
Abstract
Recently, COVID-19, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and its variants, caused > 6 million deaths. Symptoms included respiratory strain and complications, leading to severe pneumonia. SARS-CoV-2 attaches to the ACE-2 receptor of the host cell membrane to enter. Targeting the SARS-CoV-2 entry may effectively inhibit infection. Acid sphingomyelinase (ASMase) is a lysosomal protein that catalyzes the conversion of sphingolipid (sphingomyelin) to ceramide. Ceramide molecules aggregate/assemble on the plasma membrane to form “platforms” that facilitate the viral intake into the cell. Impairing the ASMase activity will eventually disrupt viral entry into the cell. In this review, we identified the metabolism of sphingolipids, sphingolipids' role in cell signal transduction cascades, and viral infection mechanisms. Also, we outlined ASMase structure and underlying mechanisms inhibiting viral entry 40 with the aid of inhibitors of acid sphingomyelinase (FIASMAs). In silico molecular docking analyses of FIASMAs with inhibitors revealed that dilazep (S = − 12.58 kcal/mol), emetine (S = − 11.65 kcal/mol), pimozide (S = − 11.29 kcal/mol), carvedilol (S = − 11.28 kcal/mol), mebeverine (S = − 11.14 kcal/mol), cepharanthine (S = − 11.06 kcal/mol), hydroxyzin (S = − 10.96 kcal/mol), astemizole (S = − 10.81 kcal/mol), sertindole (S = − 10.55 kcal/mol), and bepridil (S = − 10.47 kcal/mol) have higher inhibition activity than the candidate drug amiodarone (S = − 10.43 kcal/mol), making them better options for inhibition.
Similar content being viewed by others


Introduction
Recently, the world and public health organizations directed resources to curb the outbreak of coronavirus disease (COVID-19) caused by SARS-CoV-2 and its mutated strains [1,2,3,4,5,6]. Symptoms of COVID-19 infection included respiratory system complications and severe pneumonia, where patients needed intensive medical care and ventilator treatment [5, 7, 8]. The death rate from COVID-19 is about 0.66%, which rises sharply to 7.8% in patients over 80 years old [9]. Severe cases are characterized by a high incidence of cytokine storms and excessive inflammation with high levels of interleukin (IL)-6, IL-8, IL-10, IL-2R, and tumor necrosis factor (TNF)-alpha. The SARS-CoV-2 infects cells by attachment to its particular cellular receptor ACE-2 via a surface unit (S1) of the viral spike glycoprotein [8, 10]. Transmembrane serine protease 2 (TMPRSS2) or cathepsin L cleaves the viral spike protein after entry. When a virus enters host cells, SARS-CoV-2 RNA is released, translation of viral RNA genome into polyproteins is followed by viral release, and then replicate-transcriptase complex is brought together following protein cleavage to promote viral transcription and replication [11].
Previous membrane and cellular changes facilitating SARS-CoV-2 entry may be a promising target to minimize and inhibit viral infection. Lysosomal acid sphingomyelinase is one of the significant signalling molecules in the outer cell membrane and lysosomes [12]. This review focused on sphingomyelinase (ASMase), which converts the sphingolipid (sphingomyelin) into ceramide, which substantially affects the biophysical characteristics of the plasma membrane [13].
Acid sphingomyelinase and ceramide are essential in receptor signalling and infection biology. The acid sphingomyelinase is a glycoprotein lysosomal hydrolase enzyme that catalyzes the degradation of sphingomyelin to phosphorylcholine and ceramide. Although acid sphingomyelinase is found in lysosomes, it is recycled to the plasma membrane because these compartments constantly recycle to the plasma membrane. The activity of acid sphingomyelinase induces ceramide formation in the outer leaflet of the cell membrane. Ceramide molecules generation within the outer leaflet alters the biophysical properties of the plasma membrane because the very hydrophobic ceramide molecules spontaneously associate with each other to form small ceramide-enriched membrane domains that fuse and form large, highly hydrophobic, tightly packed, gel-like ceramide-enriched membrane domains [14].
The conversion of the sphingomyelin in rafts to ceramide can result in raft enlargement, receptor clustering, membrane invagination, and macropinosome formation, all of which promote the uptake of particles, including viruses, into cells and increase viral infectivity. Furthermore, ceramide-enriched membrane domains can bind to proteins and promote viral infectivity. SARS-CoV-2 docks onto ACE2, which is a lipid raft protein. After binding to ACE2, the S protein in the viral envelope undergoes enzymatic activation by TMPRSS2 or furin, likely located in lipid rafts. Subsequent endocytosis of SARS-CoV-2 occurs using a raft-dependent endocytic pathway.
SARS-CoV-2 induces the activity of ASMase and releases the ceramide content in lipid rafts, resulting in the virus's attachment to its receptors and increasing the concentration of virus attachment in lipid rafts domains and viral infectivity. Several reports show that the ASMase/ceramide system controls viral infection. Viruses including Rhinovirus, Ebola, and measles encephalitis [15,16,17], and bacteria like Pseudomonas aeruginosa, Staphylococcus aureus, Salmonella typhi, and Neisseria gonorrhoeae [18,19,20,21,22,23], stimulate the viral ASMase/ceramide system inducing the development of platform domains rich in ceramide, which facilitate viral entry and host cell infection. As with other viruses, SARS-CoV-2 activates the ASMase/ceramide system, inducing ceramide-enriched-platform formation and facilitating viral entry by clustering ACE-2, resulting in host cell infection [24]. Since 1970, research has shown that weak bases constrain ASMase activity [25]. Weak bases are protonated and diffused into lysosomes, where they are trapped, accumulating intra-lysosomal weakly basic molecules [26].
FIASMA are weak bases and accumulate in acidic compartments like the lysosome because they become protonated at the acidic pH. Due to the positive charge, they can no longer cross the membrane (acidic trapping). Consequently, lysosomal ASMase is displaced from the inner lysosomal membrane, and ASMase is proteolyzed. The ASMase/ceramide system is considered a treatment option in patients with respiratory COVID-19 or mutated strains [27]. This review demonstrates the metabolism and importance of sphingolipids responsible for viral infection. The function of ASMase in viral entry and infection is clarified. Accordingly, this review categorizes types of ASMase inhibitors, the functional inhibitors of acid sphingomyelinase (FIASMA) that potentially block viral entry. Additionally, molecular docking in silico of ASMase/ceramide system inhibitors is performed to predict the prospective efficacy of inhibitors as anti-SARS-CoV-2 medication.
Structure of Sphingomyelinase
Human acid sphingomyelinase is a cellular phosphodiesterase or phospholipase C (PLC), which causes sphingomyelin to hydrolyze into ceramide and phosphocholine by cleavage of the phosphodiester bond. The SMPD1 gene encodes human ASMase in the chromosomal region 11p15.4 with 6 exons, as shown in Fig. 1 (1). The 1890 bp open reading frame of the whole cDNA for ASMASE codes for 629 amino acids. A monomeric glycoprotein with a protein core of 64 kDa makes up the mature ASMase enzyme. The ASMase enzyme contains 8 disulfide bridges, 5 N-glycosylation sites are occupied, and one N-glycosylation site is not occupied [28, 29], as shown in Figs. 1 (2) and (3).

Acid Human Sphingomyelinase (ASMase) exons (1), cDNA (2), and protein structure (3)
According to the UniProt blast site, mature ASMase has numerous active domains, including a signal peptide (amino acids 1–46), a Sap-domain (amino acids 89–165), a proline-rich linker domain (amino acids 166–198), the catalytic metallo-phosphatase domain (amino acids 199–461), and the C-terminal domain (amino acids 462–629) [30]. Even in the absence of exogenous sphingolipid activator proteins, the basic sphingomyelinase cleaving activity of the ASMase polypeptide is maintained by its N-terminal Sap-domain [31]. Sphingomyelin attaches to the active site of the catalytic metallo-phosphatase domain, which has a binuclear zinc core, to activate the hydrolysis process and cleave the phosphodiester bond. The ASMase activity depends on the Sap-domain [30, 32, 33].
Human sphingomyelinase is produced in the endoplasmic reticulum as a pre-pro-enzyme with a core protein of 75 kDa, which is quickly cleaved into 72 kDa pro-ASMase in the endoplasmic reticulum-Golgi complex. After cleavage, the pro-ASM is transmitted by the secretory pathway to the extracellular space or endolysosomal compartments. ASMase and numerous other lysosomal hydrolases are transported from the trans-Golgi network (TGN) to late endosomes and lysosomes by the mannose-6-phosphate receptor (M6PR). The ASM lipid-binding proteins, prosaposin, and GM2AP have an alternative route depending on sortilin [34,35,36].
Sphingolipid Metabolism
Sphingoid bases are the basic structure of sphingolipids, including sphingosine, an 18-carbon unsaturated amino alcohol, the most common among mammals, amid links fatty acids to sphingosine, resulting in ceramide [37]. Sphingomyelin is produced when ceramide is phosphocholine esterified, while glycosylceramides are produced when ceramide is glycolyzed. Sialic acid residues result in ganglioside synthesis, as shown in Fig. 2. These are important cell membrane molecules, and the pathway intermediates for sphingolipid production and breakdown modify processes like apoptosis and T-cell trafficking [37, 38].

Illustration of sphingolipid metabolism pathway including denovo synthesis and breakdown
Sphingosine, phytosphingosine, and digydrosphingosine represent the first step in creating complex molecules. Sphingosine 1-phosphate, phytosphingosine 1-phosphate, and dihydrosphingosine-1-phosphate are three crucial signalling molecules broken down by phosphorylation of the C1 hydroxyl group. The glycosphingolipids contain a wide range of sphingolipids that differ by the type and arrangement of sugar residues linked to their head groups.
The sphingolipid metabolic pathway is a vital cellular process where ceramide plays an important role in other molecules' metabolism, catabolism, and biosynthesis. Through de novo synthesis, sphingolipids are produced via serine and palmitoyl CoA condensation. This process is catalyzed by serine palmitoyl transferase, which results in 3-keto-dihydrosphingosine [39]. Hydrolysis of sphingomyelin by sphingomyelinase into ceramide keeps the membranes in homeostasis conditions. Thus, sphingolipid metabolism is complicated but involves the de novo biosynthesis of ceramide in the endoplasmic reticulum. Ceramide is the key product in the breakdown of sphingomyelin or their de novo synthesis, which is the process by which sphingolipids are metabolically processed. The de novo synthesis could begin with serine palmitoyl-transferase, serine condensation, and palmitoyl–coenzyme A to 3-keto di hydrosphingosine [37]. Then, the reduction of 3-ketodihydrosphingosine into sphinganine is carried out by 3-ketodihydrosphingosine reductase.
Ceramide synthase adds acyl fatty acids to sphinganine, leading to dihydroceramide production. In the endoplasmic reticulum, dihydroceramide D4 saturates and desaturates into ceramide. A ceramide transfer protein transports ceramide from the endoplasmic reticulum to the Golgi apparatus. Sphingosine (2 amino-4-trans-octadecene-1,3-diol) is produced from ceramide by ceramidase enzymes. Ceramide synthase is responsible for the production of ceramide in a way opposite to ceramidase enzymes. Sphingosine kinase 1 (SPhK1) or sphingosine kinase 2 (SPhK 2) phosphorylate sphingosine to produce sphingosine 1-phosphate (S1P). Sphingosine is phosphorylated into sphingosine 1-phosphate (S1P) by either sphingosine kinase 1 (SPhK1) or sphingosine kinase 2 (SPhK 2). The S1P phosphatases transform S1P back to sphingosine, or the S1P lyase enzyme breaks down S1P into hexadecanal and phosphoryl ethanolamine [37]. Sphingolipids are generated from ceramides by sphingomyelin synthase, while sphingomyelin is converted to ceramide via sphingomyelinase. The sphingolipid metabolism pathway is illustrated in Fig. 2.
Sphingolipid Transport and Uptake
Bioactive sphingolipids interact with mediators to produce functional responses. Sphingolipids include sphingomyelin, ceramides, sphingosine, and S1P [40]. Sphingosine contains one aliphatic chain that usually has 18 carbon atoms along its length, enabling easy passage between distinct membranes. Sphingosine 1 phosphate is generated in the inner cell plasma membrane in response to tumor necrosis factor-cytokine as signalling (TNFa). Then, it transfers to the outer leaflet of the plasma membrane to bind to its S1PRs receptor [41]. The ABC transporter superfamily has two members proposed to control S1P trafficking [42]. There are two members to regulate S1P, either internalization or efflux by cystic fibrosis transmembrane regulator (CFTR) or ABCC1.
Sphingomyelin contains two aliphatic chains and a zwitter ionic head group. Thus, it has little aqueous solubility and hardly ever flip-flops across bilayers but moves laterally [43]. The movement of sphingomyelin may be hampered by interactions with sterols in cell membranes and self-aggregation [43]. The ceramide structure has two aliphatic chains and a neutral head group. Ceramide is transported from its production site in the endoplasmic reticulum to the Golgi apparatus under the control of the Ceramide Transfer Protein (CERT). Studies show neutral ceramide easily flip-flopping across cell membranes [43].
It is unknown if the organization of ceramide into microdomains prevents ceramide from flipping from the outer leaflet to the inner leaflet of the plasma membrane or whether ceramide can flip-flop as effectively in complex biological membranes [43]. Limiting the flipping of ceramide could impact its signalling functions significantly.
Role of Sphingolipids in Viral Entry
Lipid rafts are particular regions of the host cell membrane that are profuse in lipids like cholesterol, sphingolipids, and gangliosides [44]. These lipid-rich domains are characterized by containing well-organized lipid molecules stacked tightly. Studies show lipid rafts are key in viral infection cycles, including HIV, poliovirus, hepatitis C, and coronaviruses [45,46,47]. The SARS-CoV-2 virus uses lipid rafts and caveolae-mediated endocytosis for viral entry [48]. Thorp and Gallagher (2004) observed that methyl-β-cyclodextrin triggers cholesterol depletion and inhibits viral entry and infection. This observation supports a lipid raft’s functional role in viral entry [49]. Coronaviridae, including SARS-CoV, use lipid rafts to enter and host infection. In addition to the minor envelope protein and membrane protein, the SARS-CoV-2 envelope contains spike protein (S) [50]. A viral spike (S) comprises S protein trimmers, which act in viral fusion with host cellular membranes and constitute two subunits (S1 and S2). After viral binding, spike protein is cleaved by host protease transmembrane serine protease 2 with furin pre-cleavage to facilitate viral entry [11, 51, 52]. SARS-CoV-2 entry is receptor-mediated endocytosis through a specific host receptor (ACE-2). Viral S protein binds with ACE-2, enabling proteolysis of viral S1 protein by host proteases, which may be attached to caveolae, including TMPRSS2 and Cathepsin L [53].
Moreover, SARS-CoV-2's ability to enter and cause host infection depends on its interaction with specific gln493 residue of the ACE-2 receptor [54]. Viral entrance may be mediated by the host ACE-2 receptor or by sialic acids interacting with host cell surface ganglioside binding domains. This domain (111–158) is a well-conserved sequence causing viral attachment to lipid rafts, which makes it easier for SARS-CoV-2 to infect the host's ACE-2 receptor [54]. ACE-2 must colocalize with the raft markers GM1 and caveolin-1. Lipid rafts are a key platform that can concentrate host ACE-2 receptors interacting with viral S protein. Viral particles can bind to the surface of the host cell membrane because ACE-2 clusters in certain positions in the cell membrane. In this approach, lipid raft microdomains boost the efficacy of viral infection. These results agree with cholesterol depletion and reduce, but do not prevent, the susceptibility to viral infection [55].
Lipid rafts are considered targets for inhibiting viral infection. Drugs such as methyl-β-cyclodextrin cause disruption of lipid rafts, resulting in viral entry inhibition [56]. Pathogen-host interactions probably aid the development of focal adhesions and lipid raft clustering during endocytosis. Table 1 shows other inhibitors of lipid rafts such as propofol, isoflurane, pentobarbital, aspirin, naproxen, perifosine cisplatin, azithromycin, daunorubicin, doxorubicin, quercetin, and luteolin. These inhibitors may be used as antiviral drugs against SARS-CoV-2. Thus, research on lipid rafts should be included in developing antiviral drugs.
The Acid Sphingomyelinase/Ceramide System in Viruses
Scientific studies showed severe consequences and harsh symptoms resulting from acute respiratory syndrome coronavirus 2 (SARS-COV-2). Virus infectivity and spread have been extensively studied. Interestingly, SARS-CoV-2 infectivity occurs by attachment to the host cell receptor via S proteins. This results in virus priming by proteases, facilitating viral entry through endocytosis and completing the viral life cycle. The sphingolipid family is the most common lipid along the cell membrane, including sphingosine and ceramide. Such lipids can interfere with virus uptake into epithelial cells and in cultures of human nasal cells. With the different mechanisms of action, sphingosine is blocked, while ceramide enables viral infection. The well-known acid sphingomyelinase (ASMase) is essential to produce ceramide, and drug inhibition, like amitriptyline, reduces entry into epithelial cells.
Consequently, a key prognostic marker for assessing the severity of COVID-19 is S1P [89]. ASMase transforms sphingomyelin into ceramide, found either on the cell membrane surfaces or attaches to the outer surface of plasma membranes. Acid sphingomyelinase surfaces function as signalling molecules and produce ceramide in the outer parts of plasma membranes. The ceramide molecules are hydrophobic and form small membrane domains that rearrange to form larger platforms. These domains recognize 1-integrin, CD95, CD40, DR5, and other activated receptor molecules. Ceramide platforms mediate bacterial or viral infection and other stress stimuli [24].
When viruses enter cell membranes, sphingolipids function as bioactive lipids that transmit signals inside and outside cells. So, limiting viral replication by targeting the host cell's sphingolipid metabolism may give a chance for more therapeutic approaches. Host cell viral infection begins with endocytosis, then un-coating, exocytosis, and discharge of nucleocapsids into the cytoplasm. These previous actions are affected by membrane microdomains. Subsequently, the interactions between viruses and cells promote different signal cascades affecting cellular uptake, intracellular trafficking, and viral replication [90].
The ASMase activity is implicated in other viruses like Ebola’s early infection stages. Acid sphingomyelinase activation is crucial for Ebola virus endocytosis, making Niemann-Pick C protein 1 (NPC1), an endo/lysosomal cholesterol transporter, virus particle-accessible. To facilitate the fusing of the Ebola virus and endosomal membranes, NPC1 is essential for viral absorption. Thus, NPC1 acts as a receptor for the proteolytically activated viral envelope protein in an intracellular compartment rather than at the plasma membrane.
Acid sphingomyelinase activation is also recognized after the interaction of dendritic cells with the measles virus. Viral glycoproteins interact with DC-SIGN on the cell surface, which induces the activation of sphingomyelinase and the release of ceramide molecules. Then, measles virus receptor CD150 entry is translocated from an intracellular storage compartment to the cell surface, favouring viral infection of dendritic cells [90].
Trafficking Process utilized in viral entry
The trafficking or endocytosis process enables cells to internalize macromolecules, nutrients, or viruses into the cell [91]. The endocytosis process is classified into receptor-mediated endocytosis, caveolae uptake, or clathrin-independent endocytosis, including the CLIC/GEEC pathway [92,93,94]. Internalized macromolecules are categorized by endosomes, which are a pleiomorphic series of tubulovesicular compartments [95]. Internalized macromolecules are processed in various ways, including back-recycling to the cellular plasma membrane, degradation by delivery to the lysosomal molecules, or to polarized cells through transcytosis [96]. Several events accompany the maturation of endosomal compartments, including luminal pH decrease, significant phosphatidylinositol lipid alterations via regulating lipid kinases and phosphatases, and activation and differential Rab-family GTPase recruitment. The trafficking or endocytosis process has critical cellular functions. Functions include cellular communication between cells and the environment, controlling cellular homeostasis and regulating essential surface proteins, and viral or bacterial entrance [97]. Moreover, the process regulates cell signalling through G-protein coupled receptors and receptor tyrosine kinases [98, 99]. This review focuses on clathrin and dynamin-independent pathways, especially lipid raft entry, and their role in SARS-CoV-2 entry.
Clathrin and dynamin-independent pathways utilized in viral entry
Receptor-independent endocytosis (CIE) includes the CLIC/GEEC pathway responsible for cellular functions. For instance, cell signalling, adhesion, nutrient receptors, and regulation of the expression of certain membrane transporters. The endocytic vesicles/tubules of CIE are characterized by having no distinct coat. The CIE was discovered using inhibitors blocking clathrin-mediated and caveolae-mediated endocytosis [92, 93, 100]. Small GTPases Rac1 and Cdc42 involved in clathrin- and dynamin-independent pathways are responsible for actin formation-dependent clathrin-independent carriers (CLICs) [101]. The GPI-AP enriched endosomal compartments are specific early endosomal compartments generated by the fusion of CLICs (GEECs) [102, 103]. This process, called the CLIC/GEEC pathway, depends on specific proteins, including GTPase and Arf6, and is responsible for taking and recycling the major Histocompatibility Antigen I [104].
Small protein Arf6 triggers the activation of phosphatidylinositol-4-phosphate 5-kinase, resulting in PI(4,5)P2, which induces actin assembly and drives endocytosis [105]. Another endocytosis pathway is the flotillin pathway, which depends on curvature-generating and membrane-anchored proteins [106, 107]. In vitro HeLa cells undergo CLIC/GEEC and a flotillin-dependent pathway, taking up PI-anchored protein and CD59. The CLIC/GEEC pathway and the Arf6-pathway are both involved in the uptake of the transmembrane protein CD44 [108]. Several types of CLIC/GEEC pathways play a role in rapidly recycling cell membranes. The CLIC/GEEC pathway is responsible for nutrient and toxin uptake and is considered a portal for viral infection [109].
SARS-CoV-2 Entry by Lipid Rafts
Viral entry and infection depend on endocytosis pathways, especially sphingolipids and lipid rafts. Many viruses utilize lipid rafts to enter the host cell and facilitate infection, including hepatitis C viruses [47], human herpes virus 6 [110], poliovirus [46], and simian virus 40. Coronaviruses, including SARS-CoV-2, interact with lipid rafts to enter host cells and cause infection [111, 112]. Studies by Thorp and Gallagher (2004) supported the function of sphingolipids and cholesterol in viral infections, where cholesterol reduction prevents viral entry [49].
The virus is made up of an envelope that includes spike protein (S), membrane protein (M), and minor envelope protein (E). Transmembrane serine protease 2 (TMPRSS2), with the help of furin, triggers cleavage of the viral spike (S1 and S2) [51]. The Golgi apparatus contains a predominant amount of furin; the other part is found on the cell surface [52]. Once the viral spike and its structural proteins bind to ACE-2, it is activated and promotes viral entry into the host cell.
The host cell receptor is angiotensin-converting enzyme-2 that binds to the S proteins in the virus [113], enabling proteolysis of viral surface S1 subunit by a plasma-membrane-bound serine protease (TMPRSS2) and Cathepsin L (CatL), which may be associated with caveolae [114]. Once SARS-CoV-2 is attached to caveolae and enters intracellular endosomes, cathepsin L emerges as the main protease of the virus [115].
Viral gateway into the host cell or ACE-2 receptor exists on the surface of several types of cells, including kidney, respiratory, and intestinal epithelial and endothelial cells. Respiratory SARS-CoV-2 attaches to ACE-2 by gln493 residue, enabling viral entry. Viral S protein not only attaches to ACE-2 but also binds to host cell surface gangliosides. A new type of ganglioside-binding domain (111–158) was identified within the N-terminal domain of the SARS-CoV-2 S protein, facilitating attachment of viral spike to lipid rafts and attachment to host cell receptors [54]. The ACE-2 is colocalized with SARS-CoV-2, entering and infecting host cells by direct membrane fusion or by host cell ACE-2. Lipid rafts are key in both viral entrance methods, enabling the concentration of the endocytic proteins for endocytosis and fusion, as shown in Fig. 3. When endocytic proteins concentrate and interact within lipid rafts, the frequency of interprotein collisions by protein partitioning into lipid rafts increases [116]. As a result, lipid rafts act as plasma membrane "chambers" that facilitate protein interactions on the plasma membrane, promote the rate of molecule collisions, and consequently improve the efficacy of membrane reactions.

Representation of SARS-CoV-2 Entry mechanism by interacting of spike protein in RBD of the virus with host receptor ACE-2 and consequently internalized into the cell through endocytosis process by helping lipid rafts
Lipid rafts play a role in viral infection by providing appropriate platforms that concentrate host cell receptor ACE-2 on the cell membrane, where they attach with viral S protein. The receptor binding domain (RBD) is the vital part of the virus that engages the protease domain (PD) of ACE-2, resulting in a complex containing a dimeric ACE-2 with two S protein trimers [117]. Multivalent binding of virus particles to the cell surface occurs by host cell receptors clustering. Microdomains of the host cell membrane improve the efficiency of viral infection and facilitate viral endocytosis. Several studies confirm that methyl-β-cyclodextrin (MβCD) inhibits infectious bronchitis virus infection by disrupting lipid rafts, indicating that lipid rafts play a role in viral entry [56, 118]. After the viral S protein attaches to ACE-2 and the virus and host receptor complex have formed, lipid raft and focal adhesions are clustered during endocytosis. Therefore, lipid rafts are hypothesized to be significant during the early stages of coronavirus infection.
Lipid Raft Distribution Reduces SARS-CoV-2 Infectivity
Some drugs affect lipid rafts and thus play an important role as antiviral drugs. Lipid raft distribution and cholesterol depletion by methyl-β-cyclodextrin (MβCD) minimizes the infectivity of the influenza virus [119]. Lipid raft distribution reduces viral infectivity and holes in the viral envelope, which disturb the viral structure and affect viral protein release. Several studies observed that cholesterol depletion reduces the infectivity of SARS-CoV-2 [120,121,122]. Inhibiting of viral biosynthesis and infection occurs using drugs such as lovastatin or squalestatin that promote cholesterol depletion. A significant viral ASM/ceramide system in SARS-CoV-2 is important for viral infection. Entry of SARS-CoV-2 and clustering with host cell receptors are facilitated by stimulation of the ASM/ceramide system, subsequently forming membrane domains rich in ceramide platforms on the cell membrane [24].
In this context, the ASM/ceramide system is considered an antiviral target to reduce viral infection. FIASMAs are antiviral drugs used against the ASM/ceramide system in SARS-CoV-2 that inhibit the formation of ceramide-enriched membrane domains, thereby preventing SARS-CoV-2 infection (Table 2). Ceramide has several functions, including clustering of ACE-2 in large membrane domains and amplifying signaling via ACE-2, which is also required for host cell ACE-2 internalization of the virus into the endosome [123]. Cathepsins in the endosome interact with ceramide produced inside endosomes or on the cell membrane's outer leaflet, promoting spike-protein priming and membrane fusion [124]. As a result, FIASMAs inhibit the formation of domains enriched with ceramide and viral entry and infection. In this context, viral infection is inhibited by the down-regulation of the genetic expression of ASMase. FIASMAs change the pH of the endosome, enabling lysosomes to target the endosome and make the virus more susceptible to lysosomal degradation. Therefore, our review suggests FIASMA medications as antiviral therapeutics by targeting lipid raft domains.
Functional Inhibitors of Acid Sphingomyelinase FIASMAs’ Mechanism of Action
Specific electrostatic forces bind lysosomal acid sphingomyelinase to the intra-lysosomal membranes and thus remain protected against proteolytic activity. FIASMAs inhibit ASMase by an indirect mechanism [26] (Figs. 4). The intra-lysosomal space maintains a low pH by an ATP-driven proton pump, which retains the attachment of the ASMase to the intra-lysosomal membranes. The lysosomal membrane is characterized by low permeability towards the protonated bases compared to uncharged ones (lysomotropism). Therefore, with the intake of FIASMAs and other weak bases (lysosomal accumulation), the intra-lysosomal pH raises and diminishes the electrostatic interactions between the lysosomal membrane and the ASMase, resulting in ASMase detachment [127].

Representation of targeting FIASMAs on the catalytic domain of ASMase protein
Following the detachment, ASMase is cleaved and degraded within the lysosomes by proteolytic degradation [27, 167]. Notably, inhibition of ASMase by certain drugs has long been recognized, but systematic studies describing FIASMA inhibition are fairly new [127] (Figs. 5, 6).

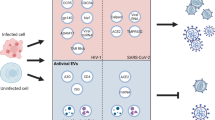
Representation of FIASMAs is antiviral drugs used against ASM/ceramide system in SARS-CoV-2 inhibits the formation of ceramide-enriched membrane domains, thereby preventing infection with SARS-CoV-2

Schematic representation of the mode of action of FIASMAs (functional inhibitors of acid sphingomyelinase). A Eukaryotic cell display cellular organelles. B A magnified version of lysozyme exhibiting FIASMs indirect inhibition of ASM (acid sphingomyelinase)
In Vitro Docking of Potent Antiviral Compounds Based on Sphingolipid Inhibition
Molecular Operating Environment (MOE) software was used to perform docking analyses [168] of acid sphingomyelinase inhibitors to quantify their inhibitory effect on SARS-CoV-2 uptake. Their binding modes with mammalian acid sphingomyelinase’s binding site (PDB code: 5FI9) and their interaction with key amino acids were compared to the candidate drug amiodarone [169]. All structure minimizations were performed until an RMSD gradient of 0.05 kcal∙mol−1 Å−1 with MMFF94x force field, and partial charges were automatically calculated. All intervening water molecules were removed from the structure, and then the target protein was prepared for docking using Protonate 3D protocol in MOE with default parameters. The co-crystalized ligand was used to define the binding site for docking simulation. The Triangle Matcher Placement method and London dG scoring function were employed for docking and scoring. The docking protocol was first validated by self-docking the co-crystallized ligand near the protein's binding site. The ligand-receptor interactions at the protein binding site were studied with the validated docking protocol (RMSD < 2) for the reported inhibitors to predict their binding mode and binding affinity.
Validation and endorsement of the docking protocol were achieved by self-docking of the co-crystallized (1-azanyl-1-phosphono-decyl) phosphonic acid (APPA) within the acid sphingomyelinase active site with an energy score (S) of –28.75 kcal/mol and RMSD of 1.49 Å, and with reproducing all interactions of APPA with the binding site of the enzyme (Fig. 7A). Reported inhibitors interacted with the key amino acids in the acid sphingomyelinase active site, indicating their inhibition activities as confirmed by their docking scores (S) and binding modes compared to that of the candidate drug amiodarone (Figures. 7B and Table 3).

A (i) Superimposition of the docking pose (green) and the co-crystallized (blue) of (1-azanyl-1-phosphono-decyl) phosphonic acid (APPA) in the acid sphingomyelinase active site with RMSD of 1.49 Å. (ii) 2D interaction diagram showing APPA docking pose interactions with the hot spots in the enzyme active site. B. 2D diagrams of the candidate drug (amiodarone) and the reported inhibitors. 1. Alverine, 2. Ambroxol, 3. Amiodarone, 4. Amitriptyline, 5. Amlodipine, 6. Aprindine, 7. Astemizole,8.Benztropine, 9. Bepridil, 10. Biperidene, 11. Camylofine, 12. Carvedilol, 13. Cepharanthine, 14. Chlorpromazine, 15. Chlorprothixene, 16. Clemastine, 17. Clofazimine, 18. Clomiphene, 19. Clomipramine, 20. Cloperastine, 21. Conessine, 22. Cyclobenzaprine, 23. Cyproheptadine, 24. Desipramine, 25. Desloratadine, 26. Dicycloverine, 27. Dilazep, 28. Dimebon, 29. Doxepine, 30. Drofenine, 31. Emetine, 32. Fendeline, 33. Flunarizine, 34. Fluoxetine, 35. Flupenthixol, 36. Fluphenazine, 37. Fluvoxamine, 38. Hydroxyzin, 39. Imipramine, 40. Loperamide, 41. Loratadine, 42. Maproteline, 43. Mebeverine, 44. Mebhydrolin, 45. Melatonin, 46. Mibefradil, 47. Norfluoxetine, 48. Nortriptyline, 49. Paroxetine, 50. Perphenazine, 51. Pimozide, 52. Profenamine, 53. Promazine, 54. Promethazine, 55. Protriptyline, 56. Quinacrine, 57.Sertindole, 58. Sertraline, 59. Solasodine, 60. Suloctidil, 61. Tamoxifene, 62. Terfenadine, 63. Thioridazine, 64. Tomatidine, 65. Trifluoperazine, 66. Triflupromazine, 67. Trimipramine, 68. Zolantidine showing their interaction with the key amino acids in the acid sphingomyelinase
The docking simulation studies revealed that dilazep (S = − 12.58 kcal/mol), emetine (S = − 11.65 kcal/mol), pimozide (S = − 11.29 kcal/mol), carvedilol (S = − 11.28 kcal/mol), mebeverine (S = − 11.14 kcal/mol), cepharanthine (S = − 11.06 kcal/mol), hydroxyzine (S = − 10.96 kcal/mol), astemizole (S = − 10.81 kcal/mol), sertindole (S = − 10.55 kcal/mol), and bepridil (S = − 10.47 kcal/mol) had higher inhibition activity than the candidate drug amiodarone (S = − 10.43 kcal/mol) towards the acid sphingomyelinase. In addition, dilazep (S = − 12.58 kcal/mol) was the most effective inhibitor. Additionally, we provided a comprehensive Table 4 for publicly available inhibitors of ASMase in vitro and in vivo in previous studies to give insight into experimental data regarding ASMase inhibitors.
Conclusion and limitations
Nevertheless, dilazep showed the most promising in silico results against ASMase with (S = − 12.58 kcal/mol); we couldn’t find a correlation with experimental data; however, our pre-elementary docking can be validated through in vitro and in vivo future experimental data. Interestingly, emetine had (S = − 11.65 kcal/mol), consistent with its in vitro capacity against SARS-CoV-2 virus in Vero E6 cells with the estimated 50% effective concentration at 0.46 μM [148]. Pimozide pointed out (S = − 11.29 kcal/mol) can be correlated with its IC50 potency of 42 ± 2 µM and its potent inhibitory infection by pseudotyped viruses with minimal effects on cell viability [143, 178]. While carvedilol had (S = − 11.28 kcal/mol), a previous cohort study didn’t confirm its role as a significant player against SARS-CoV-2 [130]. Mebeverine showed (S = − 11.14 kcal/mol); however, to our knowledge, the inhibitor hasn’t been tested experimentally. Furthermore, cepharanthine, which pointed out (S = − 11.06 kcal/mol), had potential antiviral activities against SARS-CoV-2 [179]. Hydroxyzine (S = − 10.96 kcal/mol) had previously shown a significant impact against SARS-CoV-2 in vitro and in vivo approaches [154, 193]. Astemizole had (S = − 10.81 kcal/mol) given by its ability to bind to the ACE2 receptor and inhibit the invasion of SARS-COV-2 Spike pseudoviruses [170]. Sertindole had (S = − 10.55 kcal/mol) results, which is in agreement with its showed in vitro inhibition of acid sphingomyelinase [174]. Bepridil (S = − 10.47 kcal/mol) was found to be a significant inhibitor against SARS-CoV-2 activity in both Vero E6 and A459/ACE2 cells in a dose-dependent manner with low micromolar effective concentration, 50% (EC50) values [178].
Limitations
Our work can be considered pre-elementary screening for ASMase inhibitors, leading to several candidates that should be tested in vitro and in vivo. Also, FIASMAs binding to the active site of ASMase wasn’t significantly robust as compared to crystallographic ligand (S = –28.75 kcal/mol), which can be attributed to the indirect work of FIASMAs through lysosomal accumulation and raising intra-lysosomal pH causing reduced the electrostatic interactions between the lysosomal membrane and the ASMase, resulting in ASMase detachment. Also, our in silico framework depended only on MOE software, which didn’t reveal the standard deviations of binding energies, so we recommend using additional software to validate the results further.
Availability of data and materials
The data supporting this study's findings are available from the corresponding author upon reasonable request.
References
Hoertel N, Blachier M, Blanco C, Olfson M, Massetti M, Rico MS, Limosin F, Leleu H. A stochastic agent-based model of the SARS-CoV-2 epidemic in France. Nat Med. 2020;26(9):1417–21.
Ghosh S, Bornman C, Zafer MM. Antimicrobial Resistance Threats in the emerging COVID-19 pandemic: Where do we stand? J Infect Public Health. 2021;14(5):555–60.
Alhouri A, Salloum A, Harfouch RM, Soumya G. Possible side effects of using detergents during the Covid19 pandemic in Syria. Ann Clin Cases. 2020;1(4):1023.
Meskini M, Rami MR, Maroofi P, Ghosh S, Siadat SD, Sheikhpour M. An overview on the epidemiology and immunology of COVID-19. J Infect Public Health. 2021;14(10):1284–98.
Ghosh S, Al-Sharify ZT, Maleka MF, Onyeaka H, Maleke M, Maolloum A, Godoy L, Meskini M, Rami MR, Ahmadi S, et al. Propolis efficacy on SARS-COV viruses: a review on antimicrobial activities and molecular simulations. Environ Sci Pollut Res. 2022l;29(39):58628–47.
Hessien M, Donia T, Tabll AA, Adly E, Abdelhafez TH, Attia A, Alkafaas SS, Kuna L, Glasnovic M, Cosic V. Mechanistic-Based Classification of Endocytosis-Related Inhibitors: Does It Aid in Assigning Drugs against SARS-CoV-2? Viruses. 2023;15(5):1040.
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X. Clinical features of patients infected with 2019 novel coronavirus in Wuhan China. The lancet. 2020;395(10223):497–506.
Alkafaas SS, Abdallah AM, Hussien AM, Bedair H, Abdo M, Ghosh S, Elkafas SS, Apollon W, Saki M, Loutfy SA. A study on the effect of natural products against the transmission of B.1.1. 529 Omicron. Virol J. 2023;20(1):191.
Mahase E. Covid-19: death rate is 0.66% and increases with age, study estimates. BMJ. 2020;369:1327.
Tang Y, Liu J, Zhang D, Xu Z, Ji J, Wen C. Cytokine storm in COVID-19: the current evidence and treatment strategies. Front Immunol. 2020;11:1708.
Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu N-H, Nitsche A. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271–280. e278.
Hurwitz R, Ferlinz K, Vielhaber G, Moczall H, Sandhoff K. Processing of human acid sphingomyelinase in normal and I-cell fibroblasts. J Biol Chem. 1994;269(7):5440–5.
Schissel SL. Jiang X-c, Tweedie-Hardman J, Jeong T-s, Camejo EH, Najib J, Rapp JH, Williams KJ, Tabas I: Secretory sphingomyelinase, a product of the acid sphingomyelinase gene, can hydrolyze atherogenic lipoproteins at neutral pH: implications for atherosclerotic lesion development. J Biol Chem. 1998;273(5):2738–46.
Grassmé H, Jekle A, Riehle A, Schwarz H. Berger Jr, Sandhoff K, Kolesnick R, Gulbins E: CD95 signaling via ceramide-rich membrane rafts. J Biol Chem. 2001;276(23):20589–96.
Miller ME, Adhikary S, Kolokoltsov AA, Davey RA. Ebolavirus requires acid sphingomyelinase activity and plasma membrane sphingomyelin for infection. J Virol. 2012;86(14):7473–83.
Avota E, Gulbins E, Schneider-Schaulies S. DC-SIGN mediated sphingomyelinase-activation and ceramide generation is essential for enhancement of viral uptake in dendritic cells. PLoS Pathog. 2011;7(2): e1001290.
Tani H, Shiokawa M, Kaname Y, Kambara H, Mori Y, Abe T, Moriishi K, Matsuura Y. Involvement of ceramide in the propagation of Japanese encephalitis virus. J Virol. 2010;84(6):2798–807.
Esen M, Schreiner B, Jendrossek V, Lang F, Fassbender K, Grassme H, Gulbins E. Mechanisms of Staphylococcus aureus induced apoptosis of human endothelial cells. Apoptosis. 2001;6(6):431–9.
Grassmé H, Gulbins E, Brenner B, Ferlinz K, Sandhoff K, Harzer K, Lang F, Meyer TF. Acidic sphingomyelinase mediates entry of N. gonorrhoeae into nonphagocytic cells. Cell. 1997;91(5):605–15.
Hauck CR, Grassmé H, Bock J, Jendrossek V, Ferlinz K, Meyer TF, Gulbins E. Acid sphingomyelinase is involved in CEACAM receptor-mediated phagocytosis of Neisseria gonorrhoeae. FEBS Lett. 2000;478(3):260–6.
Akinyemi KO, Al-Khafaji NS, Al-Alaq FT, Fakorede CO, Al-Dahmoshi HO, Iwalokun BA, Akpabio I, Alkafaas SS, Saki M. Extended-spectrum Beta-lactamases Encoding Genes among Salmonella Enterica serovar Typhi Isolates in Patients with Typhoid Fever from four Academic Medical Centers in Lagos. Nigeria Revista de investigacion clinica. 2022;74(3):165–71.
Alduhaidhawi AHM, AlHuchaimi SN, Al-Mayah TA, Al-Ouqaili MT, Alkafaas SS, Muthupandian S, Saki M. Prevalence of CRISPR-Cas Systems and Their Possible Association with Antibiotic Resistance in Enterococcus faecalis and Enterococcus faecium Collected from Hospital Wastewater. Infection and Drug Resistance. 2022;15:1143.
Raheem HQ, Hussein EF, Ghosh S, AlKafaas SS, Bloemfontein SA. Resistance of Klebsiella pneumoniae from Different Clinical Samples to Penicillin, Cephalosporin. Carbapenem and Fluoroquinolone Teikyo Medical Journal. 2021;44(06):1–8.
Kornhuber J, Hoertel N, Gulbins E. The acid sphingomyelinase/ceramide system in COVID-19. Mol Psychiatry. 2022;27(1):307–14.
Sakuragawa N, Sakuragawa M, Kuwabara T, Pentchev PG, Barranger JA, Brady RO. Niemann-Pick disease experimental model: sphingomyelinase reduction induced by AY-9944. Science. 1977;196(4287):317–9.
Kölzer M, Werth N, Sandhoff K. Interactions of acid sphingomyelinase and lipid bilayers in the presence of the tricyclic antidepressant desipramine. FEBS Lett. 2004;559(1–3):96–8.
Kornhuber J, Muehlbacher M, Trapp S, Pechmann S, Friedl A, Reichel M, Mühle C, Terfloth L, Groemer TW, Spitzer GM. Identification of novel functional inhibitors of acid sphingomyelinase. PLoS ONE. 2011;6(8): e23852.
Lansmann S, Schuette CG, Bartelsen O, Hoernschemeyer J, Linke T, Weisgerber J, Sandhoff K. Human acid sphingomyelinase: Assignment of the disulfide bond pattern. Eur J Biochem. 2003;270(6):1076–88.
Ferlinz K, Hurwitz R, Moczall H, Lansmann S, Schuchman EH, Sandhoff K. Functional characterization of the N-glycosylation sites of human acid sphingomyelinase by site-directed mutagenesis. Eur J Biochem. 1997;243(1–2):511–7.
Xiong Z-J, Huang J, Poda G, Pomès R, Privé GG. Structure of human acid sphingomyelinase reveals the role of the saposin domain in activating substrate hydrolysis. J Mol Biol. 2016;428(15):3026–42.
Henseler M, Klein A, Glombitza GJ, Suziki K, Sandhoff K. Expression of the three alternative forms of the sphingolipid activator protein precursor in baby hamster kidney cells and functional assays in a cell culture system. J Biol Chem. 1996;271(14):8416–23.
Gorelik A, Illes K, Heinz LX, Superti-Furga G, Nagar B. Crystal structure of mammalian acid sphingomyelinase. Nat Commun. 2016;7(1):1–9.
Zhou Y-F, Metcalf MC, Garman SC, Edmunds T, Qiu H, Wei RR. Human acid sphingomyelinase structures provide insight to molecular basis of Niemann-Pick disease. Nat Commun. 2016;7(1):1–10.
Lefrancois S, Zeng J, Hassan AJ, Canuel M, Morales CR. The lysosomal trafficking of sphingolipid activator proteins (SAPs) is mediated by sortilin. EMBO J. 2003;22(24):6430–7.
Wähe A, Kasmapour B, Schmaderer C, Liebl D, Sandhoff K, Nykjaer A, Griffiths G, Gutierrez MG. Golgi-to-phagosome transport of acid sphingomyelinase and prosaposin is mediated by sortilin. J Cell Sci. 2010;123(14):2502–11.
Ni X, Morales CR. The lysosomal trafficking of acid sphingomyelinase is mediated by sortilin and mannose 6-phosphate receptor. Traffic. 2006;7(7):889–902.
Gault CR, Obeid LM, Hannun YA: An overview of sphingolipid metabolism: from synthesis to breakdown. Sphingolipids as signaling and regulatory molecules 2010:1–23.
Alkafaas SS, Elsalahaty MI, Ismail DF, Radwan MA, Elkafas SS, Loutfy SA, Elshazli RM, Baazaoui N, Ahmed AE, Hafez W, Diab M, Sakran M, El-Saadony MT, El-Tarabily KA, Kamal HK, HEssien M. The emerging roles of sphingosine 1-phosphate and SphK1 in cancer resistance: a promising therapeutic target. Cancer cell Interna. 2024. https://doi.org/10.1186/s12935-024-03221-8.
Pralhada Rao R, Vaidyanathan N, Rengasamy M, Mammen Oommen A, Somaiya N, Jagannath MR: Sphingolipid Metabolic Pathway: An Overview of Major Roles Played in Human Diseases. J Lipids. 2013;2013:1–12.
Xia P, Gamble JR, Rye K-A, Wang L, Hii CS, Cockerill P, Khew-Goodall Y, Bert AG, Barter PJ, Vadas MA. Tumor necrosis factor-α induces adhesion molecule expression through the sphingosine kinase pathway. Proc Natl Acad Sci. 1998;95(24):14196–201.
Mitra P, Oskeritzian CA, Payne SG, Beaven MA, Milstien S, Spiegel S. Role of ABCC1 in export of sphingosine-1-phosphate from mast cells. Proc Natl Acad Sci. 2006;103(44):16394–9.
Fugmann T, Hausser A, Schöffler P, Schmid S, Pfizenmaier K, Olayioye MA. Regulation of secretory transport by protein kinase D–mediated phosphorylation of the ceramide transfer protein. J Cell Biol. 2007;178(1):15–22.
Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008;9(2):139–50.
Sorice M, Misasi R, Riitano G, Manganelli V, Martellucci S, Longo A, Garofalo T, Mattei V. Targeting lipid rafts as a strategy against coronavirus. Front Cell Dev Biol. 2021;8: 618296.
Viard M, Parolini I, Sargiacomo M, Fecchi K, Ramoni C, Ablan S, Ruscetti FW, Wang JM, Blumenthal R. Role of cholesterol in human immunodeficiency virus type 1 envelope protein-mediated fusion with host cells. J Virol. 2002;76(22):11584–95.
Danthi P, Chow M. Cholesterol removal by methyl-β-cyclodextrin inhibits poliovirus entry. J Virol. 2004;78(1):33–41.
Aizaki H, Lee K-J. Sung VM-H, Ishiko H, Lai MM: Characterization of the hepatitis C virus RNA replication complex associated with lipid rafts. Virology. 2004;324(2):450–61.
Hu W, Zhu L, Yang X, Lin J, Yang Q. The epidermal growth factor receptor regulates cofilin activity and promotes transmissible gastroenteritis virus entry into intestinal epithelial cells. Oncotarget. 2016;7(11):12206.
Thorp EB, Gallagher TM. Requirements for CEACAMs and cholesterol during murine coronavirus cell entry. J Virol. 2004;78(6):2682–92.
Yang J, Lv J, Wang Y, Gao S, Yao Q, Qu D, Ye R. Replication of murine coronavirus requires multiple cysteines in the endodomain of spike protein. Virology. 2012;427(2):98–106.
Tay MZ, Poh CM, Rénia L, MacAry PA, Ng LFP. The trinity of COVID-19: immunity, inflammation and intervention. Nat Rev Immunol. 2020;20(6):363–74.
Coutard B, Valle C, De Lamballerie X, Canard B, Seidah NG, Decroly E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral Res. 2020;176: 104742.
Tay MZ, Poh CM, Rénia L, MacAry PA, Ng LFP: The trinity of COVID-19: immunity, inflammation and intervention. Nature Reviews Immunology 2020, 20(6):363–374.
Fantini J, Di Scala C, Chahinian H, Yahi N. Structural and molecular modelling studies reveal a new mechanism of action of chloroquine and hydroxychloroquine against SARS-CoV-2 infection. Int J Antimicrob Agents. 2020;55(5): 105960.
Mañes S, del Real G, Martínez-a C. Pathogens: raft hijackers. Nat Rev Immunol. 2003;3(7):557–68.
Wang H, Yuan X, Sun Y, Mao X, Meng C, Tan L, Song C, Qiu X, Ding C, Liao Y. Infectious bronchitis virus entry mainly depends on clathrin mediated endocytosis and requires classical endosomal/lysosomal system. Virology. 2019;528:118–36.
Grim K, Abcejo A, Barnes A, Sathish V, Smelter D, Ford G, Thompson M, Prakash Y, Pabelick C. Caveolae and propofol effects on airway smooth muscle. Br J Anaesth. 2012;109(3):444–53.
Patel J, Chowdhury EA, Noorani B, Bickel U, Huang J. Isoflurane increases cell membrane fluidity significantly at clinical concentrations. Biochimica et Biophysica Acta (BBA)-Biomembranes. 2020;1862(2):183140.
Turkyilmaz S, Almeida PF, Regen SL. Effects of isoflurane, halothane, and chloroform on the interactions and lateral organization of lipids in the liquid-ordered phase. Langmuir. 2011;27(23):14380–5.
Sierra-Valdez FJ, Ruiz-Suárez J, Delint-Ramirez I. Pentobarbital modifies the lipid raft-protein interaction: A first clue about the anesthesia mechanism on NMDA and GABAA receptors. Biochimica et Biophysica Acta (BBA)-Biomembranes. 2016;1858(11):2603–10.
Kamata K, Manno S, Ozaki M, Takakuwa Y. Functional evidence for presence of lipid rafts in erythrocyte membranes: Gsα in rafts is essential for signal transduction. Am J Hematol. 2008;83(5):371–5.
Tsuchiya H, Mizogami M. Interaction of drugs with lipid raft membrane domains as a possible target. Drug Target Insights. 2020;14:34.
Hu S, Zhao T, Li H, Cheng D, Sun Z. Effect of tetracaine on dynamic reorganization of lipid membranes. Biochimica et Biophysica Acta (BBA)-Biomembranes. 2020;1862(9):183351.
Yoshida K, Takashima A, Nishio I. Effect of dibucaine hydrochloride on raft-like lipid domains in model membrane systems. MedChemComm. 2015;6(8):1444–51.
Tsuchiya H, Mizogami M, Takakura K. Reversed-phase liquid chromatographic retention and membrane activity relationships of local anesthetics. J Chromatogr A. 2005;1073(1–2):303–8.
Mizogami M, Tsuchiya H. Membrane interactivity of anesthetic adjuvant dexmedetomidine discriminable from clonidine and enantiomeric levomedetomidine. Journal of Advances in Medicine and Medical Research. 2019;29(11):1–15.
Heron DS, Shinitzky M, Zamir N, Samuel D. Adaptive modulations of brain membrane lipid fluidity in drug addiction and denervation supersensitivity. Biochem Pharmacol. 1982;31(14):2435–8.
Zhou Y, Cho K-J, Plowman SJ, Hancock JF. Nonsteroidal anti-inflammatory drugs alter the spatiotemporal organization of Ras proteins on the plasma membrane. J Biol Chem. 2012;287(20):16586–95.
Ausili A, Torrecillas A, Aranda FJ, Mollinedo F, Gajate C, Corbalan-Garcia S, de Godos A, Gomez-Fernandez JC. Edelfosine is incorporated into rafts and alters their organization. J Phys Chem B. 2008;112(37):11643–54.
Gomide AB, Thomé CH, Dos Santos G, Ferreira G, Faça VM, Rego EM, Greene LJ, Stábeli RG, Ciancaglini P, Itri R. Disrupting membrane raft domains by alkylphospholipids. Biochimica et Biophysica Acta (BBA)-Biomembranes. 2013;1828(5):1384–9.
Castro BM, Fedorov A, Hornillos V, Delgado J, Acuña AU, Mollinedo F, Prieto M. Edelfosine and miltefosine effects on lipid raft properties: membrane biophysics in cell death by antitumor lipids. J Phys Chem B. 2013;117(26):7929–40.
Wnętrzak A, Łątka K, Makyła-Juzak K, Zemla J, Dynarowicz-Łątka P. The influence of an antitumor lipid–erucylphosphocholine–on artificial lipid raft system modeled as Langmuir monolayer. Mol Membr Biol. 2015;32(5–8):189–97.
Węder K, Mach M, Hąc-Wydro K, Wydro P. Studies on the interactions of anticancer drug-Minerval-with membrane lipids in binary and ternary Langmuir monolayers. Biochimica et Biophysica Acta (BBA)-Biomembranes. 2018;1860(11):2329–36.
Lacour S, Hammann A, Grazide S, Lagadic-Gossmann D, Athias A, Sergent O, Laurent G, Gambert P, Solary E, Dimanche-Boitrel M-T. Cisplatin-induced CD95 redistribution into membrane lipid rafts of HT29 human colon cancer cells. Can Res. 2004;64(10):3593–8.
Rebillard A, Tekpli X, Meurette O, Sergent O, LeMoigne-Muller G, Vernhet L, Gorria M, Chevanne M, Christmann M, Kaina B. Cisplatin-induced apoptosis involves membrane fluidification via inhibition of NHE1 in human colon cancer cells. Can Res. 2007;67(16):7865–74.
Berquand A, Fa N, Dufrene Y, Mingeot-Leclercq M-P. Interaction of the macrolide antibiotic azithromycin with lipid bilayers: effect on membrane organization, fluidity, and permeability. Pharm Res. 2005;22:465–75.
Alves AC, Ribeiro D, Horta M, Lima JL, Nunes C, Reis S. A biophysical approach to daunorubicin interaction with model membranes: relevance for the drug’s biological activity. J R Soc Interface. 2017;14(133):20170408.
Alves AC, Magarkar A, Horta M, Lima JL, Bunker A, Nunes C, Reis S. Influence of doxorubicin on model cell membrane properties: insights from in vitro and in silico studies. Sci Rep. 2017;7(1):6343.
溝上真樹: Plant components exhibit pharmacological activities and drug interactions by acting on lipid membranes. Pharmacognosy Communications 2012;2:58–71.
Kaneko M, Takimoto H, Sugiyama T, Seki Y, Kawaguchi K, Kumazawa Y. Suppressive effects of the flavonoids quercetin and luteolin on the accumulation of lipid rafts after signal transduction via receptors. Immunopharmacol Immunotoxicol. 2008;30(4):867–82.
Tsukamoto S, Hirotsu K, Kumazoe M, Goto Y, Sugihara K, Suda T, Tsurudome Y, Suzuki T, Yamashita S, Kim Y. Green tea polyphenol EGCG induces lipid-raft clustering and apoptotic cell death by activating protein kinase Cδ and acid sphingomyelinase through a 67 kDa laminin receptor in multiple myeloma cells. Biochemical Journal. 2012;443(2):525–34.
Verstraeten SV, Oteiza PI, Fraga CG. Membrane effects of cocoa procyanidins in liposomes and Jurkat T cells. Biol Res. 2004;37(2):293–300.
Verstraeten SV, Jaggers GK, Fraga CG, Oteiza PI. Procyanidins can interact with Caco-2 cell membrane lipid rafts: involvement of cholesterol. Biochimica et Biophysica Acta (BBA)-Biomembranes. 2013;1828(11):2646–53.
Meng G, Liu Y, Lou C, Yang H. Emodin suppresses lipopolysaccharide-induced pro-inflammatory responses and NF-κB activation by disrupting lipid rafts in CD14-negative endothelial cells. Br J Pharmacol. 2010;161(7):1628–44.
Yi J-S, Choo H-J, Cho B-R, Kim H-M, Kim Y-N, Ham Y-M, Ko Y-G. Ginsenoside Rh2 induces ligand-independent Fas activation via lipid raft disruption. Biochem Biophys Res Commun. 2009;385(2):154–9.
Wei Z, Wang J, Shi M, Liu W, Yang Z, Fu Y. Saikosaponin a inhibits LPS-induced inflammatory response by inducing liver X receptor alpha activation in primary mouse macrophages. Oncotarget. 2016;7(31):48995.
Bakillah A, Hejji FA, Almasaud A, Jami HA, Hawwari A, Qarni AA, Iqbal J, Alharbi NK. Lipid raft integrity and cellular cholesterol homeostasis are critical for SARS-CoV-2 entry into cells. Nutrients. 2022;14(16):3417.
Rodrigues-Diez RR, Tejera-Muñoz A, Marquez-Exposito L, Rayego-Mateos S, Santos Sanchez L, Marchant V, Tejedor Santamaria L, Ramos AM, Ortiz A, Egido J. Statins: could an old friend help in the fight against COVID-19? Br J Pharmacol. 2020;177(21):4873–86.
Törnquist K, Asghar MY, Srinivasan V, Korhonen L, Lindholm D. Sphingolipids as modulators of SARS-CoV-2 infection. Front Cell Dev Biol. 2021;9:1574.
Schneider-Schaulies J, Schneider-Schaulies S. Sphingolipids in viral infection. Biol Chem. 2015;396(6–7):585–95.
Alkafaas SS, Loutfy SA, Diab T, Hessien M. Vasopressin induces apoptosis but does not enhance the antiproliferative effect of dynamin 2 or PI3K/Akt inhibition in luminal A breast cancer cells. Med Oncol. 2022;40(1):35.
Moya M, Dautry-Varsat A, Goud B, Louvard D, Boquet P. Inhibition of coated pit formation in Hep2 cells blocks the cytotoxicity of diphtheria toxin but not that of ricin toxin. J Cell Biol. 1985;101(2):548–59.
Hansen SH, Sandvig K, van Deurs B. The preendosomal compartment comprises distinct coated and noncoated endocytic vesicle populations. J Cell Biol. 1991;113(4):731–41.
Alkafaas SS, Abdallah AM, Ghosh S, Loutfy SA, ElKaffas SS, Abdel Fattah NF, Hessien M: Insight into the role of clathrin‐mediated endocytosis inhibitors in SARS‐CoV‐2 infection. Reviews in Medical Virology 2022:e2403.
Scott CC, Vacca F, Gruenberg J. Endosome maturation, transport and functions. Semin Cell Dev Biol. 2014;31:2–10.
Preston JE, Abbott NJ, Begley DJ. Transcytosis of macromolecules at the blood–brain barrier. Adv Pharmacol. 2014;71:147–63.
Antonescu CN, McGraw TE, Klip A. Reciprocal regulation of endocytosis and metabolism. Cold Spring Harb Perspect Biol. 2014;6(7): a016964.
Irannejad R, von Zastrow M. GPCR signaling along the endocytic pathway. Curr Opin Cell Biol. 2014;27:109–16.
Goh LK, Sorkin A. Endocytosis of receptor tyrosine kinases. Cold Spring Harb Perspect Biol. 2013;5(5): a017459.
Sandvig K, Van Deurs B. Selective modulation of the endocytic uptake of ricin and fluid phase markers without alteration in transferrin endocytosis. J Biol Chem. 1990;265(11):6382–8.
Huotari J, Helenius A. Endosome maturation. EMBO J. 2011;30(17):3481–500.
Mayor S, Parton RG, Donaldson JG. Clathrin-independent pathways of endocytosis. Cold Spring Harb Perspect Biol. 2014;6(6): a016758.
Kirkham M, Fujita A, Chadda R, Nixon SJ, Kurzchalia TV, Sharma DK, Pagano RE, Hancock JF, Mayor S, Parton RG. Ultrastructural identification of uncoated caveolin-independent early endocytic vehicles. J Cell Biol. 2005;168(3):465–76.
Donaldson JG, Jackson CL. ARF family G proteins and their regulators: roles in membrane transport, development and disease. Nat Rev Mol Cell Biol. 2011;12(6):362–75.
Radhakrishna H, Donaldson JG. ADP-ribosylation factor 6 regulates a novel plasma membrane recycling pathway. J Cell Biol. 1997;139(1):49–61.
Glebov OO, Bright NA, Nichols BJ. Flotillin-1 defines a clathrin-independent endocytic pathway in mammalian cells. Nat Cell Biol. 2006;8(1):46–54.
Frick M, Bright NA, Riento K, Bray A, Merrified C, Nichols BJ. Coassembly of flotillins induces formation of membrane microdomains, membrane curvature, and vesicle budding. Curr Biol. 2007;17(13):1151–6.
Maldonado-Báez L, Williamson C, Donaldson JG. Clathrin-independent endocytosis: a cargo-centric view. Exp Cell Res. 2013;319(18):2759–69.
Kumari S, Mg S, Mayor S. Endocytosis unplugged: multiple ways to enter the cell. Cell Res. 2010;20(3):256–75.
Huang H, Li Y, Sadaoka T, Tang H, Yamamoto T, Yamanishi K, Mori Y. Human herpesvirus 6 envelope cholesterol is required for virus entry. J Gen Virol. 2006;87(2):277–85.
Nomura R, Kiyota A, Suzaki E, Kataoka K, Ohe Y, Miyamoto K, Senda T, Fujimoto T. Human coronavirus 229E binds to CD13 in rafts and enters the cell through caveolae. J Virol. 2004;78(16):8701–8.
Choi KS, Aizaki H, Lai MM. Murine coronavirus requires lipid rafts for virus entry and cell-cell fusion but not for virus release. J Virol. 2005;79(15):9862–71.
Mathewson AC, Bishop A, Yao Y, Kemp F, Ren J, Chen H, Xu X, Berkhout B, van der Hoek L, Jones IM. Interaction of severe acute respiratory syndrome-coronavirus and NL63 coronavirus spike proteins with angiotensin converting enzyme-2. J Gen Virol. 2008;89(Pt 11):2741.
Gopal P, Rehman RU, Chadha KS, Qiu M, Colella R. Matrigel influences morphology and cathepsin B distribution of prostate cancer PC3 cells. Oncol Rep. 2006;16(2):313–20.
Liu C, Zhou Q, Li Y, Garner LV, Watkins SP, Carter LJ, Smoot J, Gregg AC, Daniels AD, Jervey S. Research and development on therapeutic agents and vaccines for COVID-19 and related human coronavirus diseases. In.: ACS Publications; 2020.
Nicolau DV Jr, Burrage K, Parton RG, Hancock JF. Identifying optimal lipid raft characteristics required to promote nanoscale protein-protein interactions on the plasma membrane. Mol Cell Biol. 2006;26(1):313–23.
Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Base estructural para el reconocimiento de SARS-CoV-2 por la enzima convertidora de angiotensina 2 (ACE2) humana completa. Science. 2020;367:1444–8.
Guo H, Huang M, Yuan Q, Wei Y, Gao Y, Mao L, Gu L, Tan YW, Zhong Y, Liu D. The important role of lipid raft-mediated attachment in the infection of cultured cells by coronavirus infectious bronchitis virus beaudette strain. PLoS ONE. 2017;12(1): e0170123.
Barman S, Nayak DP. Lipid raft disruption by cholesterol depletion enhances influenza A virus budding from MDCK cells. J Virol. 2007;81(22):12169–78.
Bailly C, Vergoten G. Glycyrrhizin: An alternative drug for the treatment of COVID-19 infection and the associated respiratory syndrome? Pharmacol Ther. 2020;214: 107618.
Fecchi K, Anticoli S, Peruzzu D, Iessi E, Gagliardi MC, Matarrese P, Ruggieri A. Coronavirus interplay with lipid rafts and autophagy unveils promising therapeutic targets. Front Microbiol. 1821;2020:11.
Radenkovic D, Chawla S, Pirro M, Sahebkar A, Banach M. Cholesterol in relation to COVID-19: should we care about it? J Clin Med. 2020;9(6):1909.
Kornhuber J, Muehlbacher M, Trapp S, Pechmann S, Friedl A, Reichel M, Mühle C, Terfloth L, Groemer TW, Spitzer GM, et al. Identification of Novel Functional Inhibitors of Acid Sphingomyelinase. PLOS ONE. 2011;6(8):1–13.
Heinrich M, Wickel M, Schneider-Brachert W, Sandberg C, Gahr J, Schwandner R, Weber T, Brunner J, Krönke M, Schütze S. Cathepsin D targeted by acid sphingomyelinase-derived ceramide. EMBO J. 1999;18(19):5252–63.
Carpinteiro A, Gripp B, Hoffmann M, Pöhlmann S, Hoertel N, Edwards MJ, Kamler M, Kornhuber J, Becker KA, Gulbins E. Inhibition of acid sphingomyelinase by ambroxol prevents SARS-CoV-2 entry into epithelial cells. J Biol Chem. 2021;296:1–12.
Pitson SM. Regulation of sphingosine kinase and sphingolipid signaling. Trends Biochem Sci. 2011;36(2):97–107.
Kornhuber J, Tripal P, Reichel M, Terfloth L, Bleich S, Wiltfang J, Gulbins E. Identification of new functional inhibitors of acid sphingomyelinase using a structure− property− activity relation model. J Med Chem. 2008;51(2):219–37.
Villoutreix BO, Krishnamoorthy R, Tamouza R, Leboyer M, Beaune P. Chemoinformatic analysis of psychotropic and antihistaminic drugs in the light of experimental anti-SARS-CoV-2 activities. Advances and applications in bioinformatics and chemistry: AABC. 2021;14:71.
Hoertel N, Sánchez-Rico M, Gulbins E, Kornhuber J, Carpinteiro A, Abellán M, de la Muela P, Vernet R, Beeker N, Neuraz A. Association between FIASMA psychotropic medications and reduced risk of intubation or death in individuals with psychiatric disorders hospitalized for severe COVID-19: an observational multicenter study. Transl Psychiatry. 2022;12(1):1–11.
Zhou Y, Hou Y, Shen J, Mehra R, Kallianpur A, Culver DA, Gack MU, Farha S, Zein J, Comhair S, et al. A network medicine approach to investigation and population-based validation of disease manifestations and drug repurposing for COVID-19. PLoS Biol. 2020;18(11): e3000970.
Weston S, Coleman CM, Haupt R, Logue J, Matthews K, Li Y, Reyes HM, Weiss SR, Frieman MB. Broad anti-coronavirus activity of food and drug administration-approved drugs against SARS-CoV-2 in vitro and SARS-CoV in vivo. J Virol. 2020;94(21):e01218–01220.
Loas G, Le Corre P. Update on functional inhibitors of acid sphingomyelinase (FIASMAs) in SARS-CoV-2 infection. Pharmaceuticals. 2021;14(7):691.
Ruan Z, Liu C, Guo Y, He Z, Huang X, Jia X, Yang T. SARS-CoV-2 and SARS-CoV: Virtual screening of potential inhibitors targeting RNA-dependent RNA polymerase activity (NSP12). J Med Virol. 2021;93(1):389–400.
Sauvat A, Ciccosanti F, Colavita F, Di Rienzo M, Castilletti C, Capobianchi MR, Kepp O, Zitvogel L, Fimia GM, Piacentini M. On-target versus off-target effects of drugs inhibiting the replication of SARS-CoV-2. Cell Death Dis. 2020;11(8):1–11.
Chen CZ, Shinn P, Itkin Z, Eastman RT, Bostwick R, Rasmussen L, Huang R, Shen M, Hu X, Wilson KM. Drug repurposing screen for compounds inhibiting the cytopathic effect of SARS-CoV-2. Front Pharmacol. 2021;11:592737.
Yang L. Pei R-j, Li H, Ma X-n, Zhou Y, Zhu F-h, He P-l, Tang W, Zhang Y-c, Xiong J: Identification of SARS-CoV-2 entry inhibitors among already approved drugs. Acta Pharmacol Sin. 2021;42(8):1347–53.
Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, O’Meara MJ, Rezelj VV, Guo JZ, Swaney DL. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583(7816):459–68.
Le Corre P, Loas G. Repurposing functional inhibitors of acid sphingomyelinase (fiasmas): an opportunity against SARS-CoV-2 infection? J Clin Pharm Ther. 2021;46(5):1213–9.
Jade D, Ayyamperumal S, Tallapaneni V, Nanjan CMJ, Barge S, Mohan S, Nanjan MJ. Virtual high throughput screening: Potential inhibitors for SARS-CoV-2 PLPRO and 3CLPRO proteases. Eur J Pharmacol. 2021;901:174082.
Le BL, Andreoletti G, Oskotsky T, Vallejo-Gracia A, Rosales R, Yu K, Kosti I, Leon KE, Bunis DG, Li C. Transcriptomics-based drug repositioning pipeline identifies therapeutic candidates for COVID-19. Sci Rep. 2021;11(1):1–14.
Imamura K, Sakurai Y, Enami T, Shibukawa R, Nishi Y, Ohta A, Shu T, Kawaguchi J, Okada S, Hoenen T. iPSC screening for drug repurposing identifies anti-RNA virus agents modulating host cell susceptibility. FEBS Open Bio. 2021;11(5):1452–64.
Xiong H-L, Cao J-L, Shen C-G, Ma J, Qiao X-Y, Shi T-S, Ge S-X, Ye H-M, Zhang J, Yuan Q. Several FDA-approved drugs effectively inhibit SARS-CoV-2 infection in vitro. Front Pharmacol. 2021;11:609592.
Fred SM, Kuivanen S, Ugurlu H, Casarotto PC, Levanov L, Saksela K, Vapalahti O, Castrén E. Antidepressant and Antipsychotic Drugs Reduce Viral Infection by SARS-CoV-2 and Fluoxetine Shows Antiviral Activity Against the Novel Variants in vitro. Front Pharmacol. 2021;12: 755600.
Hoertel N, Sánchez-Rico M, Vernet R, Beeker N, Jannot A-S, Neuraz A, Salamanca E, Paris N, Daniel C, Gramfort A. Association between antidepressant use and reduced risk of intubation or death in hospitalized patients with COVID-19: results from an observational study. Mol Psychiatry. 2021;26(9):5199–212.
Carpinteiro A, Edwards MJ, Hoffmann M, Kochs G, Gripp B, Weigang S, Adams C, Carpinteiro E, Gulbins A, Keitsch S. Pharmacological inhibition of acid sphingomyelinase prevents uptake of SARS-CoV-2 by epithelial cells. Cell Reports Medicine. 2020;1(8):100142.
Gelemanović A, Vidović T, Stepanić V, Trajković K. Identification of 37 heterogeneous drug candidates for treatment of COVID-19 via a rational transcriptomics-based drug repurposing approach. Pharmaceuticals. 2021;14(2):87.
Ge S, Lu J, Hou Y, Lv Y, Wang C, He H. Azelastine inhibits viropexis of SARS-CoV-2 spike pseudovirus by binding to SARS-CoV-2 entry receptor ACE2. Virology. 2021;560:110–5.
Gurung AB, Ali MA, Lee J, Farah MA, Al-Anazi KM. The potential of Paritaprevir and Emetine as inhibitors of SARS-CoV-2 RdRp. Saudi J Biol Sci. 2021;28(2):1426–32.
Das S, Sarmah S, Lyndem S, Singha Roy A. An investigation into the identification of potential inhibitors of SARS-CoV-2 main protease using molecular docking study. J Biomol Struct Dyn. 2021;39(9):3347–57.
Hajjo R, Tropsha A. A systems biology workflow for drug and vaccine repurposing: identifying small-molecule BCG mimics to reduce or prevent COVID-19 mortality. Pharm Res. 2020;37(11):1–15.
Klutzny S, Lesche R, Keck M, Kaulfuss S, Schlicker A, Christian S, Sperl C, Neuhaus R, Mowat J, Steckel M, et al. Functional inhibition of acid sphingomyelinase by Fluphenazine triggers hypoxia-specific tumor cell death. Cell Death Dis. 2017;8(3):e2709.
Lenze EJ, Mattar C, Zorumski CF, Stevens A, Schweiger J, Nicol GE, Miller JP, Yang L, Yingling M, Avidan MS. Fluvoxamine vs placebo and clinical deterioration in outpatients with symptomatic COVID-19: a randomized clinical trial. JAMA. 2020;324(22):2292–300.
Nazeam J, Mohammed EZ, Raafat M, Houssein M, Elkafoury A, Hamdy D, Jamil L. Based on principles and insights of covid-19 epidemiology, genome sequencing, and pathogenesis: Retrospective analysis of sinigrin and prolixinrx (fluphenazine) provides off-label drug candidates. Slas Discovery. 2020;25(10):1123–40.
Reznikov LR, Norris MH, Vashisht R, Bluhm AP, Li D, Liao YJ, Brown A, Butte AJ, Ostrov DA. Identification of antiviral antihistamines for COVID-19 repurposing. Biochem Biophys Res Commun. 2021;538:173–9.
Norinder U, Tuck A, Norgren K, Kos VM. Existing highly accumulating lysosomotropic drugs with potential for repurposing to target COVID-19. Biomed Pharmacother. 2020;130: 110582.
Schloer S, Brunotte L, Goretzko J, Mecate-Zambrano A, Korthals N, Gerke V, Ludwig S, Rescher U. Targeting the endolysosomal host-SARS-CoV-2 interface by clinically licensed functional inhibitors of acid sphingomyelinase (FIASMA) including the antidepressant fluoxetine. Emerging microbes & infections. 2020;9(1):2245–55.
Jeon S, Ko M, Lee J, Choi I, Byun SY, Park S, Shum D, Kim S. Identification of antiviral drug candidates against SARS-CoV-2 from FDA-approved drugs. Antimicrob Agents Chemother. 2020;64(7):e00819–00820.
Hou Y, Ge S, Li X, Wang C, He H, He L. Testing of the inhibitory effects of loratadine and desloratadine on SARS-CoV-2 spike pseudotyped virus viropexis. Chem Biol Interact. 2021;338: 109420.
Chavarría AP, Vázquez RRV, Cherit JGD, Bello HH, Suastegui HC, Moreno-Castañeda L, Estrada GA, Hernández F, González-Marcos O, Saucedo-Orozco H. Antioxidants and pentoxifylline as coadjuvant measures to standard therapy to improve prognosis of patients with pneumonia by COVID-19. Comput Struct Biotechnol J. 2021;19:1379–90.
Jehi L, Ji X, Milinovich A, Erzurum S, Rubin BP, Gordon S, Young JB, Kattan MW. Individualizing Risk Prediction for Positive Coronavirus Disease 2019 Testing: Results From 11,672 Patients. Chest. 2020;158(4):1364–75.
Plaze M, Attali D, Prot M, Petit A-C, Blatzer M, Vinckier F, Levillayer L, Chiaravalli J, Perin-Dureau F, Cachia A. Inhibition of the replication of SARS-CoV-2 in human cells by the FDA-approved drug chlorpromazine. Int J Antimicrob Agents. 2021;57(3): 106274.
Vatansever EC, Yang KS, Drelich AK, Kratch KC, Cho C-C, Kempaiah KR, Hsu JC, Mellott DM, Xu S. Tseng C-TK: Bepridil is potent against SARS-CoV-2 in vitro. Proc Natl Acad Sci. 2021;118(10): e2012201118.
O’Donovan SM, Imami A, Eby H, Henkel ND, Creeden JF, Asah S, Zhang X, Wu X, Alnafisah R, Taylor RT. Identification of candidate repurposable drugs to combat COVID-19 using a signature-based approach. Sci Rep. 2021;11(1):1–12.
Xiao X, Wang C, Chang D, Wang Y, Dong X, Jiao T, Zhao Z, Ren L, Dela Cruz CS, Sharma L. Identification of potent and safe antiviral therapeutic candidates against SARS-CoV-2. Front Immunol. 2020;11:586572.
Udrea A-M, Avram S, Nistorescu S, Pascu M-L, Romanitan MO. Laser irradiated phenothiazines: New potential treatment for COVID-19 explored by molecular docking. J Photochem Photobiol, B. 2020;211:111997.
Chen CZ, Xu M, Pradhan M, Gorshkov K, Petersen JD, Straus MR, Zhu W, Shinn P, Guo H, Shen M. Identifying SARS-CoV-2 entry inhibitors through drug repurposing screens of SARS-S and MERS-S pseudotyped particles. ACS Pharmacol Transl Sci. 2020;3(6):1165–75.
Hurwitz R, Ferlinz K, Sandhoff K. The tricyclic antidepressant desipramine causes proteolytic degradation of lysosomal sphingomyelinase in human fibroblasts. Bio Chem Hoppe-Seyler. 1994;375(7):447–50.
MOE V. Chemical Computing Group Inc., Montreal, Canada. In.; 2010.
Al-Qaisi ZH, Al-Garawi ZS, Al-Karawi AJM, Hammood AJ, Abdallah AM, Clegg W, Mohamed GG. Antiureolytic activity of new water-soluble thiadiazole derivatives: Spectroscopic, DFT, and molecular docking studies. Spectrochim Acta Part A Mol Biomol Spectrosc. 2022;272: 120971.
Wang X, Lu J, Ge S, Hou Y, Hu T, Lv Y, Wang C, He H. Astemizole as a drug to inhibit the effect of SARS-COV-2 in vitro. Microb Pathog. 2021;156: 104929.
Janabi AHD. Molecular Docking Analysis of Anti-Severe Acute Respiratory Syndrome-Coronavirus 2 Ligands against Spike Glycoprotein and the 3-Chymotrypsin-Like Protease. J Med Signals Sens. 2021;11(1):31–6.
Darquennes G, Le Corre P, Le Moine O, Loas G. Association between Functional Inhibitors of Acid Sphingomyelinase (FIASMAs) and Reduced Risk of Death in COVID-19 Patients: A Retrospective Cohort Study. Pharmaceuticals (Basel). 2021;14(3):226.
Castaldo N, Aimo A, Castiglione V, Padalino C, Emdin M, Tascini C. Safety and Efficacy of Amiodarone in a Patient With COVID-19. JACC Case Rep. 2020;2(9):1307–10.
Hoertel N, Sánchez-Rico M, Gulbins E, Kornhuber J, Carpinteiro A, Lenze EJ, Reiersen AM, Abellán M, de la Muela P, Vernet R, et al. Association Between FIASMAs and Reduced Risk of Intubation or Death in Individuals Hospitalized for Severe COVID-19: An Observational Multicenter Study. Clin Pharmacol Ther. 2021;110(6):1498–511.
i Y, Chen J, Pang L, Chen C, Ye J, Liu H, Chen H, Zhang S, Liu S, Liu B, et al. The Acid Sphingomyelinase Inhibitor Amitriptyline Ameliorates TNF-α-Induced Endothelial Dysfunction. Cardiovasc Drugs Ther. 2022:1–14.
Sencanski M, Perovic V, Pajovic SB, Adzic M, Paessler S, Glisic S. Drug Repurposing for Candidate SARS-CoV-2 Main Protease Inhibitors by a Novel In Silico Method. Molecules. 2020;25(17):3830.
Loas G, Le Corre P. Update on Functional Inhibitors of Acid Sphingomyelinase (FIASMAs) in SARS-CoV-2 Infection. Pharmaceuticals (Basel). 2021;14(7):691.
Vatansever EC, Yang KS, Drelich AK, Kratch KC, Cho CC, Kempaiah KR, Hsu JC, Mellott DM, Xu S, Tseng CK, et al. Bepridil is potent against SARS-CoV-2 in vitro. Proc Natl Acad Sci U S A. 2021;118(10):1–8.
Jeon S, Ko M, Lee J, Choi I, Byun SY, Park S, Shum D, Kim S. Identification of Antiviral Drug Candidates against SARS-CoV-2 from FDA-Approved Drugs. Antimicrob Agents Chemother. 2020;64(7):1–9.
Jade D, Ayyamperumal S, Tallapaneni V, Joghee Nanjan CM, Barge S, Mohan S, Nanjan MJ. Virtual high throughput screening: Potential inhibitors for SARS-CoV-2 PL(PRO) and 3CL(PRO) proteases. Eur J Pharmacol. 2021;901: 174082.
Yuan S, Yin X, Meng X, Chan JF, Ye ZW, Riva L, Pache L, Chan CC, Lai PM, Chan CC, et al. Clofazimine broadly inhibits coronaviruses including SARS-CoV-2. Nature. 2021;593(7859):418–23.
Sauvat A, Ciccosanti F, Colavita F, Di Rienzo M, Castilletti C, Capobianchi MR, Kepp O, Zitvogel L, Fimia GM, Piacentini M, et al. On-target versus off-target effects of drugs inhibiting the replication of SARS-CoV-2. Cell Death Dis. 2020;11(8):656.
Barnard DL, Day CW, Bailey K, Heiner M, Montgomery R, Lauridsen L, Jung KH, Li JK, Chan PK, Sidwell RW. Is the anti-psychotic, 10-(3-(dimethylamino)propyl)phenothiazine (promazine), a potential drug with which to treat SARS infections? Lack of efficacy of promazine on SARS-CoV replication in a mouse model. Antiviral Res. 2008;79(2):105–13.
Weston S, Coleman CM, Haupt R, Logue J, Matthews K, Li Y, Reyes HM, Weiss SR, Frieman MB. Broad Anti-coronavirus Activity of Food and Drug Administration-Approved Drugs against SARS-CoV-2 In Vitro and SARS-CoV In Vivo. J Virol. 2020;94(21):e01218–20.
Chen CZ, Shinn P, Itkin Z, Eastman RT, Bostwick R, Rasmussen L, Huang R, Shen M, Hu X, Wilson KM, et al. Drug Repurposing Screen for Compounds Inhibiting the Cytopathic Effect of SARS-CoV-2. Front Pharmacol. 2020;11: 592737.
Carpinteiro A, Edwards MJ, Hoffmann M, Kochs G, Gripp B, Weigang S, Adams C, Carpinteiro E, Gulbins A, Keitsch S, et al. Pharmacological Inhibition of Acid Sphingomyelinase Prevents Uptake of SARS-CoV-2 by Epithelial Cells. Cell Rep Med. 2020;1(8): 100142.
Morin-Dewaele M, Bartier S, Berry F, Brillet R, López-Molina DS, Nguyễn CT, Maille P, Sereno K, Nevers Q, Softic L, et al. Desloratadine, an FDA-approved cationic amphiphilic drug, inhibits SARS-CoV-2 infection in cell culture and primary human nasal epithelial cells by blocking viral entry. Sci Rep. 2022;12(1):21053.
Le BL, Andreoletti G, Oskotsky T, Vallejo-Gracia A, Rosales R, Yu K, Kosti I, Leon KE, Bunis DG, Li C, et al. Transcriptomics-based drug repositioning pipeline identifies therapeutic candidates for COVID-19. Sci Rep. 2021;11(1):12310.
Ge S, Wang X, Hou Y, Lv Y, Wang C, He H. Repositioning of histamine H(1) receptor antagonist: Doxepin inhibits viropexis of SARS-CoV-2 Spike pseudovirus by blocking ACE2. Eur J Pharmacol. 2021;896:173897.
Choy KT, Wong AY, Kaewpreedee P, Sia SF, Chen D, Hui KPY, Chu DKW, Chan MCW, Cheung PP, Huang X, et al. Remdesivir, lopinavir, emetine, and homoharringtonine inhibit SARS-CoV-2 replication in vitro. Antiviral Res. 2020;178:104786.
Duarte RRR, Copertino DC Jr, Iñiguez LP, Marston JL, Bram Y, Han Y, Schwartz RE, Chen S, Nixon DF, Powell TR. Identifying FDA-approved drugs with multimodal properties against COVID-19 using a data-driven approach and a lung organoid model of SARS-CoV-2 entry. Mol Med. 2021;27(1):105.
Sahoo BM, Bhattamisra SK, Das S, Tiwari A, Tiwari V, Kumar M, Singh S. Computational Approach to Combat COVID-19 Infection: Emerging Tools for Accelerating Drug Research. Curr Drug Discov Technol. 2022;19(3):40–53.
Norinder U, Tuck A, Norgren K, Munic Kos V. Existing highly accumulating lysosomotropic drugs with potential for repurposing to target COVID-19. Biomed Pharmacother. 2020;130: 110582.
Cheng F, Rao S, Mehra R. COVID-19 treatment: Combining anti-inflammatory and antiviral therapeutics using a network-based approach. Cleve Clin J Med. 2020:1–6.
Zhou Y, Hou Y, Shen J, Huang Y, Martin W, Cheng F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020;6:14.
Gelemanović A, Vidović T, Stepanić V, Trajković K. Identification of 37 Heterogeneous Drug Candidates for Treatment of COVID-19 via a Rational Transcriptomics-Based Drug Repurposing Approach. Pharmaceuticals (Basel). 2021;14(2):87.
Naz A, Asif S, Alwutayd KM, Sarfaraz S, Abbasi SW, Abbasi A, Alenazi AM, Hasan ME. Repurposing FIASMAs against Acid Sphingomyelinase for COVID-19: A Computational Molecular Docking and Dynamic Simulation Approach. Molecules. 2023;28(7):2989.
Liu DY, Liu JC, Liang S, Meng XH, Greenbaum J, Xiao HM, Tan LJ, Deng HW. Drug Repurposing for COVID-19 Treatment by Integrating Network Pharmacology and Transcriptomics. Pharmaceutics. 2021;13(4):545.
Touret F, Gilles M, Barral K, Nougairède A, van Helden J, Decroly E, de Lamballerie X, Coutard B. In vitro screening of a FDA approved chemical library reveals potential inhibitors of SARS-CoV-2 replication. Sci Rep. 2020;10(1):13093.
O’Donovan SM, Imami A, Eby H, Henkel ND, Creeden JF, Asah S, Zhang X, Wu X, Alnafisah R, Taylor RT, et al. Identification of candidate repurposable drugs to combat COVID-19 using a signature-based approach. Sci Rep. 2021;11(1):4495.
Udrea AM, Avram S, Nistorescu S, Pascu ML, Romanitan MO. Laser irradiated phenothiazines: New potential treatment for COVID-19 explored by molecular docking. J Photochem Photobiol B. 2020;211:111997.
Daisy AH, Daniel JBC, Rasmus M, Yuling H, Liuliu Y, Megan LW, Alexander L, Kasopefoluwa YO, Christian S, Benhur L, et al. Modulating the transcriptional landscape of SARS-CoV-2 as an effective method for developing antiviral compounds. bioRxiv. 2020:1–27.
Acknowledgements
Not applicable.
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB). Open access funding provided by Te Science, Technology & Innovation Funding Authority (STDF) in cooperation with Te Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
SSA: Conceptualization; supervision; validation and software; data curation. A.M.A: Molecular docking; All authors wrote and edited the original draft. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Financial interest
All authors declare that they have no financial or non-financial interests that are directly or indirectly related to the work submitted for publication.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Alkafaas, S.S., Abdallah, A.M., Hassan, M.H. et al. Molecular docking as a tool for the discovery of novel insight about the role of acid sphingomyelinase inhibitors in SARS- CoV-2 infectivity. BMC Public Health 24, 395 (2024). https://doi.org/10.1186/s12889-024-17747-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12889-024-17747-z