring insulin or other medication apart from metformin;
-
c.
warfarin use;
-
d.
stroke or cardiac event in previous 6 months;
-
e.
unstable cardiovascular condition;
-
f.
fluid intake restriction;
-
g.
renal (kidney) problems (unless clearance is obtained from GP, including GP confirmation that estimated glomerular filtration rate > 30 mL/min/1.73m2);
-
xxiv)
have any neurological condition affecting lower limbs;
-
xxv)
are pregnant or planning pregnancy;
-
xxvi)
have vegan dietary requirements (due to the complexity of delivering a nutritionally complete diet within the ketogenic diet regime).
Procedure
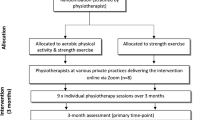
The trial phases are outlined in Fig. 1. Volunteers will initially be screened by an online form, then over the phone by research staff at The University of Melbourne. Volunteers may also call a telephone number to proceed directly to phone screening if preferred to completing the online screening. Additional clearance to participate will be sought from a general practitioner for anyone who does not pass the pre-exercise screening questions and/or indicates that they suffer with renal dysfunction. During the telephone screening process the Trial Coordinator will provide a verbal description of the project to ensure that participants fully understand the trial procedures. If participants pass the eligibility criteria assessed during the telephone screening process, they will receive the Plain Language Statement (PLS) and Consent Form (see Additional File 2) in the post or by email. After reading the PLS, and if they give their consent to participate, consent will be obtained online using REDCap or they will sign a paper-based consent form and return it via a reply-paid envelope in the post or by scanning and emailing the document to the Trial Coordinator. Screening information and study consent forms will be stored within a secure data collection platform (Qualtrics or REDCap) and accessible only by password to the researchers. Potentially eligible participants will undergo an antero-posterior supine hip x-ray. Participants who have undergone an antero-posterior supine hip x-ray in the prior 12 months and can provide the images and reports to the research staff for screening will not undergo new x-rays. It is deemed unnessary to expose these partcipants to additional radiation. For participants with bilaterally eligible hips, the most symptomatic hip will be deemed the study hip with respect to outcome measurement.

Flow diagram of study phases
Data collection and management
Body composition scans and x-rays will be collected at clinics in Melbourne and Sydney and will be stored on a secure password-protected server. Other participant-reported or clinician reported outcomes will be collected electronically via computer or paper (if preferred) at all times. If questionnaires are not completed or returned, the participant will be contacted by email and/or phone to prompt completion and return, or as a last resort, to obtain primary outcome data over the phone. Paper questionnaires will be stored in locked filing cabinets, and the data will be manually entered into REDCap by the Trial Coordinator. Electronic copies will be stored in the REDCap website, accessible only to the researchers by password protection.
Randomisation and allocation concealment
Eligible participants will be randomised to receive either i) diet and exercise or ii) exercise. An independent biostatistician has prepared the randomisation schedule (random permuted blocks) stratified by site (Melbourne or Sydney) and sex (male or female). The schedule will be stored on a password-protected website (REDCap) at The University of Melbourne maintained by a researcher not involved in either participant recruitment or administration of outcome measures. Each participant will receive a unique study ID code, and this will be documented in the participant’s record/database in addition to all study documents. Group allocation will be revealed after completion of baseline outcomes.
Although participants will not be informed about the study hypothesis until the study is completed, the participants will be unblinded. Participants will be informed that we are comparing the two interventions (described below) to determine which intervention achieves the most benefits. The primary outcomes and some of the secondary outcomes are participant-reported, so participants are also the assessors. Physiotherapists and dietitians will not be blinded to group allocation or study hypothesis. Staff conducting assessments for some of the secondary outcomes (e.g. body composition) will be blinded. Statistical analyses will be performed blinded.
Interventions
The interventions are based on a diet and exercise program developed by the researchers for people with knee OA [28], with input from various stakeholders, and are reported according to TIDIeR recommendations [29]. Clinical consultations are informed by the Behaviour Change Wheel [30] and based on research into exercise [28, 31] and weight management [22] for hip and knee OA. Prior to the first consultation with the physiotherapist and dietitian, participants will complete a pre-consultation survey asking about their main problems and goals, brief history of their hip symptoms and other health issues and previous weight management if they are allocated to the weight loss and exercise group.
Exercise
In addition to receiving hardcopy educational resources (Table 1) via post, participants in both groups will have 5 videoconference individual consultations with a physiotherapist over 6 months. The timing of these consultations is recommended in weeks 1, 3, 9, 15 and 21, but will be negotiated between the participant and their physiotherapist. The first consultation will last for approximately 45 min, with the remaining consultations to last approximately 30 min. Participants will receive four exercise resistance bands of varying resistance (yellow, green, red and blue), an ankle cuff-weight (1.0 to 5.0 kg), a booklet of exercise instructions (along with progressions) and an exercise logbook to record their program and what exercises they complete. The exercise logbooks are used as motivational tool for participants and for their physiotherapist to assess their progress through the exercise program. Physiotherapists will i) support participants to engage in a structured and progressive muscle strengthening program; ii) support participants to engage in a physical activity plan to increase incidental and general physical activity; iii) identify barriers to exercise and approaches to overcome these; iv) advise how to self-monitor and manage flare-ups, and v) review, progress and modify program as appropriate.
The strengthening exercise program is to be completed 3 times per week and includes exercises that target the hip extensors, hip flexors, hip abductors, hip adductors and knee extensors. Physiotherapists will prescribe 4–6 exercises (Table 2), at an intensity where participants feel that is “hard to very hard” to complete a full set of each exercise (equivalent to 5–7 on the Borg CR 10 Rating of Perceived Exertion) [32]. The volume (i.e. set and repetitions) will be negotiated between the physiotherapist and participant. During the consultations, physiotherapists will i) review the logbook to assess adherence and progress; ii) modify the strengthening program as needed; iii) provide feedback on technique and; iv) discuss any flare-ups or adverse events. For participants in the weight loss and exercise group, physiotherapists will also prescribe arm strengthening exercises to minimise loss of muscle mass, which can occur with weight loss [33]. For the physical activity plan, physiotherapists will encourage the participant to increase their general and incidental levels of physical/aerobic activity based on their individual need and goals, as well as their current level of activity. Participants will be encouraged to keep going with the exercise program after consultations with the physiotherapists finish.
Diet
Participants in the diet and exercise group will also receive six individual consultations with a dietitian via videoconference over 6 months. The timing of these consultations is recommended in weeks 1, 3, 6, 10, between 14 and 17 weeks, and between 19 and 23 weeks, but will be negotiated between the participant and their dietitian. The first consultation will occur 2–3 days following the first consultation with the physiotherapist and will last for approximately 45 min. The remaining consultations will last approximately 30 min. For each participant, dietitians will individualise a dietary intervention involving a ketogenic VLCD. Our pilot study using a ketogenic VLCD demonstrated high acceptability among people with hip OA [22]. Participants will be encouraged to lose at least 10% of their body weight. Five percent weight loss is recommended for improvement in OA symptoms [8] albeit based on trials in knee OA but a dose response is apparent with greater benefits found for 10% or more weight loss. We have demonstrated that 10% weight loss is achievable in our pilot study using a ketogenic VLCD [22].
Prior to their first consultation, participants will receive via post a variety of Optifast meal replacements (Nestlé Health Science, Rhodes, Australia) equivalent to two meals per day for 1 month and educational materials to support weight loss (Table 1). Participants will receive their preferred Optifast meal replacements over the subsequent 5 months. Participants will be asked to weigh themselves at home weekly to record their weight in their logbook throughout the duration of the trial. The purpose of tracking weekly weight is help to motivate participants.
During the first consultation, a personalised management plan will be developed, including appropriate weight loss goals and target weight. Participants will be asked to replace two meals each day, with Optifast meal replacement products and consume a low-carbohydrate meal consisting of protein (e.g. meat, seafood, eggs or tofu), non-starchy vegetables or salad and a tablespoon of oil/fat (if gall bladder in situ). The diet consists of approximately 800 kcal (3280 kJ) per day. Modifications of the diet will be permitted as necessary. Throughout the remaining five consultations, dietitians will discuss progress and use motivational interviewing principles and techniques to help motivation, self-efficacy and overcome barriers preventing participants completing their weight loss plan. Dietitians will also discuss the resource booklets sent to participants, to help adhere to their weight loss plan such as choosing a support person and planning for unforeseen events.
Once participants achieve at least 10% weight loss, they will choose whether to transition to a weight maintenance phase or continue with the ketogenic VLCD to reduce weight further. The transition phase will last at least 2 weeks and involves reducing meal replacements to one meal per day and reintroducing low glycaemic index carbohydrates. Upon completion of the transition phase, participants will enter the maintenance phase and be asked to consume a healthy diet in accordance with the Commonwealth Scientific and Industrial Research Organisation Total Wellbeing diet of high protein, low glycaemic index carbohydrate and low fat. Participants who regain a minimum of 2 kg or more will be advised to again engage in the ketogenic VLCD for 1–2 weeks. Online consultation notes will be used by dietitians. Participants will have access to meal replacements from enrolment to 6 months at no cost to themselves, after which if they opt to re-engage with the ketogenic VLCD, meal replacements will be self-funded. Long term weight maintenance strategies will be addressed during the program to help participants maintain their weight loss after their dietitian support finishes.
Physiotherapist and dietitian training
Eight musculoskeletal physiotherapists with experience managing people with hip OA, and current registration with Australian Health Practitioner Regulation Agency will deliver the exercise program. All physiotherapists attended a two-hour training session and were provided with a treatment manual describing the exercise program. Five accredited dietitians, with clinical experience in weight management will deliver the diet intervention. All dietitians will complete i) training in best-practice OA management delivered by researchers; ii) motivational interviewing skills delivered by Health and Wellbeing Training Consultants over 2-days; iii) ketogenic VLCD delivered by researchers via 1-h webinar. Further details on training have been published elsewhere [28]. Telephone meetings will be conducted between the research team, physiotherapists and dietitians to address any study issues that arise.
Treatment fidelity
Throughout the trial, clinicians will be encouraged to discuss any issues that arise delivering the interventions with the researchers. Treatment notes from consultations will be assessed for dietitian and physiotherapist adherence to trial protocol.
Outcome measures
Table 3 summarises the outcome measures and assessment time-point for each outcome measure. The primary outcome is overall average hip pain severity in the last week measured on an 11-point numeric rating scale (NRS), where 0 = ‘no pain’ and 10 = ‘worst pain possible’ [34] measured at baseline, 6 months and 12 months. This is a validated measure of pain that has been recommended for use in OA clinical trials [35]. Conclusions regarding efficacy will be based on the 6-month change in our primary outcome.
Secondary outcomes
-
i)
Body weight, measured in kilograms. Participants will be asked to use the same set of scales across the time-points;
-
ii)
Body mass index, measured in kg/m2;
-
iii)
Visceral fat mass, measured in grams and percentage of total body mass;
-
iv)
Total body fat mass, measured in grams and percentage of total body mass;
-
v)
Hip pain, measured as the proportion who meet or exceed the minimal clinical important difference (MCID) in NRS pain (at least 1.8 units of improvement) [36];
-
vi)
Hip Osteoarthritis Outcome Scale (HOOS) pain subscale, normalised to 0–100 scale (100 indicating no symptoms and 0 indication extreme symptoms) [37];
-
vii)
HOOS activities of daily living subscale, normalised to 0–100 scale (100 indicating no difficulty and 0 indication extreme difficulty) [37];
-
viii)
HOOS quality of life subscale, normalised to 0–100 scale (100 indicating better quality of life and 0 indication lower quality of life) [37];
-
ix)
Health-related quality of life, measured using the Assessment of Quality of Life Instrument (AQoL-8D) [38];
-
x)
Global rating of change in physical activity, scored on a 7-point Likert scale for “change in your physical activity levels since you began the study” from “much less” to “much more”;
-
xi)
Global rating of overall change in hip problem, scored on a 7-point Likert scale for “change in your hip problem since you began the study” from “much worse” to “much better” when compared to baseline.
Descriptive measures
Descriptive measures include age; body height, sex; duration of symptoms; geographical location; education level; current employment status; problems such as pain, aching discomfort or stiffness in other joints; radiographic disease severity using the Kellgren and Lawrence scale [39]; comorbidities assessed using the Self-Administered Comorbidity Questionnaire [40]; and treatment expectation assessed on a 5-point ordinal scale.
Other measures
Other measures will be collected to describe total body lean mass; depression, anxiety and stress [41]; pain-related fear [42], and self-control for eating [43] (Table 3). These measures will not be used to determine treatment efficacy.
Treatment adherence
At 6 months, participants will report the number of strengthening exercise sessions performed over the prior 2 weeks. At 6 months, adherence to the prescribed physical activity programs, and weight management plan, if applicable, will be self-reported by participants using a 5-point NRS (0 = none of the time; 5 = all of the time). Physiotherapists and dietitians will record the number of attended consultations and the duration of the consultations throughout the 6-months for each participant. The number of weeks participants use meal replacements (diet and exercise group) will also be recorded.
Adverse events
Adverse events will be defined as any untoward medical occurrence, irrespective of whether it is related to the study interventions or not. Participants are advised to report any adverse events to their study physiotherapist or dietitian as soon as they can. If necessary, treatment is discontinued and further medical advice is arranged. Clinicians are instructed to report any adverse events to the Trial Coordinator and record the event in their consultation notes. Participants are provided with a “help” email address to contact if they have any issues with their exercise/physical activity or weight management program in between consultations. The email is monitored daily by research staff and, if necessary, staff can seek further advice/guidance from the participant’s physiotherapist and/or dietitian. Adverse events will be acquired from participants at 6 and 12 months. Participants are requested to provide details on the nature of the event, if they sought treatment from a doctor or any other health professional, if the event was life-threatening or required hospitalisation, and whether they considered the event related to any component of the intervention they are receiving as part of the study. The Chief Investigator along with other study investigators will determine causality. If the event is related to the trial, it will be deemed a related adverse event. A serious adverse event will be defined as any untoward medical occurrence that; i) results in death; ii) is life-threatening; iii) requires hospitalisation or prolongation of existing inpatients hospitalisation; iv) results in persistent or significant disability or incapacity; v) is a congenital anomaly or birth defect, or; vi) any other important medical condition which, although not included in the above, may require medical or surgical intervention to prevent one of the outcomes listed.
Co-interventions
Participants will indicate whether they have taken common arthritis medications over the past month for their study hip at baseline. At 6 and 12 months, participants will indicate whether they used oral opioid medication, paracetamol or paracetamol combinations and oral anti-inflammatories for their study hip ‘less often’, ‘more often’ or the ‘same as before’ (over the past month). At baseline, 6 and 12 months, participants will complete a customised questionnaire regarding prescription and non-prescription medications including injections, over the past 6 months.
Sample size calculation
We aim to detect a MCID between groups of 1.8 NRS units in change in overall hip pain severity (baseline minus 6 months) [36]. We have performed the sample size calculation taking into account differential clustering between arms. As the physiotherapists treat participants in both arms of the trial, only the clustering by dietitians was accounted for in the sample size calculation. We have assumed a conservative standard deviation at baseline across all participants of 2.5 units and a conservative correlation between baseline and 6-month scores of 0.25, as guided by our other research [44]. We have also assumed an intra-cluster correlation of 0.05 and that there will be 5 dietitians treating approximately 8 patients each. Given these parameters, we need 40 per arm to achieve 80% power to detect the MCID in pain at a 0.05 significance level. Allowing for 20% attrition, we will recruit 50 people per arm in total (n = 100).
Statistical analyses
A biostatistician will analyse blinded data. Main comparative analyses between groups will be performed using intention-to-treat. Multiple imputation will be used if the primary outcome has greater than 5% missing data. For the primary hypothesis, differences in mean change in pain (baseline minus follow-up) will be compared between groups using a mixed-effects linear regression model including all data from 6 and 12 months for each participant with random effects for participants, and accounting for clustering by physiotherapist and dietitian as appropriate. The model will be adjusted for baseline values and the stratifying variables of site (Sydney/Melbourne) and sex. Similar mixed-effects linear regression models will be used for continuous secondary outcomes measured at baseline, 6 months, and 12 months. The proportion of participants in each group that show an improvement that reaches or exceeds the MCID in pain (≥1.8 units) will also be calculated. For this and other binary outcomes, groups will be compared using risk differences and risk ratios, calculated from logistic regression models adjusted for the stratifying variables of site location and sex, and fit using generalized estimating equations to account for clustering. All effect sizes, 95% confidence intervals and p-values will be reported and used to interpret the results. Standard diagnostic plots will be used to check model assumptions. A full statistical analysis plan, including a description of secondary analyses (e.g. mediation analyses and/or health economic analysis) will be published prior to undertaking the formal analysis of collected data.
Timelines
Ethical approval was obtained from the Human Research Ethics Committee of The University of Melbourne in March 2021. Recruitment commenced in May 2021 and will be completed in May 2022, pending COVID-related restrictions. The trial is due for completion in May 2023 when all participants will have completed 12-month data.
Patient and public involvement
The trial has been endorsed by the Australia and New Zealand Musculoskeletal (ANZMUSC) Clinical Trial Network indicating its high priority and quality, importance to consumers/patients, clinicians and policy makers, and its potential to improve patient outcomes. The ANZMUSC endorsement process included an assessment of the protocol by a scientific and consumer advisory group that supported the design of the trial.
Dissemination
Study findings will be disseminated through publication in peer-reviewed journals and conference presentations.
Discussion
This protocol presented the rationale, theoretical foundation, and protocol for a two-arm comparative effectiveness clinical trial comparing diet and exercise to exercise only for people with hip OA. Clinical guidelines recommend weight loss for hip OA in the absence of robust evidence on the effectiveness of diet interventions for hip OA. High quality trials specific to hip OA have been highlighted by leading international organisations as a priority [5]. Findings from the ECHO trial will assist clinicians and patients with hip OA in determining whether a diet intervention in addition to exercise is beneficial for hip OA symptoms compared to exercise alone.
Availability of data and materials
The datasets used and/or analyses including statistical code will be available from the corresponding author (k.bennell@unimelb.edu.au) on reasonable request once the study has been completed.
Abbreviations
- ANZMUSC:
-
Australia & New Zealand Musculoskeletal Clinical Trials Network
- ANCOVA:
-
Analysis of Covariance
- AQoL-6d:
-
Assessment of Quality of Life instrument − 6 dimension
- BMI:
-
Body Mass Index
- CHESM:
-
Centre for Health, Exercise and Sports Medicine
- CI:
-
Confidence Interval
- MCID:
-
Minimal Clinically Important Difference
- NRS:
-
Numeric Rating Scale
- OA:
-
Osteoarthritis
- OARSI:
-
Osteoarthritis Research Society International
- PLS:
-
Plain Language Statement
- RCT:
-
Randomised Controlled Trial
- SD:
-
Standard Deviation
- VLCD:
-
Very Low-Calorie Diet
References
Murphy LB, Helmick CG, Schwartz TA, Renner JB, Tudor G, Koch GG, et al. One in four people may develop symptomatic hip osteoarthritis in his or her lifetime. Osteoarthr Cartil. 2010;18(11):1372–9.
Ackerman IN, Bohensky MA, de Steiger R, Brand CA, Eskelinen A, Fenstad AM, et al. Lifetime risk of primary Total hip replacement surgery for osteoarthritis from 2003 to 2013: a multinational analysis using National Registry Data. Arthritis Care Res (Hoboken). 2017;69(11):1659–67.
McAlindon TE, Bannuru RR, Sullivan MC, Arden NK, Berenbaum F, Bierma-Zeinstra SM, et al. OARSI guidelines for the non-surgical management of knee osteoarthritis. Osteoarthr Cartil. 2014;24(14):00016–8.
The Royal Australian College of General Practitioners. Guideline for the management of knee and hip osteoarthritis. 2nd edn. East Melbourne, Vic: RACGP; 2018.
Bannuru RR, Osani MC, Vaysbrot EE, Arden NK, Bennell K, Bierma-Zeinstra SMA, et al. OARSI guidelines for the non-surgical management of knee, hip, and polyarticular osteoarthritis. Osteoarthr Cartil. 2019;27(11):1578–89.
Fernandes L, Hagen KB, Bijlsma JWJ, Andreassen O, Christensen P, Conaghan PG, et al. EULAR recommendations for the non-pharmacological core management of hip and knee osteoarthritis. Ann Rheum Dis. 2013;72(7):1125–35.
National Clinical Guideline Centre. Osteoarthritis. Care and management in adults. NICE clinical guideline CG177. London; National Institute for Health and Clinical Excellence.
Kolasinski SL, Neogi T, Hochberg MC, Oatis C, Guyatt G, Block J, et al. 2019 American College of Rheumatology/Arthritis Foundation guideline for the Management of Osteoarthritis of the hand, hip, and knee. Arthritis Rheumatol. 2020;72(2):220–33.
Moseng T, Dagfinrud H, Smedslund G, Østerås N. The importance of dose in land-based supervised exercise for people with hip osteoarthritis. A systematic review and meta-analysis. Osteoarthr Cartil. 2017;25(10):1563–76.
Moseng T, Dagfinrud H, Smedslund G, Østerås N. Corrigendum to ‘The importance of dose in land-based supervised exercise for people with hip osteoarthritis. A systematic review and meta-analysis’. Osteoarthritis Cartilage. 2017;25:1563–76 In: 2018 ; Great Britain: Elsevier Science B.V., Amsterdam; 2018: 707.
Loureiro A, Mills PM, Barrett RS. Muscle weakness in hip osteoarthritis: a systematic review. Arthritis Care Res (Hoboken). 2013;65(3):340–52.
Veronese N, Cereda E, Maggi S, Luchini C, Solmi M, Smith T, et al. Osteoarthritis and mortality: a prospective cohort study and systematic review with meta-analysis. Semin Arthritis Rheum. 2016;46(2):160–7.
Wallis JA, Webster KE, Levinger P, Taylor NF. What proportion of people with hip and knee osteoarthritis meet physical activity guidelines? A systematic review and meta-analysis. Osteoarthr Cartil. 2013;21(11):1648–59.
Svege I, Nordsletten L, Fernandes L, Risberg MA. Exercise therapy may postpone total hip replacement surgery in patients with hip osteoarthritis: a long-term follow-up of a randomised trial. Ann Rheum Dis. 2015;74(1):164–9.
Reyes C, Leyland KM, Peat G, Cooper C, Arden NK, Prieto-Alhambra D. Association between overweight and obesity and risk of clinically diagnosed knee, hip, and hand osteoarthritis: a population-based cohort study. Arthritis Rheumatol (Hoboken, NJ). 2016;68(8):1869–75.
Ross R, Bradshaw AJ. The future of obesity reduction: beyond weight loss. Nat Rev Endocrinol. 2009;5(6):319–25.
Hall M, Castelein B, Wittoek R, Calders P, Van Ginckel A. Diet-induced weight loss alone or combined with exercise in overweight or obese people with knee osteoarthritis: a systematic review and meta-analysis. Semin Arthritis Rheum. 2019;48(5):765–77.
Purcell K, Sumithran P, Prendergast LA, Bouniu CJ, Delbridge E, Proietto J. The effect of rate of weight loss on long-term weight management: a randomised controlled trial. Lancet Diabetes Endocrinol. 2014;2(12):954–62.
Castellana M, Conte E, Cignarelli A, Perrini S, Giustina A, Giovanella L, et al. Efficacy and safety of very low calorie ketogenic diet (VLCKD) in patients with overweight and obesity: a systematic review and meta-analysis. Rev Endocr Metab Disord. 2020;21(1):5–16.
Haywood CJ, Prendergast LA, Purcell K, Le Fevre L, Lim WK, Galea M, et al. Very low calorie diets for weight loss in obese older adults-a randomized trial. J Gerontol A Biol Sci Med Sci. 2017;73(1):59–65.
Lawford BJ, Bennell KL, Jones SE, Keating C, Brown C, Hinman RS. “It’s the single best thing I’ve done in the last 10 years”: a qualitative study exploring patient and dietitian experiences with, and perceptions of, a multi-component dietary weight loss program for knee osteoarthritis. Osteoarthr Cartil. 2021;29(4):507–17.
Hall M, Spiers L, Knox G, Hinman RS, Sumithran P, Bennell KL. Feasibility of exercise and weight management for people with hip osteoarthritis and overweight or obesity: a pilot study. Osteoarthritis Cartilage Open. 2021;3(3):100174.
Schulz KF, Altman DG, Moher D. CONSORT 2010 statement: updated guidelines for reporting parallel group randomised trials. Int J Surg. 2011;9(8):672–7.
Chan AW, Tetzlaff JM, Altman DG, Laupacis A, Gotzsche PC, Krleza-Jeric K, et al. SPIRIT 2013 statement: defining standard protocol items for clinical trials. Ann Intern Med. 2013;158(3):200–7.
Altman R, Alarcón G, Appelrouth D, Bloch D, Borenstein D, Brandt K, et al. The American College of Rheumatology criteria for the classification and reporting of osteoarthritis of the hip. Arthritis Rheum. 1991;34(5):505–14.
Grima M, Dixon JB. Obesity recommendations for management in general practice and beyond. Aust Fam Physician. 2013;42(8):532–41.
Australia ESS: Adult Pre-Exercise Screening System (APSS). 2019.
Bennell KL, Keating C, Lawford BJ, Kimp AJ, Egerton T, Brown C, et al. Better knee, better me™: effectiveness of two scalable health care interventions supporting self-management for knee osteoarthritis - protocol for a randomized controlled trial. BMC Musculoskelet Disord. 2020;21(1):160–0.
Hoffmann TC, Glasziou PP, Boutron I, Milne R, Perera R, Moher D, et al. Better reporting of interventions: template for intervention description and replication (TIDieR) checklist and guide. BMJ. 2014;348:g1687.
Michie S, Ashford S, Sniehotta FF, Dombrowski SU, Bishop A, French DP. A refined taxonomy of behaviour change techniques to help people change their physical activity and healthy eating behaviours: the CALO-RE taxonomy. Psychol Health. 2011;26(11):1479–98.
Bennell KL, Nelligan R, Dobson F, Rini C, Keefe F, Kasza J, et al. Effectiveness of an internet-delivered exercise and pain-coping skills training intervention for persons with chronic knee pain: a randomized trial. Ann Intern Med. 2017;166(7):453–62.
Borg G. Borg’s perceived exertion and pain scales. Champaign: Human Kinetics; 1998.
Frimel TN, Sinacore DR, Villareal DT. Exercise attenuates the weight-loss-induced reduction in muscle mass in frail obese older adults. Med Sci Sports Exerc. 2008;40(7):1213–9.
Bellamy N. Osteoarthritis clinical trials: candidate variables and clinimetric properties. J Rheumatol. 1997;24(4):768–78.
Bellamy N. Outcome measurement in osteoarthritis clinical trials. J Rheumatol Suppl. 1995;43:49–51.
Bellamy N, Carette S, Ford P, Kean W, le Riche N, Lussier A, et al. Osteoarthritis antirheumatic drug trials. II. Tables for calculating sample size for clinical trials. J Rheumatol. 1992;19(3):1992.
Nilsdotter AK, Lohmander LS, Klässbo M, Roos EM. Hip disability and osteoarthritis outcome score (HOOS)--validity and responsiveness in total hip replacement. BMC Musculoskelet Disord. 2003;4:10–0.
Osborne RH, Hawthorne G, Lew EA, Gray LC. Quality of life assessment in the community-dwelling elderly: validation of the assessment of quality of life (AQoL) instrument and comparison with the SF-36. J Clin Epidemiol. 2003;56(2):138–47.
Kellgren JH, Jeffrey MR, Ball J. The epidemiology of chronic rheumatism: atlas of standard radiographs, vol. 2. Oxford: Blackwell Scientific; 1963.
Sangha O, Stucki G, Liang MH, Fossel AH, Katz JN. The self-administered comorbidity questionnaire: a new method to assess comorbidity for clinical and health services research. Arthritis Rheum. 2003;49(2):156–63.
Henry JD, Crawford JR. The short-form version of the depression anxiety stress scales (DASS-21): construct validity and normative data in a large non-clinical sample. Br J Clin Psychol. 2005;44(Pt 2):227–39.
Shelby RA, Somers TJ, Keefe FJ, DeVellis BM, Patterson C, Renner JB, et al. Brief fear of movement scale for osteoarthritis. Arthritis Care Res. 2012;64(6):862–71.
Ames GE, Heckman MG, Grothe KB, Clark MM. Eating self-efficacy: development of a short-form WEL. Eat Behav. 2012;13(4):375–8.
Bennell KL, Nelligan RK, Rini C, Keefe FJ, Kasza J, French S, et al. Effects of internet-based pain coping skills training before home exercise for individuals with hip osteoarthritis (HOPE trial): a randomised controlled trial. Pain. 2018;159(9):1833–42.
Acknowledgements
Not applicable.
Funding
This study is funded by the National Health and Medical Research Council Investigator Grant (#1774431) and Centre of Research Excellence Grant (#1079078). MH is supported by a NHMRC Investigator Grant Emerging Leader 1 (#1172928). PS is supported by an NHMRC Investigator Grant (#1178482). RSH is supported by NHMRC Senior Research Fellowship (#1154217). KLB is supported by NHMRC Investigator Grant Leadership 2 (#1174431). DJH is supported by an NHMRC Investigator Grant Leadership 2 (#1194737). FMC is supported by an NHMRC Investigator Grant (APP1194829). The funding sources had no role in the design of this study and will not have any role during its execution, analyses, interpretation of the data, or decision to submit results.
Author information
Authors and Affiliations
Contributions
MH, RSH, KLB conceived the idea for the study and KLB is leading the trial. KLB obtained funding for the study. MH, RSH, GK, LS, KLB designed the trial protocol with input from NJM, FC, DJH, PS, FM, KEL and SPM. MH drafted the manuscript with input from RSH, GK, LS, PS, NJM, FM, KEL, FC, DJH, SPM and KLB. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical approval has been obtained from the University of Melbourne Human Research Ethics Committee (HREC 20516). All participants will provide written informed consent.
Consent for publication
Not applicable.
Competing interests
KB received payments from Wolters Kluwer for UpToDate clinical Osteoarthritis guidelines.
PS reports fees from Novo Nordisk for participation in advisory boards and a lecture unrelated to the submitted work. DJH provides consulting advice on scientific advisory boards for Pfizer, Lilly, TLCBio, Novartis, Tissuegene, Biobone.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
ECHO study protocol version 2.
Additional file 2.
ECHO study participant consent form.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hall, M., Hinman, R.S., Knox, G. et al. Effects of adding a diet intervention to exercise on hip osteoarthritis pain: protocol for the ECHO randomized controlled trial. BMC Musculoskelet Disord 23, 215 (2022). https://doi.org/10.1186/s12891-022-05128-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12891-022-05128-9