
Abstract
Background and objective
Here, we present real-world data on the incidence and risk factors of acute exacerbation (AE) in patients with chronic fibrotic interstitial pneumonia (CFIP) treated with antifibrotic agents, which has been previously poorly documented.
Methods
We retrospectively examined clinical characteristics, incidence and risk factors of AE in a cohort of 100 patients with CFIP (n = 75, idiopathic pulmonary fibrosis [IPF]; n = 25, other conditions), all of whom received antifibrotic agents in a real-world setting.
Results
The median follow-up was 17.4 months (interquartile range [IQR], 6.6 to 26.7 months). During the follow-up periods, 21 patients experienced AE after starting antifibrotic agents. The estimated 1-, 2-, and 3-year AE incidence rates were 11.4% (95% confidence interval [95%CI], 6.2–20.3%), 32% (95%CI, 20.7–47.4%), and 36.3% (95%CI 23.5–53.1%), respectively. Decreased baseline lung function (forced vital capacity and carbon monoxide diffusing capacity of the lung), existence of pulmonary hypertension estimated from an echocardiogram, higher Interstitial Lung Disease-Gender, Age, and Physiology (ILD-GAP) score, supplementary oxygen, and concomitant corticosteroid and proton-pump inhibitor (PPI) use upon starting the antifibrotic agent were risk factors of AE. Concomitant corticosteroid and PPI use and corticosteroid dose were risk factor of AE in a multivariate Cox regression hazard model adjusting for ILD-GAP score.
Conclusion
AE of CFIP is more common in patients with physiologically and functionally advanced disease under antifibrotic agents. Prudent use of corticosteroids and PPIs when initiating antifibrotic agents may be recommended. Further studies are warranted.
Similar content being viewed by others

Introduction
Chronic fibrotic interstitial pneumonia (CFIP) is associated with substantially reduced health-related quality of life and survival. Idiopathic pulmonary fibrosis (IPF) is a type of CFIP. The disease course of progressive CFIPs, such as IPF, is variable and unpredictable; however, the median survival time after IPF diagnosis is 3–5 years [1,2,3].
Until recently, no effective pharmacological options existed for IPF; however, two drugs are now approved for antifibrotic therapy: pirfenidone [4] and nintedanib [5]. Antifibrotic therapy can slow declining lung function [5, 6] and reduce the risk of death from IPF [6,7,8], but it cannot stop the disease course. International guidelines recommend using antifibrotic agents for IPF patients [9].
Confidently diagnosing CFIP is challenging [10]. Recent international clinical guidelines recommend performing surgical lung biopsies when physicians cannot confidently diagnose IPF [11]. However, in real-world practice, performing surgical lung biopsies and bronchoscopies can be difficult because of age, advanced disease, or patient refusal. In these cases, physicians sometimes prescribe antifibrotic agents when computed tomography imaging and pulmonary function tests show fibrotic processes in the lungs indicating that IPF is likely. Clinical trials evaluating antifibrotic agent efficacy and safety in patients with progressive fibrosing lung diseases other than IPF are ongoing [12, 13].
Most IPF patients have relatively slow clinical courses, but some experience acute respiratory worsening, namely acute exacerbation (AE) [14]. A recent epidemiological survey of Japanese patients with IPF revealed that the most common cause of death was AE-IPF [2]. Several studies found that AE occurs in other CFIPs as well [15, 16].
Previous studies found that AE-IPF is more common in patients with physiologically and functionally advanced disease [14]. Recent clinical trial data on AE incidence among patients treated with antifibrotics revealed that antifibrotics may reduce the probability of AE-IPF [5, 17]. However, real-world data on AE incidence and risk factors among patients treated with antifibrotics are poorly documented. Most data on AE risk factors are derived from the pre-antifibrotic agent era; thus, the risk factors for AE-CFIP in patients treated with antifibrotic agents are not well known. To address these clinical questions, this study determined the AE incidence and risk factors in CFIP patients treated with antifibrotic agents in a real-world clinical setting.
Methods
The institutional review board of Saiseikai Kumamoto Hospital approved this study, which was conducted in accordance with the Declaration of Helsinki. Because the study was retrospective, informed consent from participants was unrequired per the “Ethical Guidelines for Medical and Health Research Involving Human Subjects” presented by the Japanese Ministry of Health, Labour and Welfare.
Patients
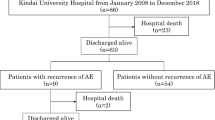
Patients with progressive CFIP treated with antifibrotic agents (pirfenidone or nintedanib) from 01 Aug 2015 to 31 Aug 2018 with no history of AE were identified at Saiseikai Kumamoto Hospital. All patients received antifibrotic agents as the standard-of-care. Follow-up included an inpatient visit or phone call by a research team member. Date of last follow-up was identified as either date of death, last in-person visit or last research phone call. Data were locked on 30 Sept 2018. Subjects were censored if they [1] experienced no events by 30 Sept 2018 or [2] were lost to follow-up.
Demographic, clinical, and pathological data were collected from electronic medical records. Patients’ pharmacy records were reviewed to determine whether patients were documented as having received corticosteroids, anticoagulant and/or antiplatelet drugs, and H2-blockers or proton-pump inhibitors upon initiating the antifibrotic agent. Corticosteroid dose was expressed as total daily milligrams of prednisone equivalents. The following clinical characteristics were collected for all patients: age upon antifibrotic initiation, gender, time from 1st visit to antifibrotic initiation (months), surgical lung biopsy (yes or no), clinical diagnosis upon starting antifibrotic agents (IPF vs others: chronic hypersensitivity pneumonitis [CHP], collagen vascular disease-associated IP, and unclassifiable), long-term oxygen therapy (no, therapy started prior to antifibrotic agent initiation, or therapy introduced simultaneously with antifibrotic agents), serum KL-6 level, serum lactate dehydrogenase (LDH), Interstitial Lung Disease-Gender, Age, and Physiology (ILD-GAP) score [18], smoking history, and updated Charlson comorbidity index [19]. Baseline pulmonary function testing, right ventricular systolic pressure estimated by echocardiogram (> or ≤ 40 mmHg), serum KL-6, and serum LDH levels at baseline were performed on the day the antifibrotic agent was initiated or within 14 days before the first antifibrotic agent treatment. ILD-GAP scores were calculated from data obtained at the start of antifibrotic agent use. Using the ILD-GAP index, we classified patients into one of three categories based on enrollment values: stage 1 (0–3 points), stage 2 (4–5 points), and stage 3 (6–8 points).
Diagnoses of acute exacerbations of interstitial pneumonia were made in accordance with the definition of acute exacerbation of IPF proposed by Collard et al. [14], which includes both triggered and idiopathic acute exacerbations. IPF was diagnosed by the diagnostic criteria proposed by the American Thoracic Society, European Respiratory Society, Japanese Respiratory Society, and Latin American Thoracic Association [20] and also included cases confirmed to have features of possible usual interstitial pneumonia (UIP) and traction bronchiectasis on high-resolution computed tomography with no surgical lung biopsy as previously described [21]. Non-IPF was diagnosed after discussing and integrating the clinical, radiological, bronchoalveolar lavage, and pathological findings.
Statistical analysis
Patients’ baseline characteristics were summarized using percentages for categorical variables and medians and interquartile ranges for continuous variables. Between-group comparisons were performed using the Mann-Whitney rank-sum and Fisher’s exact tests. Time-to-event endpoints were defined as the time from antifibrotic initiation to the first admission of AE. Time-to-end endpoints were estimated using the Kaplan-Meier method based on events and compared with the log-rank test (univariable analysis). Risk factors for time to first AE development and univariate survival were analyzed using the Cox proportional hazards model to determine hazard ratios (HR) and 95% confidence intervals (95%CI). For concomitant medications, multivariable logistic regression was performed for AE risk factors using an a priori covariable of ILD-GAP scores in some covariate.
We used the inverse probability of treatment-weighting analysis using the propensity score calculated from the data at the start of treatment to analyze the pharmacological treatment effect for the AE risk as a sensitivity analysis. All tests were two-sided and performed at a significance level of 0.05. All statistical analyses were performed using EZR (Saitama Medical Center, Jichi Medical University, Saitama, Japan) [22], a graphical user interface for R, version 3.2.2 (The R Foundation for Statistical Computing, Vienna, Austria).
Results
Baseline characteristics
One hundred patients treated with antifibrotic agents without previous history of AE were recruited. Three patients who started antifibrotics after AE were identified and excluded from this analysis.
Table 1 shows the patients’ baseline demographics and clinical characteristics. The study cohort included 75 men and 25 women, and the patients’ median age was 68 years (interquartile range [IQR] 64–73 years). The median follow-up was 17.4 months (IQR 6.6–6.7 months). The median time from first visit to starting antifibrotics was 12 months (IQR 6–36 months). The clinical diagnoses upon starting antifibrotics were IPF (n = 75) and others (n = 25; CHP = 19; collagen-vascular disease-associated [rheumatoid arthritis] =2; unclassified = 4). The baseline median values for percent predicted forced vital capacity (FVC) and percent predicted diffusing capacity of the lung for carbon monoxide (DLCO) were 70.8% (IQR 58.7–82.4%) and 55.9% (IQR 45.1–68.4%), respectively. Patients who had been diagnosed as not-IPF before commencing anti-fibrotic agents were more frequently prescribed corticosteroids than those with an IPF diagnosis at the time of starting anti-fibrotic agents (IPF n = 15 [20%] vs. non-IPF n = 11 [44%]; p = 0.033).
AE incidence
During the follow-up periods, 21 patients experienced AE after antifibrotic agent introduction. Figure 1 shows the cumulative incidence of AE-CFIP. The estimated 1-, 2-, and 3-year AE incidences were 11.4% (95%CI, 6.2–20.3%), 32% (95%CI, 20.7–47,4%), and 36.3% (95%CI, 23.5–53.1%), respectively. Table 2 compares the baseline characteristics and patient outcomes with and without AE. Sex, smoking history, clinical diagnosis, and comorbidity index distributions did not significantly differ between groups. Patients with AE were slightly younger than those without AE, but the percentage of elderly patients did not differ between groups. There was a tendency for patients who developed AEs to have had SLBs (47.6 versus 24.1% p = 0.06); however, the difference was not statistically significant. No AEs were associated with surgical lung biopsy. Patients with AE had decreased lung functions; higher ILD-GAP scores; more frequent use of corticosteroids, PPIs, and long-term oxygen therapy; and longer times from diagnosis to starting antifibrotic agents than did the non-AE group.

Cumulative incidence of acute exacerbation of chronic fibrotic interstitial pneumonia
Additional file 1: Table S1 lists details of each patient’s baseline characteristics. AE varied seasonally and appeared more frequently during winter.
Risk factors for AE
Table 3 lists risk factors of AE. Decreased baseline lung function (FVC, DLCO), estimated right ventricular systolic pressure over 40 mmHg by echocardiogram, and higher ILD-GAP score and stage were risk of AE. Patients receiving long-term oxygen therapy prior to starting antifibrotics had higher risks of AE (HR 4.8; 95%CI 1.6–14.7; P = 0.006) than did patients who had not received prior oxygen therapy or patients who began both simultaneously. Patients receiving corticosteroids upon beginning antifibrotics had higher risks of AE (adjusted HR 11.3; 95%CI 4.1–32.0; p < 0.0001) than those not receiving corticosteroids, independent of underlying disease severity. Additionally, AE was increased in patients receiving > 10 mg or 1–10 mg prednisone compared with patients not receiving corticosteroids (Fig. 2b).

(a) Cumulative incidence of acute exacerbation based on concomitant corticosteroid (with[w/] or without[w/o] PSL) use at antifibrotic agent initiation; (b) Cumulative incidence of acute exacerbation based on baseline corticosteroid dose at antifibrotic agent initiation; (c) Cumulative incidence of acute exacerbation based on concomitant use of proton-pump inhibitors (PPI)
Patients receiving PPIs also had a greater risk of AE than those not on PPIs (Figure2c), independent of underlying disease severity (adjusted HR 5.1; 95%CI 1.2–21.9; p = 0.03). Concomitantly using H2 blockers and anticoagulant and/or antiplatelet drugs with antifibrotic agents was not an AE risk.
The AE incidence rates in patients on nintedanib and pirfenidone were 15.4% (95%CI 7.3–27.2%) and 17.4% (95%CI 9.3–28.4%) per 100 patient-years, respectively, but this was statistically insignificant (Incidence rate ratio 0.89; 95%CI 0.40–1.95; P = 0.76).
To analyze sensitivity, we performed the inverse probability of the treatment-weighting analysis using the propensity score calculated from the data taken upon starting antifibrotic treatment to analyze the pharmacological treatment effect on AE risk. In this analysis, corticosteroid and PPI use at baseline were a risk factor of AE in patients treated with antifibrotics. The adjusted HRs estimated using the propensity score were 4.2 (95%CI 1.4–13.3; P = 0.013) for corticosteroid use and 6.7 (95%CI 1.5–30.1; P = 0.013) for PPI use.
Discussion
This study demonstrated the incidence and risk factors of acute exacerbation (AE) in patients with chronic fibrotic interstitial pneumonia (CFIP) treated with antifibrotic agents. AE-CFIP treated with antifibrotic agents was more common in patients with physiologically and functionally advanced diseases, as previously reported. Concomitantly using corticosteroids and PPIs may be a risk factor of AE in patients with CFIP treated with antifibrotics.
The estimated 1-, 2-, and 3-year AE incidence rates were 11.4, 32, and 36.3%, respectively (for CIs, see results section), which was consistent with a previous report that AE occurs in approximately 5–15% of IPF patients [23]. A recent clinical trial found that AE occurs in 5–10% of patients on nintedanib [17]. AE incidence in this study cohort may have been slightly higher than that of recent clinical trial data from patients treated with antifibrotics. However, previous cohort studies have also reported higher AE-IPF incidences than have clinical trials, possibly because the real-world data included patients with more advanced disease.
A previous report revealed that AE-IPF is more common in patients with physiologically and functionally advanced disease [14]. Our results also indicated that AE was more common in patients with advanced disease, even when treated with antifibrotics, and that the ILD-GAP model [18] could better predict AE-CFIP. The ILD-GAP model modified the GAP model [24] to apply to IPF and other interstitial lung diseases based on the following variables: interstitial lung disease (ILD) subtype, gender, age, and two lung physiological variables (FVC and DLCO) [18]. GAP models were previously reported as good predictors of AE-IPF [25, 26]. Our results suggest that the ILD-GAP model could be used as a simple screening tool to predict AE-CFIP patients treated with antifibrotics.
We demonstrated that using corticosteroids upon antifibrotic initiation is dose-dependently associated with increased AE risk. Antifibrotic treatment has been a significant advance for IPF patients. As these agents have become a standard of care, common practices that may affect the antifibrotic’s efficacy must be recognized and studied. Recent clinical guidelines recommend not using corticosteroids for IPF either alone or combined with other medications [20, 27]. Thus, corticosteroid use is being gradually reduced. However, recent reports have shown that approximately 20% of patients still receive corticosteroids [8, 28,29,30], even in the antifibrotic-agent era. This percentage is consistent with our study’s findings. Corticosteroids are commonly administered for various indications (e.g., decreased appetite, fatigue, cough). Analysis of the INPULSIS trials demonstrated that corticosteroid use at baseline did not influence nintedanib’s effectiveness or the rate of FVC decline [31]. However, our study demonstrated that corticosteroid use when simultaneously initiating antifibrotics is associated with increased AE risk even dose-dependently at low doses, and AE greatly impacted CFIP mortality. Our results suggest that prudent corticosteroid use when initiating antifibrotics may be warranted.
In our analysis, PPI use was associated with higher AE incidences after adjusting for disease severity. Previous retrospective studies revealed that antacid use was associated with a slower decline in FVC over time [32] and possibly fewer AEs in patients with IPF [33]. The 2015 treatment guidelines for IPF conditionally recommended antacid use for treating IPF rather than gastroesophageal reflux disease [9] based on the aforementioned studies. Multiple studies published on antacid therapy in IPF patients present different conclusions regarding whether antacid therapy is beneficial. A recent study suggested antacids do not benefit the IPF clinical course [34, 35], and another suggested that baseline antacid use was most strongly associated with AE risk among IPF patients treated with nintedanib [36]. Our results indicated that PPIs were associated with an increased AE risk, but H2 blockers were not. PPIs are reported to be associated with increased risks of adverse events (e.g., community-acquired pneumonia and cardiovascular events) and excess risk of death [37]. PPIs are also recognized as affecting the lung microbiome [38]. A recent study discovered a change in the respiratory microbiome during an AE-IPF [39]. Thus, clinical trials are urgently needed to prospectively evaluate the efficacy of PPIs in IFP in patients taking antifibrotics.
This study had some limitations. First, it was performed at a single center and included patients with varying fibrotic ILD, disease severities, and comorbidities. However, the cohort’s demographic features and lung functions were comparable to those of other studies, and systematic history collection, physical examinations and blood tests were performed on all patients upon initiating antifibrotics. Second, because the number of patients was relatively small, we could not perform multivariate analyses including many factors. Third, the study lacked a control group not taking antifibrotics. Fourth, the study lacked detailed information on the duration and intensity of corticosteroid use while taking antifibrotics. Time-dependent covariate analyses were impossible given the limited data.
Conclusions
In conclusion, this study showed that even in patients treated with antifibrotics, acute exacerbation (AE) of chronic fibrotic interstitial pneumonia (CFIP) is more common in those with physiologically and functionally advanced disease. This study demonstrated the need for early diagnosis and early treatment choices in patients with CFIP. Corticosteroid use at baseline may be a risk factor for AE with antifibrotic treatment. Given this study’s limitations, these results should be further validated in a larger cohort. Prudent indication for concurrent corticosteroid and proton-pump inhibitor use when initiating antifibrotic agents is recommended. Further prospective data are needed to assess the impact of corticosteroid and PPI use on IPF treated with antifibrotics.
Availability of data and materials
All data generated or analysed during this study are included in this published article and its supplementary information files.
Abbreviations
- AE:
-
Acute exacerbation
- CFIP:
-
Chronic fibrotic interstitial pneumonia
- CI:
-
Confidence interval
- DLCO:
-
Diffusing capacity of the lungs for carbon monoxide
- FVC:
-
Forced vital capacity
- HR:
-
Hazard ratios
- IPF:
-
Idiopathic pulmonary fibrosis
- IQR:
-
Interquartile range
- PPI:
-
Proton-pump inhibitor
References
Raghu G, Chen SY, Yeh WS, Maroni B, Li Q, Lee YC, et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001-11. Lancet Respir Med. 2014;2(7):566–72.
Natsuizaka M, Chiba H, Kuronuma K, Otsuka M, Kudo K, Mori M, et al. Epidemiologic survey of Japanese patients with idiopathic pulmonary fibrosis and investigation of ethnic differences. Am J Respir Crit Care Med. 2014;190(7):773–9.
Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet. 2017;389(10082):1941–52.
King TE, Jr., Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. The New England journal of medicine. 2014;370(22):2083–2092.
Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071–82.
Noble PW, Albera C, Bradford WZ, Costabel U, du Bois RM, Fagan EA, et al. Pirfenidone for idiopathic pulmonary fibrosis: analysis of pooled data from three multinational phase 3 trials. Eur Respir J. 2016;47(1):243–53.
Nathan SD, Albera C, Bradford WZ, Costabel U, Glaspole I, Glassberg MK, et al. Effect of pirfenidone on mortality: pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir Med. 2017;5(1):33–41.
Jo HE, Glaspole I, Grainge C, Goh N, Hopkins PM, Moodley Y, et al. Baseline characteristics of idiopathic pulmonary fibrosis: analysis from the Australian idiopathic pulmonary fibrosis registry. Eur Respir J. 2017;49(2).
Raghu G, Rochwerg B, Zhang Y, Garcia CA, Azuma A, Behr J, et al. An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 clinical practice guideline. Am J Respir Crit Care Med. 2015;192(2):e3–e19.
Morell F, Villar A, Montero M-Á, Muñoz X, Colby TV, Pipvath S, et al. Chronic hypersensitivity pneumonitis in patients diagnosed with idiopathic pulmonary fibrosis: a prospective case-cohort study. Lancet Respir Med. 2013;1(9):685–94.
Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, et al. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198(5):e44–68.
Flaherty KR, Brown KK, Wells AU, Clerisme-Beaty E, Collard HR, Cottin V, et al. Design of the PF-ILD trial: a double-blind, randomised, placebo-controlled phase III trial of nintedanib in patients with progressive fibrosing interstitial lung disease. BMJ Open Respir Res. 2017;4(1):e000212.
Maher TM, Corte TJ, Fischer A, Kreuter M, Lederer DJ, Molina-Molina M, et al. Pirfenidone in patients with unclassifiable progressive fibrosing interstitial lung disease: design of a double-blind, randomised, placebo-controlled phase II trial. BMJ Open Respir Res. 2018;5(1):e000289.
Collard HR, Ryerson CJ, Corte TJ, Jenkins G, Kondoh Y, Lederer DJ, et al. Acute exacerbation of idiopathic pulmonary fibrosis. An international working group report. Am J Respir Crit Care Med. 2016;194(3):265–75.
Ryerson CJ, Collard HR. Acute exacerbations complicating interstitial lung disease. Curr Opin Pulm Med. 2014;20(5):436–41.
Moua T, Westerly BD, Dulohery MM, Daniels CE, Ryu JH, Lim KG. Patients with fibrotic interstitial lung disease hospitalized for acute respiratory worsening: a large cohort analysis. Chest. 2016;149(5):1205–14.
Crestani B, Huggins JT, Kaye M, Costabel U, Glaspole I, Ogura T, et al. Long-term safety and tolerability of nintedanib in patients with idiopathic pulmonary fibrosis: results from the open-label extension study, INPULSIS-ON. Lancet Respir Med. 2018.
Ryerson CJ, Vittinghoff E, Ley B, Lee JS, Mooney JJ, Jones KD, et al. Predicting survival across chronic interstitial lung disease: the ild-gap model. Chest. 2014;145(4):723–8.
Quan H, Li B, Couris CM, Fushimi K, Graham P, Hider P, et al. Updating and validating the Charlson comorbidity index and score for risk adjustment in hospital discharge abstracts using data from 6 countries. Am J Epidemiol. 2011;173(6):676–82.
Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788–824.
Raghu G, Wells AU, Nicholson AG, Richeldi L, Flaherty KR, Le Maulf F, et al. Effect of Nintedanib in subgroups of idiopathic pulmonary fibrosis by diagnostic criteria. Am J Respir Crit Care Med. 2017;195(1):78–85.
Kanda Y. Investigation of the freely available easy-to-use software 'EZR' for medical statistics. Bone Marrow Transplant. 2013;48(3):452–8.
Song JW, Hong SB, Lim CM, Koh Y, Kim DS. Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir J. 2011;37(2):356–63.
Ley B, Ryerson CJ, Vittinghoff E, Ryu JH, Tomassetti S, Lee JS, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med. 2012;156(10):684–91.
Kishaba T, Shimaoka Y, Fukuyama H, Nagano H, Nei Y, Yamashiro S, et al. Clinical characteristics of idiopathic pulmonary fibrosis patients with gender, age, and physiology staging at Okinawa Chubu hospital. J Thorac Dis. 2015;7(5):843–9.
Kakugawa T, Sakamoto N, Sato S, Yura H, Harada T, Nakashima S, et al. Risk factors for an acute exacerbation of idiopathic pulmonary fibrosis. Respir Res. 2016;17(1):79.
Homma S, Bando M, Azuma A, Sakamoto S, Sugino K, Ishii Y, et al. Japanese guideline for the treatment of idiopathic pulmonary fibrosis. Respir Investig. 2018;56(4):268–91.
Brunnemer E, Walscher J, Tenenbaum S, Hausmanns J, Schulze K, Seiter M, et al. Real-world experience with Nintedanib in patients with idiopathic pulmonary fibrosis. Respiration; international review of thoracic diseases. 2018;95(5):301–9.
Yoon HY, Kim DS, Song JW. Efficacy and safety of Pirfenidone in advanced idiopathic pulmonary fibrosis. Respiration; international review of thoracic diseases. 2018:1–10.
Yoon HY, Park S, Kim DS, Song JW. Efficacy and safety of nintedanib in advanced idiopathic pulmonary fibrosis. Respir Res. 2018;19(1):203.
Costabel U, Inoue Y, Richeldi L, Collard HR, Tschoepe I, Stowasser S, et al. Efficacy of Nintedanib in idiopathic pulmonary fibrosis across Prespecified subgroups in INPULSIS. Am J Respir Crit Care Med. 2016;193(2):178–85.
Lee JS, Ryu JH, Elicker BM, Lydell CP, Jones KD, Wolters PJ, et al. Gastroesophageal reflux therapy is associated with longer survival in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;184(12):1390–4.
Lee JS, Collard HR, Anstrom KJ, Martinez FJ, Noth I, Roberts RS, et al. Anti-acid treatment and disease progression in idiopathic pulmonary fibrosis: an analysis of data from three randomised controlled trials. Lancet Respir Med. 2013;1(5):369–76.
Costabel U, Behr J, Crestani B, Stansen W, Schlenker-Herceg R, Stowasser S, et al. Anti-acid therapy in idiopathic pulmonary fibrosis: insights from the INPULSIS(R) trials. Respir Res. 2018;19(1):167.
Kreuter M, Spagnolo P, Wuyts W, Renzoni E, Koschel D, Bonella F, et al. Antacid therapy and disease progression in patients with idiopathic pulmonary fibrosis who received Pirfenidone. Respiration; international review of thoracic diseases. 2017;93(6):415–23.
Collard HR, Richeldi L, Kim DS, Taniguchi H, Tschoepe I, Luisetti M, et al. Acute exacerbations in the INPULSIS trials of nintedanib in idiopathic pulmonary fibrosis. Eur Respir J. 2017;49(5).
Xie Y, Bowe B, Li T, Xian H, Yan Y, Al-Aly Z. Risk of death among users of proton pump inhibitors: a longitudinal observational cohort study of United States veterans. BMJ Open. 2017;7(6):e015735.
Rosen R, Hu L, Amirault J, Khatwa U, Ward DV, Onderdonk A. 16S community profiling identifies proton pump inhibitor related differences in gastric, lung, and oropharyngeal microflora. J Pediatr. 2015;166(4):917–23.
Molyneaux PL, Cox MJ, Wells AU, Kim HC, Ji W, Cookson WO, et al. Changes in the respiratory microbiome during acute exacerbations of idiopathic pulmonary fibrosis. Respir Res. 2017;18(1):29.
Acknowledgments
We thank Drs. Shigeo Hiroshige, Hiroyuki Muranaka, Yoshitomo Eguchi, Naoki Shingu, Yoshihiko Sakata, Junpei Hisanaga, Tatuya Nitawaki, Yuko Sekido, Miwa iio, Aiko Nakano, and Aoi Teruya for their clinical assistance. Editorial assistance was provided by Traci Raley, MS, ELS from Edanz Group (www.edanzediting.com/ac). This study received no funding.
Funding
Self-funded.
Author information
Authors and Affiliations
Contributions
KK and KI planned the project. KK analyzed the data and wrote the manuscript. KK,KI, KA, YY, and HI collected data. KK, KI, MS, HI, and TS interpreted the results and helped write the manuscript. All authors reviewed, revised and approved the manuscript for submission.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This single-center retrospective study was approved by the institutional review board of Saiseikai Kumamoto Hospital(IRB.No.502). Because the study was retrospective, informed consent from participants was unrequired per the “Ethical Guidelines for Medical and Health Research Involving Human Subjects” presented by the Japanese Ministry of Health, Labour and Welfare.
Consent for publication
Not applicable.
Competing interests
The authors have no conflicts of interest to disclose.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Table S1. lists details of each patient’s baseline characteristics. AE varied seasonally and appeared more frequently during winter (DOCX 16 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Kawamura, K., Ichikado, K., Ichiyasu, H. et al. Acute exacerbation of chronic fibrosing interstitial pneumonia in patients receiving antifibrotic agents: incidence and risk factors from real-world experience. BMC Pulm Med 19, 113 (2019). https://doi.org/10.1186/s12890-019-0880-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12890-019-0880-0