
Abstract
Background
Progressive pulmonary fibrosis is the symptomatic, physiological, and radiological progression of interstitial lung diseases. The aim of this study was to examine the relationship between progressive pulmonary fibrosis and demographic characteristics and to evaluate the effect on clinical outcomes and mortality.
Methods
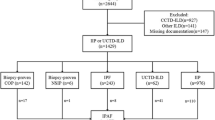
This cross-sectional study included 221 patients diagnosed with non-idiopathic pulmonary fibrosis interstitial lung diseases who were followed in the last 5 years. Patient symptoms, clinical, radiological, and demographic data were examined. Risk factors for the development of progressive pulmonary fibrosis and the relationship with clinical outcomes and mortality were examined.
Results
Of the patients, 33.0% (n = 73) had fibrotic idiopathic nonspecific interstitial pneumonia (iNSIP), 35.7% (n = 79) had fibrotic hypersensitivity pneumonia (HP), 18.1% (n = 40) had fibrotic connective tissue disease (CTD) interstitial lung diseases (ILD), and 13.1% (n = 29) had postinfectious fibrotic ILD. The progressive pulmonary fibrosis development rates of the subtypes were 46.5% iNSIP (n = 34), 86.0% fibrotic HP (n = 68), 42.5% fibrotic CTD-ILD (n = 17), and 20.7% postinfectious ILD (n = 6). The presence of progressive pulmonary fibrosis was associated with the development of respiratory failure and mortality (odds ratio [OR]: 2.70, 95% CI: 1.04–7.05 and OR: 2.13, 95% CI: 1.23–3.69). Progressive pulmonary fibrosis development was higher in hypersensitivity pneumonia patients with farmer’s lung (OR: 5.06, 95% CI: 1.02–25.18).
Conclusion
Progressive pulmonary fibrosis was more prevalent in older patients. Farming was an important risk factor in the development of hypersensitivity pneumonia-progressive pulmonary fibrosis. Respiratory failure and mortality were higher in those who developed progressive pulmonary fibrosis.
Similar content being viewed by others


Introduction
Interstitial lung diseases (ILD) are a heterogeneous group of more than 200 pulmonary diseases that affect the lung parenchyma [1]. Three categories exist for ILD that progress to fibrosis: idiopathic interstitial fibrosis (IPF), non-IPF ILD, and unclassifiable ILD. While IPF is the most well-known and well-recognized group of ILDs associated with fibrosis development, non-IPF ILDs such as idiopathic nonspecific interstitial pneumonia (iNSIP) and hypersensitivity pneumonia (HP) are also associated with fibrosis development. The other non-IPF ILD subtype is connective tissue disease (CTD) ILD. Comorbidity of ILD and CTD is a serious condition that affects morbidity and mortality. The clinical spectrum varies from progressive irreversible pulmonary fibrosis to mild, self-limiting illness in CTD-ILD [2]. Another non-IPF ILD is postinfectious ILD, which became more common after the COVID-19 pandemic [3].
Progressive pulmonary fibrosis (PPF) is the defining feature of progression in fibrotic lung diseases. It is defined as the presence of two of three characteristics: worsening respiratory symptoms, physiological evidence of disease progression, and radiological evidence of disease progression [4]. According to current guidelines [4], it is strongly advised that research should prioritize investigating the optimal timing for administering corticosteroids, immunosuppressants, and antifibrotic drugs in patients with PPF. Additionally, investigating various determinants of PPF in the ILD population, evaluating clinical outcomes, and identifying phenotypes indicating PPF will contribute considerably to the management of patients. This multifaceted approach aims to enhance our understanding of PPF and refine therapeutic strategies tailored to the diverse needs of affected individuals [4].
The variables related to disease progression, demographic risk factors, and prognostic indicators of diagnosis have not been fully determined for non-IPF ILD subgroups. Furthermore, the mechanisms and differences of PPF in different ILD subtypes are not fully understood [5]. Studies have shown that some patients in the ILD subgroup have similar features, such as decreased lung function, symptomatic deterioration, deterioration in quality of life, and increased mortality rates [6,7,8]. Previous cohort studies [9] have shown a 10% decrease in forced vital capacity (FVC)-predicted adverse effects on survival in patients with non-IPF ILD.
Although the existing literature includes studies examining PPF associated with collagen tissue disease [10, 11], there is a need for more research on this subject due to the paucity of publications on PPF resulting from other fibrotic ILDs. The aim of this study was to identify risk factors for PPF development in patients with non-IPF ILD, evaluate clinical outcomes, and determine its relationship with mortality.
Methods
Study design and population
This cross-sectional study enrolled patients with non-IPF interstitial lung diseases from our outpatient clinic within the last 5 years. Patient files were reviewed and their age, smoking status, and gender at the time of enrollment were recorded. The criteria in the current guidelines were used as the basis for diagnosis of PPF in non-IPF interstitial lung diseases [4]. Patients with unclassifiable interstitial pneumonia were excluded from the study. The progression of symptoms in patients after diagnosis and decrease in FVC or diffusing capacity for carbon monoxide (DLCO), were evaluated. An experienced radiologist of interstitial lung diseases evaluated the radiological evidence of disease progression. According to guideline criteria [4], PPF was diagnosed if at least two of the following criteria were met: worsening of respiratory symptoms, physiological evidence of progression, or radiological evidence of progression. Physiological evidence of progression was defined as either a 5% decrease in FVC or a 10% decrease in DLCO within 1 year. Radiological evidence of progression was defined as the progression of radiological findings within a 1-year period. The radiologist (an author) performed a blinded evaluation of the tomography findings, independent of the clinical data. All authors reached a consensus on the diagnosis of PPF by collectively reviewing the findings. The development of respiratory failure in patients, the number of annual exacerbations, the number of annual hospitalizations, and mortality were recorded. Respiratory failure was defined as a saturation value below 90% recorded by pulse oximetry or a partial oxygen pressure below 60 mmHg in arterial blood gas. For the definition of exacerbation, criteria similar to the acute exacerbation criteria of IPF were used [12]. Acute exacerbation was diagnosed by the presence of dyspnea lasting less than a month, ground glass or consolidation areas on CT scan, the exclusion of heart failure, and increased fluid overload. As this was a retrospective study, written consent from the patients was not required. This study received Ethics Committee approval from Adana City Training and Research Hospital, reference number 2668 and protocol number 129 (June 22, 2023).
Statistical analysis
All analyses were performed using SPSS 23 for Windows. The Chi-squared test was used for categorical data to examine the relationship between the characteristics of patients with and without PPF. The Mann–Whitney U test was used to calculate the mean, standard deviation, and differences between groups of continuous data. For risk calculations related to PPF, univariate logistic regression analysis was used. Conditional logistic regression analysis was used for ILD subtype risk evaluations. Kaplan–Meier survival analysis was used to calculate mean survival times. A p-value below 0.05 was considered statistically significant.
Results
There were 123 (55.7%) of the total cohort of 221 patients over the age of 65 years, and 85 (38.5%) were female. The mean age was 65.78 years, and the rate of those currently smoking was 43% (n = 95). Fibrotic iNSIP was present in 73 (33.0%) patients, fibrotic HP in 79 (35.7%), fibrotic CTD-ILD in 40 (18.1%), and postinfectious fibrotic ILD in 29 (13.1%) patients. Surgical biopsy was performed in 40 of 221 patients in this study (18%). During follow-up, respiratory failure developed in 95 (43.0%) patients and mortality occurred in 99 (44.8%) patients. Of the total number of patients, 125 (56.6%) were classified as PPF. The number of patients meeting the definition of PPF in the study population was 34 (46.5%) in iNSIP, 68 (86.0%) in fibrotic HP, 17 (42.5%) in fibrotic CTD-ILD, and six (20.7%) in post-infectious ILD. The age of patients who developed PPF was significantly higher than younger patients (p = 0.02). Furthermore, patients categorised as PPF had shorter survival times (Fig. 1). Demographic data and comparisons of non-PPF and PPF cases are presented in Table 1. When all cases were evaluated (Table 2), patients with age over 65 years had a significantly increased risk of PPF (OR: 2.36, 95% CI: 1.37–4.08). Patients with PPF were more susceptible to respiratory failure (OR: 1.73, 95% CI: 1.01–3.00) and mortality was associated with PPF (OR: 2.13, 95% CI: 1.23–3.69). Although being older and smoking appeared to be protective in terms of PPF in postinfectious ILD, there was no statistical significance. In the other three ILD subgroups, patients older than 65 years were at higher risk of PPF. There was no difference between males and females in terms of PPF. Smoking was associated with the development of PPF in iNSIP (OR: 2.88, 95% CI: 1.11–7.47) and CTD-ILD (OR: 4.04, 95% CI: 1.05–15.47). The presence of PPF was not associated with the number of hospitalizations or exacerbations per year. Patient characteristics of subtypes with PPF are detailed in Table 3. Of the total HP cases (Table 4), 21.5% (n = 17) had bird fancier’s lung and 49.3% (n = 39) had farmer’s lung. Engaging in a farming-related job increased the risk of PPF (OR: 5.06, 95% CI: 1.02–25.18). No relationship was found between medication and PPF development; the medication rates of the patients and their PPF development status are shown in Table 5.

Survival time of non-IPF interstitial lung diseases based on the presence or absence of progressive pulmonary fibrosis
Discussion
The incidence and prevalence of pulmonary fibrosis increase with age [13]. The pathophysiology and mechanisms of the increase in IPF cases in older adults have been investigated [14, 15]. Ineffective mucociliary clearance, mitochondrial dysfunction, cellular senescence, decreased autophagy, among other factors are thought to influence pulmonary fibrosis during the aging process [13]. In our study, we found that older patients were more susceptible to PPF. Our findings were based on non-IPF cases and support the results of existing studies on IPF [13,14,15]. There was no difference in terms of PPF development by gender. Being a current smoker was associated with PPF only in the iNSIP and fibrotic CTD-ILD subtypes. In our study population, the rate of PPF development in non-IPF was 56.6% and the subtype that most developed PPF was HP, with a rate of 86%. There is limited literature on PPF caused by fibrotic HP. Regarding geographical region, there are many farmers (eg, cotton farming, citrus farming, etc.) who are exposed to dust of organic origin, which usually causes bronchiolar reactions and hypersensitivity pneumonitis [16, 17]. Hypersensitivity pneumonitis is an immunologically mediated disease that results from exposure to a wide range of environmental antigens in genetically predisposed individuals [18, 19]. In our study, HP cases were significantly associated with PPF in those with a farming-related work history (OR: 5.06, 95% CI: 1.02–25.18). Continued occupational exposure might have had a negative effect on HP progression. Studies have shown that identifying exposures in HP and removing the relevant antigens has prognostic and therapeutic effects [20]. In addition, physiological parameters have been studied in relation to prognosis. The term progressive fibrosis was defined by George et al. previously as a relative decrease in FVC or DLCO, increasing fibrosis on tomographic findings, and progressive symptoms over a 24-month period [21]. Macaluso et al. suggested that HP might not need to change over a 24-month period for the patient to experience prognostically significant improvement and suggested that a 12-month follow-up could be meaningful [22]. They observed that age, physiological parameters, the presence of honeycombs on tomography, and pulmonary hypertension significantly reduced survival in patients with HP within a 1 year [22]. In a study of patients with HP, the mortality rate was 56% [23]. Compared with survivors, patients who died were older, more likely to smoke, had a lower predicted FVC percentage, and had a higher severity of dyspnea [23]. The second largest group of our study cohort was iNSIP (n = 73) with a PPF rate pf 46.5%. For iNSIP, old age (OR: 2.80, 95%CI: 1.06–7.38) and smoking (OR: 2.88, 95%CI: 1.11–7.47) appeared to be important risk factors for PPF. PPF was associated with respiratory failure (OR: 2.70, 95% CI: 1.04–7.05) and mortality (OR: 2.13, 95% CI: 1.23–3.69). In a study on 270 patients with ILD in Eastern Siberia, PPF rates were 23.8% for iNSIP, 72.7% for fibrotic HP, and 24.1% for CTD-ILD. When all ILD cases were examined, an age older than 65 years, male gender, and current smoking were risk factors for PPF [24]. Furthermore, the time from the first symptom to death of patients classified as PPF was 40 months, and the death rate was 59% [24]. Unlike our study, the published study included many more subgroups, including IPF, sarcoidosis, drug-induced ILD, and cryptogenic organizing pneumonia. The specific surveillance and characteristics of the diseases between the subgroups might explain the difference in the results. In our study, the PPF rate in fibrotic CTD-ILD was 42.5%, which is similar to a study with a large number of patients that was conducted in Canada [25]. We observed that older age (OR: 4.50, 95%CI: 1.16–17.37) and having a history of smoking (OR: 4.04, 95%CI: 1.05–15.47) were effective in the diagnosis of PPF in CTD-ILD. Kim et al. found no difference in age, gender, or smoking history between PPF and non-PPF groups with CTD-ILD [11]. Male gender, older age, and smoking are prognostic factors for ILD progression in systemic sclerosis and for mortality in patients with ILD involvement in rheumatoid arthritis [10]. The clinical course of CTD-ILD is variable, but patients with evidence of pulmonary fibrosis tend to have a worse prognosis. The group developing PPF was associated with greater respiratory failure and mortality [10]. In our study, patients considered to have PPF were more likely to develop respiratory failure and mortality. It is possible that such results would be more attainable in a larger study population.
The first limitation of the study is the time-dependent nature of PPF diagnosis. Evaluation for PPF can be performed after a 1-year follow-up. The duration from diagnosis to PPF development and the mortality period after PPF diagnosis were not calculated. Secondly, additional comorbidities might have an effect on mortality in chronic respiratory diseases. In our study, additional comorbid conditions that the patients might have had, could not be evaluated. Thirdly, since the study started before antifibrotics were approved for PPF, we were unable to evaluate the effect of the antifibrotic medication on the results. Since antifibrotics were added to the treatment in the last year, the results could not be evaluated; however, this should be considered in future research. The study was retrospective, and the data were based on patient files. In particular, the definition of exacerbation included tomography findings and dyspnea symptoms; therefore, we estimate that the number of exacerbations was higher, which might have created bias in the calculations regarding exacerbation.
Conclusion
In conclusion, old age was shown to be a risk factor for PPF. PPF was not associated with gender, the annual number of exacerbations, or hospitalizations. The mortality rate for the PPF patient group was higher compared with the group without PPF. For HP, exposure is important in the development of PPF. Considering the poor prognosis in these groups, careful consideration of antifibrotic therapy according to new guidelines is essential for patients with PPF.
Availability of data and materials
The datasets used and/or analysed during the current study a vailable from the corresponding author on reasonable request.
References
Brown KK, Martinez FJ, Walsh SLF, et al. The natural history of progressive fibrosing interstitial lung diseases. Eur Respir J. 2020;55(6):2000085. https://doi.org/10.1183/13993003.00085-2020. PMID: 32217654; PMCID: PMC7315005.
Hyldgaard C, Bendstrup E, Pedersen AB, et al. Interstitial lung disease in connective tissue diseases: survival patterns in a population-based cohort. J Clin Med. 2021;10(21):4830. https://doi.org/10.3390/jcm10214830. PMID:34768349;PMCID:PMC8584507.
Frix AN, Schoneveld L, Ladang A, et al. Could KL-6 levels in COVID-19 help to predict lung disease? Respir Res. 2020;21(1):309. https://doi.org/10.1186/s12931-020-01560-4. PMID: 33234132; PMCID: PMC7683867.
Raghu G, Remy-Jardin M, Richeldi L, et al. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2022;205(9):e18–47. https://doi.org/10.1164/rccm.202202-0399ST. PMID: 35486072; PMCID: PMC9851481.
Fan JJ, Gu JM, Xiao SY, et al. Risk factors for progression of pulmonary fibrosis: a single-centered, retrospective study. Front Med (Lausanne). 2024;11:1335758. https://doi.org/10.3389/fmed.2024.1335758. PMID: 38384414; PMCID: PMC10879408.
Wang P, Jones KD, Urisman A, et al. pathologic findings and prognosis in a large prospective cohort of chronic hypersensitivity pneumonitis. Chest. 2017;152(3):502–9. https://doi.org/10.1016/j.chest.2017.02.011. Epub 2017 Feb 20 PMID: 28223152.
Belloli EA, Beckford R, Hadley R, et al. Idiopathic non-specific interstitial pneumonia. Respirology. 2016;21(2):259–68. https://doi.org/10.1111/resp.12674. Epub 2015 Nov 13 PMID: 26564810.
Skolnik K, Ryerson CJ. Unclassifiable interstitial lung disease: a review. Respirology. 2016;21(1):51–6. https://doi.org/10.1111/resp.12568. Epub 2015 Jun 9 PMID: 26059704.
Pugashetti JV, Adegunsoye A, Wu Z, et al. Validation of proposed criteria for progressive pulmonary fibrosis. Am J Respir Crit Care Med. 2023;207(1):69–76. https://doi.org/10.1164/rccm.202201-0124OC. PMID: 35943866; PMCID: PMC9952866.
Molina-Molina M, Castellví I, Valenzuela C, et al. Management of progressive pulmonary fibrosis associated with connective tissue disease. Expert Rev Respir Med. 2022;16(7):765–74. https://doi.org/10.1080/17476348.2022.2107508. Epub 2022 Aug 5 PMID: 35912842.
Kim K, Lee J, Jo YS, et al. Factors for progressive pulmonary fibrosis in connective tissue disease-related interstitial lung disease. Ther Adv Respir Dis. 2023;17:17534666231212300. https://doi.org/10.1177/17534666231212301. PMID: 37991015; PMCID: PMC10666675.
Collard HR, Ryerson CJ, Corte TJ, et al. Acute exacerbation of idiopathic pulmonary fibrosis. An international working group report. Am J Respir Crit Care Med. 2016;194(3):265–75. https://doi.org/10.1164/rccm.201604-0801CI.
Gulati S, Thannickal VJ. The aging lung and idiopathic pulmonary fibrosis. Am J Med Sci. 2019;357(5):384–9. https://doi.org/10.1016/j.amjms.2019.02.008.
Selman M, López-Otín C, Pardo A. Age-driven developmental drift in the pathogenesis of idiopathic pulmonary fibrosis. Eur Respir J. 2016;48(2):538–52. https://doi.org/10.1183/13993003.00398-2016.
Raghu G, Chen SY, Hou Q, Yeh WS, Collard HR. Incidence and prevalence of idiopathic pulmonary fibrosis in US adults 18–64 years old. Eur Respir J. 2016;48(1):179–86. https://doi.org/10.1183/13993003.01653-2015.
Khan AJ, Nanchal R. Cotton dust lung diseases. Curr Opin Pulm Med. 2007;13(2):137–41. https://doi.org/10.1097/MCP.0b013e32802c7ceb. PMID: 17255805.
Jindal SK, Aggarwal AN, Gupta D. Dust-induced interstitial lung disease in the tropics. Curr Opin Pulm Med. 2001;7(5):272–7. https://doi.org/10.1097/00063198-200109000-00004. PMID: 11584175.
Vasakova M, Selman M, Morell F, et al. Hypersensitivity pneumonitis: current concepts of pathogenesis and potential targets for treatment. Am J Respir Crit Care Med. 2019;200(3):301–8. https://doi.org/10.1164/rccm.201903-0541PP. PMID: 31150272.
Selman M, Pardo A. When things go wrong: exploring possible mechanisms driving the progressive fibrosis phenotype in interstitial lung diseases. Eur Respir J. 2021;58(3):2004507. https://doi.org/10.1183/13993003.04507-2020. PMID: 33542060.
Pereira CA, Gimenez A, Kuranishi L, et al. Chronic hypersensitivity pneumonitis. J Asthma. Allergy. 2016;9:171–81. https://doi.org/10.2147/JAA.S81540. PMID: 27703382; PMCID: PMC5036552.
George PM, Spagnolo P, Kreuter M, et al. Progressive fibrosing interstitial lung disease: clinical uncertainties, consensus recommendations, and research priorities. Lancet Respir Med. 2020;8(9):925–34. https://doi.org/10.1016/S2213-2600(20)30355-6. PMID: 32890499.
Macaluso C, Boccabella C, Kokosi M, et al. Short-term lung function changes predict mortality in patients with fibrotic hypersensitivity pneumonitis. Respirology. 2022;27(3):202–8. https://doi.org/10.1111/resp.14204. Epub 2022 Jan 12. PMID: 35023231; PMCID: PMC9302621.
Fernández Pérez ER, Swigris JJ, et al. Identifying an inciting antigen is associated with improved survival in patients with chronic hypersensitivity pneumonitis. Chest. 2013;144(5):1644–51. https://doi.org/10.1378/chest.12-2685. PMID: 23828161; PMCID: PMC4694094.
Nashatyreva MS, Trofimenko IN, Chernyak BA, et al. Pulmonary fibrosis and progressive pulmonary fibrosis in a prospective registry of interstitial lung diseases in Eastern Siberia. Life (Basel). 2023;13(1):212. https://doi.org/10.3390/life13010212. PMID: 36676161; PMCID: PMC9861544.
Hambly N, Farooqi MM, Dvorkin-Gheva A, et al. Prevalence and characteristics of progressive fibrosing interstitial lung disease in a prospective registry. Eur Respir J. 2022;60(4):2102571. https://doi.org/10.1183/13993003.02571-2021. PMID: 35273032.
Acknowledgements
None.
Funding
The funder of the study had no role in the study design, data collection, data analysis, data interpretation, or writing of the report.
Author information
Authors and Affiliations
Contributions
ÖED: Conception and design, Administrative support, Provision of study materials or patients, Collection and assembly of data, Data analysis and interpretation, Manuscript writing, Final approval of manuscript; ÖG: Conception and design, Administrative support, Data analysis and interpretation, Manuscript writing, Final approval of manuscript; HA: Conception and design, Provision of study materials or patients, Collection and assembly of data, Manuscript writing, Final approval of manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study received Ethics Committee approval from Adana City Training and Research Hospital with reference number 2668 and protocol number 129 (22 June 2023). Due to the retrospective nature of the study, the obligation to obtain informed consent was waived by the Ethics Committee.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Erçen Diken, Ö., Güngör, Ö. & Akkaya, H. Evaluation of progressive pulmonary fibrosis in non-idiopathic pulmonary fibrosis-interstitial lung diseases: a cross-sectional study. BMC Pulm Med 24, 403 (2024). https://doi.org/10.1186/s12890-024-03226-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12890-024-03226-z