
Abstract
Background
Both body mass index (BMI) and genetic factors independently contribute to cardiovascular disease (CVD). However, it is unclear whether genetic risk modifies the association between BMI and the risk of incident CVD. This study aimed to investigate whether BMI categories and genetic risk jointly and interactively contribute to incident CVD events, including hypertension (HTN), atrial fibrillation (AF), coronary heart disease (CHD), stroke, and heart failure (HF).
Methods
A total of 496,851 participants from the UK Biobank with one or more new-onset CVD events were included in the analyses. BMI was categorized as normal weight (< 25.0 kg/m2), overweight (25.0–29.9 kg/m2), and obesity (≥ 30.0 kg/m2). Genetic risk for each outcome was defined as low (lowest tertile), intermediate (second tertile), and high (highest tertile) using polygenic risk score. The joint associations of BMI categories and genetic risk with incident CVD were investigated using Cox proportional hazard models. Additionally, additive interactions were evaluated.
Results
Among the 496,851 participants, 270,726 (54.5%) were female, with a mean (SD) age was 56.5 (8.1) years. Over a median follow-up (IQR) of 12.4 (11.5–13.1) years, 102,131 (22.9%) participants developed HTN, 26,301 (5.4%) developed AF, 32,222 (6.9%) developed CHD, 10,684 (2.2%) developed stroke, and 13,304 (2.7%) developed HF. Compared with the normal weight with low genetic risk, the obesity with high genetic risk had the highest risk of CVD: HTN (HR: 3.96; 95%CI: 3.84–4.09), AF (HR: 3.60; 95%CI: 3.38–3.83), CHD (HR: 2.76; 95%CI: 2.61–2.91), stroke (HR: 1.44; 95%CI: 1.31–1.57), and HF (HR: 2.47; 95%CI: 2.27–2.69). There were significant additive interactions between BMI categories and genetic risk for HTN, AF, and CHD, with relative excess risk of 0.53 (95%CI: 0.43–0.62), 0.67 (95%CI: 0.51–0.83), and 0.37 (95%CI: 0.25–0.49), respectively.
Conclusions
BMI and genetic factors jointly and interactively contribute to incident CVD, especially among participants with high genetic risk. These findings have public health implications for identifying populations more likely to have cardiovascular benefit from weight loss interventions.
Similar content being viewed by others

Background
Cardiovascular disease (CVD) remains a major cause of premature mortality and rising health care costs [1, 2]. Globally, an estimated 17.9 million people died from CVD in 2019, with the majority of these deaths occurring in adults aged 75 years and older [3, 4]. However, relatively younger individuals can benefit significantly from preventive treatment [5]. Additionally, there has been a noticeable and steady rise in the prevalence of obesity worldwide over the past 30 years [6, 7]. Body mass index (BMI) is a widely used measure of obesity and overweight [8], and higher BMI is associated with an increased risk of CVD [9]. However, findings are not entirely consistent. For example, a recent Israel cohort study found a significant association between BMI and stroke [10], whereas other studies did not report such a significant association [11, 12]. Moreover, a consensus regarding the patterns of associations between BMI and CVD risk has not yet widely emerged. Some studies have reported a linear relationship between BMI and atrial fibrillation risk [13, 14], while others have observed a non-linear relationship [15, 16]. Further studies are needed to strengthen the weak evidence regarding the associations between BMI categories and the risk of CVD [9].
Twin studies have revealed that genetic factors play an important role in the development of CVD and its related death [17, 18]. Meanwhile, increasingly large genome-wide association studies (GWASs) have identified a mass of single nucleotide variants across the genome associated with CVD [19,20,21]. Evidence has shown that single genetic variants can modify the associations between obesity and multiple diseases, such as dyslipidemia [22, 23] and hypertension [24]. However, the heritability of a single genetic variant is limited due to the complexity of disease development. Polygenic risk score (PRS) is generally more effective in measuring the cumulative effect of multiple genetic variants associated with the risk of CVD. PRS enables comprehensive genetic risk assessment, early detection of high-risk individuals, and application in precision medicine [25,26,27,28]. Previous studies have largely evaluated the associations of obesity or polygenic risk with CVD separately, leaving it unclear how PRS interacts with obesity to influence CVD risk. Understanding the joint and interactive associations between obesity and PRS might help refine risk stratification, provide more timely and personalized preventive advice, and aid in developing effective strategies for CVD prevention.
Considering the CVD burden attributable to obesity and genetic factors, the primary objective of this study was to investigate whether the BMI and genetic factors have a joint and interactive association with incident CVD events, including hypertension (HTN), atrial fibrillation (AF), coronary heart disease (CHD), stroke, and heart failure (HF)—the leading risks and causes of cardiovascular death [29]—in a prospective population-based cohort.
Methods
Study population
The UK Biobank study protocol is available online (https://www.ukbiobank.ac.uk/media/gnkeyh2q/study-rationale.pdf). More than 500,000 participants aged 40–69 years were recruited into the UK Biobank and visited one of 22 assessment centers in England, Wales, and Scotland from 2006 to 2010, where they completed a self-reported touchscreen questionnaire, which collected comprehensive personal and exposure information; underwent physical measurements; and provided blood, urine, and saliva samples that were used for various types of assays [30]. All participants provided written informed consent, and the UK Biobank study was granted ethics approval from the North West Multi-Center Research Ethical Committee.
Body mass index
BMI was calculated as weight in kilograms divided by height in meters squared according to the protocol of the UK Biobank. Standing height was measured using a Seca 202 device, and weight was measured by a variety of means during the initial assessment center visit (https://biobank.ctsu.ox.ac.uk/crystal/label.cgi?id=100010).
According to the World Health Organization (WHO) 2000 criteria, four basic categories are defined based on BMI: underweight (< 18.5 kg/m2), normal weight (18.5–24.9 kg/m2), overweight (25.0–29.9 kg/m2), and obesity (≥ 30.0 kg/m2) [31, 32]. Since the underweight was extremely rare in our analytical datasets (Overall: 0.5%; HTN: 0.6%; AF: 0.5%; CHD: 0.5%; stroke: 0.5%; HF: 0.5%), it was not considered as a separate category but was combined with the normal weight group in the main analyses [33, 34]. Therefore, participants were classified into three categories: normal weight (< 25.0 kg/m2), overweight (25.0–29.9 kg/m2), and obesity (≥ 30.0 kg/m2).
PRS construction
The genotyping and imputation procedures for the single nucleotide polymorphisms (SNPs) are described briefly in the Additional file 1: Supplemental Methods. Based on the largest HTN GWAS published recently [19], we constructed the PRS for HTN with 496 out of 901 SNPs after quality control [35,36,37] and linkage disequilibrium (LD) clumping (see Additional file 1: Fig. S1 for details). Similarly, 89 out of 166 SNPs [38], 143 out of 241 SNPs [20], 56 out of 89 SNPs [21], and 6 out of 12 SNPs [39] were used to construct PRS for AF, CHD, stroke, and HF, respectively. The SNPs used to construct the PRS are provided (Additional file 1: Table S1-S5). Details about PRS generation are shown in the Additional file 1: Supplemental Methods. The PRS for each outcome was further categorized as low (lowest tertile), intermediate (second tertile), and high (highest tertile) genetic risk (tertiles were based on the distribution of the PRS among the non-cases) [40].
Outcome ascertainment
The outcomes of this study were the first incident CVD events (HTN, AF, CHD, stroke, and HF) obtained through linkage with inpatient hospitalization, primary care records, recorded as the underlying/contributory cause of death, or self-reported at the UK Biobank assessment center visit. We defined outcomes according to the International Classification of Disease edition 10 (ICD-10) [41]: I10-I13, I15 for HTN; I48 for AF; I20-I25 for CHD; I60-I64, I69 for stroke; and I50 for HF. Participants were followed up from enrollment until the date of the first occurrence of CVD or censoring. Censoring was defined as any CVD-unrelated death, withdrawal from the study, or until the end of follow-up (August 15, 2021), whichever came first.
Covariates
A directed acyclic graph (DAG) was drawn to identify which covariates are considered confounders (Additional file 1: Fig. S2). Demographic characteristic (age, sex, and ethnicity), individual socioeconomic status (education, employment status, and household income), lifestyles (smoking status, drink frequency, healthy diet, and International Physical Activity Questionnaire (IPAQ)), area socioeconomic status (Townsend deprivation index), residential exposure (residential air and noise pollution) were all considered. The detailed information for covariates is described in the Additional file 1: Supplemental Methods.
Analytical cohort
We excluded participants with missing or outliers (BMI < 15 kg/m2 or > 60 kg/m2) of BMI data [42]. Participants with prevalent disease at baseline, defined as the date of first occurrence of an event preceding the date of first attendance at the assessment center, were excluded per outcome. Participants with missing ethnicity data were excluded since predicting the ethnicity at the individual level is problematic [43]. In the end, a total of 496,851 participants remained in at least one analytical cohort. The sample sizes were 445,907 for the HTN cohort; 488,689 for the AF cohort; 470,159 for the CHD cohort; 489,074 for the stroke cohort; and 494,230 for the HF cohort. Combined with PRS, the number of participants were 432,761, 474,140, 456,218, 474,570, and 479,535 for each outcome cohort, respectively (Fig. 1).

Study workflow. Definition of abbreviations: AF atrial fibrillation, BMI body mass index, CHD coronary heart disease, CVD cardiovascular disease, HF heart failure, HTN hypertension, IPAQ International physical activity questionnaire, PRS polygenic risk score
Statistical analyses
Characteristics of the study population were presented as mean (standard deviations [SD]), median (interquartile range [IQR]), or frequency (percentage). We imputed missing covariate data using multiple imputation by chained equations [44] (see Additional file 1: Supplemental Methods for details). Cox proportional hazard models were applied to estimate the hazard ratios (HRs) and 95% confidence intervals (CIs) for the associations of BMI categories and genetic factors with incident events of HTN, AF, CHD, stroke, and HF. Schoenfeld residual plot was used to check the proportional hazard assumption, and no violation of the assumption was observed. Cox proportional hazard models were adjusted for the confounders identified by the DAG (Additional file 1: Fig. S2), including age, sex (model 1), ethnicity, education, employment status, household income, smoking status, drink frequency, healthy diet, IPAQ, Townsend deprivation index, and the top two pollution principal components (model 2). When genetic factors were included in the models, we further adjusted for the genotyping batch and the top four genetic principal components.
In the main analyses, the results were reported based on three BMI categories: normal weight (< 25.0 kg/m2, the reference category), overweight (25.0–29.9 kg/m2), and obesity (≥ 30.0 kg/m2). The P value for trend between BMI categories and each outcome was calculated using the median value of each BMI category. Restricted cubic splines (RCSs) with the population median BMI as the reference value were used to detect any possible non-linear dependencies in the Cox models. Five knots were placed at default positions according to BMI percentiles (5th, 27.5th, 50th, 72.5th, and 95th percentiles) [45]. The deciles of PRS were also used to test for positive associations between PRS and each outcome. The tests for trend were performed by modelling the PRS (tertiles or deciles) as an ordinal variable [40].
We included the interaction term of BMI categories and PRS tertiles (genetic risk) in the models to calculate the joint HRs (95%CIs) (the normal weight with low genetic risk as the reference group). We also calculated cumulative incidence in each population with various combination of BMI category and genetic risk, standardized to the mean of adjusted covariates. The additive interaction was evaluated two indexes: the relative excess risk because of the interaction (RERI) and the attributable proportion because of the interaction (AP). The 95%CIs of the RERI and AP were computed by the multivariate delta method [46]. The Wald global test was performed to examine the overall additive interaction.
Sensitivity analyses were conducted to examine the robustness of our results. First, we removed participants with missing values for covariates and ran complete case analyses. Second, we excluded events that occurred within the first year of follow-up or restricted the analysis to participants free of any HTN, AF, CHD, stroke, and HF at baseline to minimize confounding from illness. Third, considering potential confounding rather than mediating effects of a history of diabetes, we additionally adjusted for the history of diabetes. Fourth, we evaluated the associations between time-dependent BMI categories and CVD using time-dependent Cox proportional hazard models to validate the results of our main analyses, which modeled only the baseline BMI categories. Fifth, the underweight (< 18.5 kg/m2) was considered as a separate category to enhance the validity of our results (the normal weight as the reference group).
All analyses were implemented using R Software, version 4.2.3. Multiple imputation and Cox regression were performed using the mice and survival packages, respectively. Bonferroni method was used to control the type I error rate inflation due to multiple testing (see Additional file 1: Supplemental Methods for details).
Results
Baseline characteristics
A total of 496,851 participants (mean [SD] age, 56.5 [8.1] years; 270,726 [54.5%] female) from the UK Biobank were included in this study. Participants from the following race and ethnicity groups were included: 470,385 white (94.7%) and 26,466 other (5.3%). In total, 164,289 (33.1%) individuals were of normal weight (including 0.5% underweight), 211,099 (42.5%) were overweight, and 121,463 (24.4%) were obese (Table 1). The baseline characteristics of the five analytical cohorts (Additional file 1: Table S6) are provided, as well as those grouped by BMI and PRS categories (Additional file 1: Table S7-S11). Baseline characteristics of incomplete and complete cases are also presented (Additional file 1: Table S12-S13). During a median follow-up (IQR) of 12.4 (11.5–13.1) years, 102,131 (22.9%) participants developed HTN, 26,301 (5.4%) developed AF, 32,222 (6.9%) developed CHD, 10,684 (2.2%) developed stroke, and 13,304 (2.7%) developed HF.
Association of BMI with incident CVD
There were significant associations between obesity and the risk of HTN (HR: 2.68; 95%CI: 2.63–2.73), AF (HR: 1.86; 95%CI: 1.80–1.92), CHD (HR: 1.79; 95%CI: 1.73–1.84), stroke (HR: 1.27; 95%CI: 1.21–1.34), and HF (HR: 2.27; 95%CI: 2.16–2.38). Overweight was associated with a 60% higher risk of HTN (95%CI: 57%-63%), an 18% higher risk of AF (95%CI: 14%-22%), a 32% higher risk of CHD (95%CI: 29%-36%), a 7% higher risk of stroke (95%CI: 2%-12%), and a 22% higher risk of HF (95%CI: 17%-28%). The risk of CVD increased significantly from normal weight to obesity (all Ptrend < 0.001) (Table 2). The results were robust in sensitivity analyses (Additional file 1: Table S14-S18). The underweight was not significantly associated with the risk of AF, CHD, stroke and HF, but was associated with HTN (Additional file 1: Table S19).
RCSs showed J-shaped non-linear associations between BMI and the risk of HTN and CHD, and U-shaped non-linear associations between BMI and the risk of AF, stroke and HF, with the lowest risk at around 23 kg/m2 (Pnon-linearity = 0.009 for stroke, Pnon-linearity < 0.001 for the rest). There were steeper slopes between BMI and the risk of HTN, AF and HF, whereas plateauing increased trends were observed between BMI and the risk of CHD and stroke as BMI increased (Additional file 1: Fig. S3).
Association of BMI and PRS with incident CVD
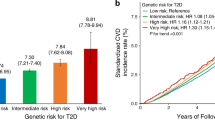
Participants with incident CVD events tended to have a higher PRS than unaffected participants (Additional file 1: Fig. S4). There was a significant increase in the risk of CVD across the deciles of the PRS (all Ptrend < 0.001; Additional file 1: Table S20, Fig. S5). Participants with intermediate and high genetic risk had higher risks for CVD than those with low genetic risk, with observable risk stratification (Additional file 1: Table S21, Fig. S6).
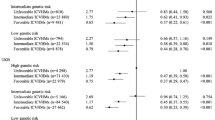
We observed joint associations of BMI categories and genetic risk on CVD risk in a dose–response manner, with the overall risk of CVD events increasing as both BMI and genetic risk increased. Specifically, compared with the normal weight with low genetic risk, the obesity with high genetic risk had the highest risk of CVD: HTN (HR: 3.96; 95%CI: 3.84–4.09), AF (HR: 3.60; 95%CI: 3.38–3.83), CHD (HR: 2.76; 95%CI: 2.61–2.91), stroke (HR: 1.44; 95%CI: 1.31–1.57), and HF (HR: 2.47; 95%CI: 2.27–2.69) (Fig. 2, Additional file 1: Fig. S7a). Similarly, a graduate increase in stratified cumulative CVD risk was observed compared with the normal weight with low genetic risk (Additional file 1: Fig. S8). The results of joint associations were robust in sensitivity analyses (Additional file 1: Table S22-S27).

Risk of incident CVD according to BMI categories and genetic risk groups. The hazard ratios for CVD according to BMI categories and PRS categories in the HTN (A), AF (B), CHD (C), Stroke (D), HF (E) cohort were estimated using Cox proportional hazard models with adjustment for age, sex, ethnicity, education, employment status, household income, smoking status, drink frequency, healthy diet, IPAQ, Townsend deprivation index, the top two pollution principal components, and the top four principal components of ancestry and genotyping batch. Definition of abbreviations: AF atrial fibrillation, CHD coronary heart disease, CVD cardiovascular disease, CI confidence interval, HF heart failure, HTN hypertension, NA not applicable
Significant positive additive interactions between BMI and genetic risk were found for HTN, AF, and CHD (all Poverall < 0.001). Specifically, for the obesity with high genetic risk, the RERI for HTN was 0.53 (95%CI: 0.43–0.62), indicating a 0.53 relative excess risk due to the additive interaction, accounting for 13% (95%CI: 11%-16%) of the risk of HTN in participants exposed to both high genetic risk and obesity. Similarly, for the obesity with high genetic risk, the RERI and AP for AF were 0.67 (95%CI: 0.51–0.83) and 19% (95%CI: 14%-23%), and for CHD were 0.37 (95%CI: 0.25–0.49) and 13% (95%CI: 9%-18%) (Table 3, Additional file 1: Fig. S7b).
Discussion
In this large, prospective cohort study, we observed that obesity and overweight were significantly associated with an increased risk of incident CVD in the UK Biobank. Intermediate and high genetic risk were also associated with higher risks of CVD. Notably, the greatest relative increases in risk were observed among the obesity with high genetic risk for CVD. Additionally, there were positive additive interactions between BMI categories and genetic risk for incident HTN, AF, and CHD.
The results of a population-based study in the USA are consistent with our findings, showing that overweight and obese adults had a higher lifetime risk of developing CVD [47]. We found similar dose–response associations in a meta-analysis of more than 2.3 million participants and in the European Prospective Investigation into Cancer and Nutrition (EPIC) study, where the risk of HTN and CHD increased continuously with increasing BMI [48, 49]. There is evidence of U-shaped non-linear associations between BMI and the risk of AF, stroke, and HF [15, 50,51,52,53]. We also found U-shaped non-linear associations at the risk of AF, stroke, and HF with BMI. However, the underweight was extremely rare and thus combined into the normal weight category in our analyses. These results should be interpreted with caution. Additionally, the risk of HTN, AF, and HF increased dramatically when BMI > 30 kg/m2 compared to the risk of CHD and stroke, suggesting that focusing on obesity and weight management might be more effective in reducing the incidence of HTN, AF, and HF. Further studies will be needed to unveil the true patterns of associations between the full spectrum of BMI and the risk of CVD.
CVD has a strong genetic component based on heritability estimates ranging between ~ 40–60% [17, 54, 55]. As shown in our study, the risk of CVD increased as PRS increased, e.g., individuals with high genetic risk had up to a 2-fold risk of AF compared to those with low genetic risk. Over the past 10 to 15 years, global interest, efforts, and controversies have surrounded the clinical utility of PRS, a promising tool for precision medicine and early risk stratification, for the primary prevention of non-communicable CVD [56, 57]. However, knowledge of genetic risk alone may lead individuals to believe they are destined to develop diseases and respond passively to other beneficial public health policies [58]. From this perspective, the results of positive additive interactions between obesity and genetic risk suggest that people with high genetic risk may benefit more from weight loss, which may promote the practical application of PRS in the primary prevention of CVD.
Previous studies mostly focused on the impact of adiposity or genetic risk on CVD risk separately, while ignoring the combined disease burden they contribute to. We observed that the overall risk of CVD events increased as both BMI and genetic risk increased. The associations between the risk of CVD and BMI categories were similar across all strata of genetic risk. Even with low genetic risk, the obesity was associated with a 2.89-fold risk of HTN, a 1.95-fold risk of AF, a 1.86-fold risk of CHD, a 1.27-fold risk of CHD, and a 2.27-fold risk of HF compared to normal weight. These results support the benefit of combating overweight and obesity for the entire population.
Additionally, there were statistically significant positive additive interactions between obesity and high genetic risk for incident HTN, AF, and CHD, suggesting that the joint association of obesity and high genetic risk was greater than the sum of the two main associations. Moreover, 19% of AF risk could be attributed to the additive interactions, the greatest proportion among the five CVDs. These results suggest that genetic risk modifies the impact of obesity on CVD, which have public health implications for identifying populations more likely to have cardiovascular benefit from weight loss interventions. Specifically, maintaining a normal weight plays an important role in the primary prevention of CVD, especially in populations with high genetic risk.
To our knowledge, this is the first study to assess the joint association and additive interaction of combined BMI and genetic risk with incident HTN, AF, CHD, stroke, and HF, enabling us to accurately determine the risk variation of incident CVD events in the overweight and obese with different susceptibility levels. Strengths of our study include the large sample size and the prospective cohort design. In addition, we adjusted for a large number of potential confounders identified by the DAG, including demographic characteristics, socioeconomic status, lifestyle factors and residential contexts, etc. We also considered various sensitivity analyses and controlled for type I error rate inflation due to multiple testing to increase the validity of the main findings.
Study limitations
The present study has several potential limitations. First, BMI is a dynamic indicator that changes with growth and aging. However, only 4.1% of participants had at least two measures of BMI in the UK Biobank, meaning that the current data would not be able to depict the trajectory of BMI change for most participants. Second, we combined the underweight with the normal weight group, but the underweight was not significantly associated with the risk of AF, CHD, stroke, and HF. There was a significant inverse association between the underweight and HTN risk. Third, we did not evaluate the relations of other adiposity-related measures with incident CVD, such as waist circumference and measures reflecting body mass distribution (e.g., percentage of fat mass, lean fat mass, etc.). Further studies including these measures may provide a more comprehensive picture of the changes in obesity status and CVD risk. Fourth, the use of multiple imputation to address missing covariates might have led to partial deviation of our estimates from true values. However, the assumption of missing at random and the fundamental theory underlying classical multiple imputation seems established, analyses restricted to complete cases yielded similar results [59]. Fifth, additional functional studies are warranted to elucidate the mechanisms underlying the modification effects of genetic factors on the associations between BMI and the risk of CVD.
Conclusions
In conclusion, findings from this large, prospective cohort study demonstrate that BMI and genetic factors jointly contribute to the development of CVD, and there are additive interactions. It is important to formulate policies to combat overweight and obesity, especially for those with high genetic risk.
Availability of data and materials
The datasets supporting the conclusions of this article are available from the UK Biobank project site, subject to registration and application process. This research has been conducted using the UK Biobank resource under application number 92675. Further details can be found at https://www.ukbiobank.ac.uk.
Abbreviations
- BMI:
-
Body mass index
- CVD:
-
Cardiovascular disease
- HTN:
-
Hypertension
- AF:
-
Atrial fibrillation
- CHD:
-
Coronary heart disease
- HF:
-
Heart failure
- SNP:
-
Single nucleotide polymorphism
- PRS:
-
Polygenic risk score
- RERI:
-
The relative excess risk because of the interaction
- AP:
-
The attributable proportion because of the interaction
References
GBD 2019 Diseases and Injuries Collaborators. Global burden of 369 diseases and injuries in 204 countries and territories, 1990-2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet. 2020;396(10258):1204–22.
Mensah GA, Roth GA, Fuster V. The Global Burden of Cardiovascular Diseases and Risk Factors: 2020 and Beyond. J Am Coll Cardiol. 2019;74(20):2529–32.
Cardiovascular diseases (CVDs). https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds). Accessed 8 Jul 2023.
GBD 2017 Causes of Death Collaborators. lobal, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2018;392(10159):1736–88.
Kim KI. Risk Stratification of Cardiovascular Disease according to Age Groups in New Prevention Guidelines: A Review. J Lipid Atheroscler. 2023;12(2):96–105.
Boutari C, Mantzoros CS. A 2022 update on the epidemiology of obesity and a call to action: as its twin COVID-19 pandemic appears to be receding, the obesity and dysmetabolism pandemic continues to rage on. Metabolism. 2022;133: 155217.
Lopez-Jimenez F, Almahmeed W, Bays H, Cuevas A, Di Angelantonio E, le Roux CW, et al. Obesity and cardiovascular disease: mechanistic insights and management strategies. A joint position paper by the World Heart Federation and World Obesity Federation. Eur J Prev Cardiol. 2022;29(17):2218–37.
Adab P, Pallan M, Whincup PH. Is BMI the best measure of obesity? BMJ. 2018;360:k1274.
Kim MS, Kim WJ, Khera AV, Kim JY, Yon DK, Lee SW, et al. Association between adiposity and cardiovascular outcomes: an umbrella review and meta-analysis of observational and Mendelian randomization studies. Eur Heart J. 2021;42(34):3388–403.
Bardugo A, Fishman B, Libruder C, Tanne D, Ram A, Hershkovitz Y, et al. Body Mass Index in 1.9 Million Adolescents and Stroke in Young Adulthood. Stroke. 2021;52(6):2043–52.
Abete I, Arriola L, Etxezarreta N, Mozo I, Moreno-Iribas C, Amiano P, et al. Association between different obesity measures and the risk of stroke in the EPIC Spanish cohort. Eur J Nutr. 2015;54(3):365–75.
Hu G, Sarti C, Jousilahti P, Silventoinen K, Barengo NC, Tuomilehto J. Leisure time, occupational, and commuting physical activity and the risk of stroke. Stroke. 2005;36(9):1994–9.
Poorthuis MHF, Sherliker P, de Borst GJ, Carter JL, Lam KBH, Jones NR, et al. Joint Associations Between Body Mass Index and Waist Circumference With Atrial Fibrillation in Men and Women. J Am Heart Assoc. 2021;10(8):e019025.
Tedrow UB, Conen D, Ridker PM, Cook NR, Koplan BA, Manson JE, et al. The long- and short-term impact of elevated body mass index on the risk of new atrial fibrillation the WHS (women’s health study). J Am Coll Cardiol. 2010;55(21):2319–27.
Feng T, Vegard M, Strand LB, Laugsand LE, Mørkedal B, Aune D, et al. Weight and weight change and risk of atrial fibrillation: the HUNT study. Eur Heart J. 2019;40(34):2859–66.
Singleton MJ, German CA, Carnethon M, Soliman EZ, Bertoni AG, Yeboah J. Race, Body Mass Index, and the Risk of Atrial Fibrillation: The Multi-Ethnic Study of Atherosclerosis. J Am Heart Assoc. 2021;10(1):e018592.
Zdravkovic S, Wienke A, Pedersen NL, Marenberg ME, Yashin AI, De Faire U. Heritability of death from coronary heart disease: a 36-year follow-up of 20 966 Swedish twins. J Intern Med. 2002;252(3):247–54.
Christophersen IE, Ravn LS, Budtz-Joergensen E, Skytthe A, Haunsoe S, Svendsen JH, et al. Familial aggregation of atrial fibrillation: a study in Danish twins. Circ Arrhythm Electrophysiol. 2009;2(4):378–83.
Evangelou E, Warren HR, Mosen-Ansorena D, Mifsud B, Pazoki R, Gao H, et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat Genet. 2018;50(10):1412–25.
Aragam KG, Jiang T, Goel A, Kanoni S, Wolford BN, Atri DS, et al. Discovery and systematic characterization of risk variants and genes for coronary artery disease in over a million participants. Nat Genet. 2022;54(12):1803–15.
Mishra A, Malik R, Hachiya T, Jürgenson T, Namba S, Posner DC, et al. Stroke genetics informs drug discovery and risk prediction across ancestries. Nature. 2022;611(7934):115–23.
Kang M, Sung J. A genome-wide search for gene-by-obesity interaction loci of dyslipidemia in Koreans shows diverse genetic risk alleles. J Lipid Res. 2019;60(12):2090–101.
Lamina C, Forer L, Schönherr S, Kollerits B, Ried JS, Gieger C, et al. Evaluation of gene-obesity interaction effects on cholesterol levels: a genetic predisposition score on HDL-cholesterol is modified by obesity. Atherosclerosis. 2012;225(2):363–9.
Xu Z, Zhang D, Lin J, Li X, Liu Y, Gao J, et al. The influence of CYP2R1 polymorphisms and gene-obesity interaction with hypertension risk in a Chinese rural population. Nutr Metab Cardiovasc Dis. 2022;32(1):241–8.
O’Sullivan JW, Raghavan S, Marquez-Luna C, Luzum JA, Damrauer SM, Ashley EA, et al. Polygenic Risk Scores for Cardiovascular Disease: A Scientific Statement From the American Heart Association. Circulation. 2022;146(8):e93–118.
Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, Choi SH, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet. 2018;50(9):1219–24.
Mars N, Koskela JT, Ripatti P, Kiiskinen TTJ, Havulinna AS, Lindbohm JV, et al. Polygenic and clinical risk scores and their impact on age at onset and prediction of cardiometabolic diseases and common cancers. Nat Med. 2020;26(4):549–57.
Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372(9):793–5.
Joseph P, Leong D, McKee M, Anand SS, Schwalm JD, Teo K, et al. Reducing the Global Burden of Cardiovascular Disease, Part 1: The Epidemiology and Risk Factors. Circ Res. 2017;121(6):677–94.
Collins R. What makes UK Biobank special? Lancet. 2012;379(9822):1173–4.
Kelly SP, Graubard BI, Andreotti G, Younes N, Cleary SD, Cook MB. Prediagnostic Body Mass Index Trajectories in Relation to Prostate Cancer Incidence and Mortality in the PLCO Cancer Screening Trial. J Natl Cancer Inst. 2017;109(3):djw225.
WHO Consultation on Obesity (1999: Geneva, Switzerland) & World Health Organization. Obesity : preventing and managing the global epidemic : report of a WHO consultation. World Health Organization. 2000. https://iris.who.int/handle/10665/42330.
Chen X, Li H, Mandic M, Hoffmeister M, Brenner H. Assessment of Body Mass Index, Polygenic Risk Score, and Development of Colorectal Cancer. JAMA Netw Open. 2022;5(12):e2248447.
Chen C, Ye Y, Zhang Y, Pan XF, Pan A. Weight change across adulthood in relation to all cause and cause specific mortality: prospective cohort study. BMJ. 2019;367:l5584.
Choi SW, Mak TS, O’Reilly PF. Tutorial: a guide to performing polygenic risk score analyses. Nat Protoc. 2020;15(9):2759–72.
Collister JA, Liu X, Clifton L. Calculating Polygenic Risk Scores (PRS) in UK Biobank: A Practical Guide for Epidemiologists. Front Genet. 2022;13:818574.
Cho SMJ, Koyama S, Ruan Y, Lannery K, Wong M, Ajufo E, et al. Measured Blood Pressure, Genetically Predicted Blood Pressure, and Cardiovascular Disease Risk in the UK Biobank. JAMA Cardiol. 2022;7(11):1129–37.
Nielsen JB, Thorolfsdottir RB, Fritsche LG, Zhou W, Skov MW, Graham SE, et al. Biobank-driven genomic discovery yields new insight into atrial fibrillation biology. Nat Genet. 2018;50(9):1234–9.
Shah S, Henry A, Roselli C, Lin H, Sveinbjörnsson G, Fatemifar G, et al. Genome-wide association and Mendelian randomisation analysis provide insights into the pathogenesis of heart failure. Nat Commun. 2020;11(1):163.
Arthur RS, Wang T, Xue X, Kamensky V, Rohan TE. Genetic Factors, Adherence to Healthy Lifestyle Behavior, and Risk of Invasive Breast Cancer Among Women in the UK Biobank. J Natl Cancer Inst. 2020;112(9):893–901.
ICD-10 : international statistical classification of diseases and related health problems : tenth revision, 2nd ed. https://apps.who.int/iris/handle/10665/42980. Accessed 18 Mar 2023.
Global BMIMC, Di Angelantonio E, Bhupathiraju Sh N, Wormser D, Gao P, Kaptoge S, et al. Body-mass index and all-cause mortality: individual-participant-data meta-analysis of 239 prospective studies in four continents. Lancet. 2016;388(10046):776–86.
Inaccuracies in Medicare's Race and Ethnicity Data Hinder the Ability To Assess Health Disparities. https://oig.hhs.gov/oei/reports/OEI-02-21-00100.pdf. Accessed 15 Apr 2023.
Azur MJ, Stuart EA, Frangakis C, Leaf PJ. Multiple imputation by chained equations: what is it and how does it work? Int J Methods Psychiatr Res. 2011;20(1):40–9.
Adamson C, Kondo T, Jhund PS, de Boer RA, Cabrera Honorio JW, Claggett B, et al. Dapagliflozin for heart failure according to body mass index: the DELIVER trial. Eur Heart J. 2022;43(41):4406–17.
Li R, Chambless L. Test for additive interaction in proportional hazards models. Ann Epidemiol. 2007;17(3):227–36.
Khan SS, Ning H, Wilkins JT, Allen N, Carnethon M, Berry JD, et al. Association of Body Mass Index With Lifetime Risk of Cardiovascular Disease and Compression of Morbidity. JAMA Cardiol. 2018;3(4):280–7.
Jayedi A, Rashidy-Pour A, Khorshidi M, Shab-Bidar S. Body mass index, abdominal adiposity, weight gain and risk of developing hypertension: a systematic review and dose-response meta-analysis of more than 2.3 million participants. Obes Rev. 2018;19(5):654–67.
Lassale C, Tzoulaki I, Moons KGM, Sweeting M, Boer J, Johnson L, et al. Separate and combined associations of obesity and metabolic health with coronary heart disease: a pan-European case-cohort analysis. Eur Heart J. 2018;39(5):397–406.
Aune D, Sen A, Schlesinger S, Norat T, Janszky I, Romundstad P, et al. Body mass index, abdominal fatness, fat mass and the risk of atrial fibrillation: a systematic review and dose-response meta-analysis of prospective studies. Eur J Epidemiol. 2017;32(3):181–92.
Shiozawa M, Kaneko H, Itoh H, Morita K, Okada A, Matsuoka S, et al. Association of Body Mass Index with Ischemic and Hemorrhagic Stroke. Nutrients. 2021;13(7):2343.
Aune D, Sen A, Norat T, Janszky I, Romundstad P, Tonstad S, et al. Body Mass Index, Abdominal Fatness, and Heart Failure Incidence and Mortality: A Systematic Review and Dose-Response Meta-Analysis of Prospective Studies. Circulation. 2016;133(7):639–49.
Kokkinos P, Faselis C, Franklin B, Lavie CJ, Sidossis L, Moore H, et al. Cardiorespiratory fitness, body mass index and heart failure incidence. Eur J Heart Fail. 2019;21(4):436–44.
Bevan S, Traylor M, Adib-Samii P, Malik R, Paul NL, Jackson C, et al. Genetic heritability of ischemic stroke and the contribution of previously reported candidate gene and genomewide associations. Stroke. 2012;43(12):3161–7.
Vinkhuyzen AA, Wray NR, Yang J, Goddard ME, Visscher PM. Estimation and partition of heritability in human populations using whole-genome analysis methods. Annu Rev Genet. 2013;47:75–95.
Klarin D, Natarajan P. Clinical utility of polygenic risk scores for coronary artery disease. Nat Rev Cardiol. 2022;19(5):291–301.
Groenendyk JW, Greenland P, Khan SS. Incremental Value of Polygenic Risk Scores in Primary Prevention of Coronary Heart Disease: A Review. JAMA Intern Med. 2022;182(10):1082–8.
Hollands GJ, French DP, Griffin SJ, Prevost AT, Sutton S, King S, et al. The impact of communicating genetic risks of disease on risk-reducing health behaviour: systematic review with meta-analysis. BMJ. 2016;352: i1102.
Sterne JA, White IR, Carlin JB, Spratt M, Royston P, Kenward MG, et al. Multiple imputation for missing data in epidemiological and clinical research: potential and pitfalls. BMJ. 2009;338: b2393.
Acknowledgements
This study was conducted using the UK Biobank Resource (Approved Research ID: 92675). We thank the investigators and participants involved in the UK Biobank for their contributions to this study. No one was financially compensated for their contribution.
Funding
This study was supported by the National Natural Science Foundation of China (Grant No. 82273738, 82473734, 82404374; China) and Qing Lan Project (Jiangsu, China).
Author information
Authors and Affiliations
Contributions
J.B., S.N., Z.Z., R.H. and X.K. contributed to the conception and design of the study. R.H., X.K. and R.G. performed the statistical analysis and drafted the manuscript. R.H., X.K., J.W., T.C., J.L., C.L., Y.W., D.Y., Y.Z., Z.Z., S.N. and J.B. revised the manuscript for important intellectual content. J.B., S.N., and Z.Z. provided financial support and study supervision. All authors contributed to the acquisition, analysis or interpretation of data. All authors read and approved the final manuscript. J.B., S.N., and Z.Z. are the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All participants provided written informed consent, and the UK Biobank study was granted ethics approval from the North West Multi-Center Research Ethical Committee (16/NW/0274).
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
12889_2024_19916_MOESM1_ESM.pdf
Additional file 1: Supplemental Methods. Genotyping and Imputation; Polygenic Risk Score (PRS); Covariates; Missing data; Multiple testing correction. Table S1. Summary results of SNPs used for PRS construction of hypertension (HTN). Table S2. Summary results of SNPs used for PRS construction of atrial fibrillation (AF). Table S3. Summary results of SNPs used for PRS construction of coronary heart disease (CHD). Table S4. Summary results of SNPs used for PRS construction of stroke. Table S5. Summary results of SNPs used for PRS construction of heart failure (HF). Table S6. Baseline characteristics of five analytical cohort. Table S7. Baseline characteristics per BMI categories and genetic risk group of HTN cohort. Table S8. Baseline characteristics per BMI categories and genetic risk group of AF cohort. Table S9. Baseline characteristics per BMI categories and genetic risk group of CHD cohort. Table S10. Baseline characteristics per BMI categories and genetic risk group of stroke cohort. Table S11. Baseline characteristics per BMI categories and genetic risk group of HF cohort. Table S12. Baseline characteristics of incomplete and complete case in HTN, AF and CHD cohort. Table S13. Baseline characteristics of incomplete and complete case in stroke and HF cohort. Table S14. Sensitivity analyses restricted to participants with complete covariates. Table S15. Sensitivity analyses after excluding incident cases during the first year of follow-up. Table S16. Sensitivity analyses restricted to participants free of all five CVDs at baseline. Table S17. Sensitivity analyses additionally adjusting for history of diabetes. Table S18. Sensitivity analyses evaluated the associations between time-dependent BMI categories and CVD. Table S19. Sensitivity analyses performed underweight considered as a separate category. Table S20. Associations between CVD risk and genetic factors according to deciles of PRS. Table S21. Associations between CVD risk and genetic factors according to risk levels of genetic risk. Table S22. Sensitivity analyses of join associations restricted to participants with complete covariates. Table S23. Sensitivity analyses of join association after excluding incident cases during the first year of follow-up. Table S24. Sensitivity analyses of join associations restricted to participants free of all five CVDs at baseline. Table S25. Sensitivity analyses of join associations additionally adjusting for history of diabetes. Table S26. Sensitivity analyses of join associations between time-dependent BMI categories and CVD. Table S27. Sensitivity analyses of joint associations when underweight was considered as a separate category. Fig. S1 The flow chart of PRS construction. Fig. S2 Directed acyclic graph of the associations between BMI and CVD. Fig. S3 The RCS between BMI and incident CVD of five analytical cohort. Fig. S4 Distribution of PRS in participants affected with or without incident CVD of five analytical cohort. Fig. S5 Risk of CVD according to deciles of PRS of five analytical cohort. Fig. S6 Risk stratification of PRS of five analytical cohorts. Fig. S7 Main findings: association of BMI and PRS with incident CVD. Fig. S8 Cumulative incidence of genetic risk and BMI categories of five analytical cohorts.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Huang, R., Kong, X., Geng, R. et al. Joint and interactive associations of body mass index and genetic factors with cardiovascular disease: a prospective study in UK Biobank. BMC Public Health 24, 2371 (2024). https://doi.org/10.1186/s12889-024-19916-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12889-024-19916-6