
Abstract
Background
Menkes disease (MD) is a rare, inherited, multisystemic copper metabolism disorder. Classical Menkes disease is characterized by low serum copper and ceruloplasmin concentrations, leading to multiple abnormalities in the whole-body, especially in connective tissue and central nervous system. However, serum copper and ceruloplasmin levels are not reliable diagnostic biomarkers due to the low concentrations in healthy newborns either. The featured imaging manifestations play an important role in diagnosing Menkes disease. To our knowledge, there are few reports on the systemic imaging manifestations of Menkes disease.
Case presentation
A 4-month-old male patient presented with recurrent seizures. He had cognitive, intellectual, growth, gross motor, precision movement, and language developmental lags. The patient’s hemoglobin and serum ceruloplasmin level were low. On MRI, increased intracranial vascular tortuosity, cerebral and cerebellar atrophy, white matter changes, and basal ganglia abnormalities were observed. Plain radiograph revealed wormian bones, rib flaring, metaphyseal spurring, and periosteal reactions in the long bones of the limbs. A pathogenic variant in ATP7A gene was identified in the patient, so he was confirmed the diagnosis of Menkes disease. His symptoms did not improve despite symptomatic and supportive treatment during his hospitalization. Unfortunately, the infant died 3 months after leaving hospital.
Conclusion
A comprehensive and intuitive understanding of the disease’s imaging manifestations can help clinicians to identify the disease and avoid delays in care.
Similar content being viewed by others


Background
Menkes disease (MD) is a rare X-linked genetic disorder with multisystem involvement due to copper transport defects caused by mutations in the ATP7A gene. This was first reported in 1962 by John H. Menkes et al. [1]. It is characterized by progressive neurological degeneration and connective tissue abnormalities. The incidence of Menkes disease is low, ranging from 1 in 100 000 to 1 in 300 000 births [2]. Affected infants often appear healthy at birth until 2–3 months of age, when they exhibit feeding difficulties, recurrent seizures, hypotonia, and delayed developmental milestones. Physical examination may reveal distinctive features, such as coarse, sparse, and lightly pigmented kinky hair; a jowly appearance with sagging cheeks; and lax skin. Tragically, without treatment, most patients die in early childhood, usually before the age of 3 [3]. Early recognition of the disease is important to facilitate early intervention and treatment. A systematic understanding of the imaging manifestations of the disease can help clinicians identify the disease in time.
Case presentation
A 4-month-old male patient presented with recurrent seizures, with squeezing of the left eye and tilting of the corner of the mouth toward the left. Each seizure lasted approximately 10 s, with approximately 10 episodes per day that resolved independently. The infant was born prematurely at 34 weeks of gestation and presented with prolonged physiological jaundice during the neonatal period. The infant was unable to hold his head upward. He had cognitive, intellectual, growth, gross motor, precision movement, and language developmental lags. His height and weight were 0.60 m and 5.8 kg, respectively (body mass index = 16.11 kg/m2), which were two standard deviations lower than the average for the same age and sex. His head circumference was 41 cm, falling within one standard deviation (1.2 cm) of the peers. He had sparse, coarse, twisted, and lightly pigmented hair on the back of the head (Fig. 1) and bilateral inguinal hernias. Unfortunately, the older brother of the infant, who was diagnosed with epilepsy, died at the age of 2 years.

The infant has short, sparse, coarse and kinky hair on the back of the head
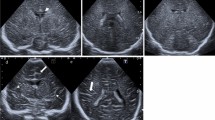
The patient’s hemoglobin level was low at 81 g/L (normal > 90 g/L), serum ceruloplasmin level low at 37 mg/L (normal 110–620 mg/L), and serum pyruvate level high at 315 µmol/L (normal 20–100 µmol/L), serum β-hydroxybutyric acid unremarkable. A cerebrospinal fluid (CSF) test revealed a high lactic acid level of 45 U/L (normal 5–35 U/L). A cranial MRI (PHILIPS Achiva 1.5T Nova Dual) performed during hospitalization revealed multiple intracranial abnormalities: (i) The patient showed white matter changes. T1-weighted image (T1WI) revealed delayed myelination in the brain. T2-weighted image (T2WI) showed massive white matter hyperintensities in both cerebral hemispheres, with tumefactive lesions in the right temporal lobe (Fig. 2a and b). (ii) The patient showed basal ganglia abnormalities. T2-fluid-attenuated inversion recovery (T2-FLAIR) showed asymmetric hyperintensity in the bilateral heads of the caudate nucleus and globus pallidus. Diffusion-weighted and apparent diffusion coefficient maps revealed asymmetric ovoid lesions with restricted diffusion in the bilateral globus pallidus (Fig. 2c and e). (iii) The patient showed cerebral and cerebellar atrophy (Fig. 2f). (iv)The patient showed intracranial blood vessel abnormalities. Magnetic Resonance Angiography (MRA) showed an increased tortuosity of the internal carotid, anterior cerebral, middle cerebral, posterior cerebral, vertebral, and basilar arteries (Fig. 2g and i)

Cranial MRI images. (a-b) Axial T1-weighted image (T1WI) shows the delayed myelination (arrows). Axial T2-weighted image (T2WI) shows the distinctive tumefactive lesion in the right temporal lobe (arrow). (c-e) Axial T2-fluid-attenuated inversion recovery (T2-FLAIR) shows asymmetric hyperintensity in bilateral head of caudate nucleus and globus pallidus (arrows). Diffusion-weighted image (DWI) (b value = 800 s/mm2) and apparent diffusion coefficient (ADC) maps show asymmetric ovoid foci of restricted diffusion in bilateral globus pallidus (arrows). (f) Sagittal T1WI shows cerebellar atrophy. (g-i) Magnetic Resonance Angiography (MRA) shows increased tortuosity of the internal carotid, anterior cerebral, middle cerebral, posterior cerebral, vertebral and basilar arteries
After discovering abnormalities in the patient’s serum and cerebrospinal fluid metabolic markers, the clinicians initially suspected a diagnosis of Menkes disease. In order to understand the overall skeletal condition, a full-body X-ray examination was conducted. Plain radiography of the entire body revealed bone abnormalities: (i) Anteroposterior and lateral skull radiographs showed numerous wormian bones in the sagittal suture, bilateral lambdoid sutures, and mastoid fontanelle (Fig. 3a and b). (ii) Anteroposterior chest radiograph showed flaring and expansion of the anterior ends of bilateral ribs (Fig. 3c). (iii) Anteroposterior upper and lower extremity radiographs showed enlargement and mineralization of the metaphysis with excessive lateral spurs involving the humerus, ulna, radius, femur, tibia, and fibula. Some mild osteopenia but no fractures were observed (Fig. 3d and f)

Plain radiograph images. (a-b) Anteroposterior and lateral skull radiograph shows numerous wormian bones along the sagittal suture, bilateral lambdoid sutures and mastoid fontanelle (arrows). (c) Anteroposterior chest radiograph shows flaring and expansion of anterior ends of bilateral ribs. (d-f) Enlargement and mineralization of metaphysis with lateral spurs involving right ulnar, left humerus, right femur and left tibia (arrows)
A variant (NM_000052.7: exon 15:c.3076delA(p.Ile1026*)) in ATP7A was identified in the patient and classified as pathogenic according to the American College of Medical Genetics and Genomics classification (ACMG) guideline [4]. At admission, the patient was treated with ceftriaxone and amoxycillin clavulanate potassium to prevent infection, with levetiracetam to control seizure, and with vitamins, levocarnitine to improve metabolic conditions. His symptoms have not improved and unfortunately died 3 months after leaving hospital.
Discussion and conclusions
Menkes disease is associated with abnormal copper metabolism in the body. Copper is an essential micronutrient required for the activity of critical metabolic reactions, signaling pathways and redox reactions [5]. The copper-transporting ATPase ATP7A acts as an important cofactor for connective tissue and neuronal copper-dependent enzymes, such as tyrosinase, peptidylαamidating monooxygenase (PAM), lysyl oxidase (LOX), dopamine-β hydroxylases (DβH), and cytochrome c oxidase (COX) [6,7,8]. In normal conditions, ATP7A enables the uptake of copper from the intestinal brush border, to be transported across cells to various tissues and organs throughout the body [9]. In Menkes disease, mutations in the ATP7A gene (including deletion ranging in size from a single exon to almost the entire gene, missense and splice-site mutations, exon duplications, and point mutations) may lead to impaired protein function, protein misfolding, and rapid degradation [7]. This results in the inadequate uptake of copper by the intestinal epithelium and impaired cross-cellular transport, ultimately leading to abnormal copper distribution with severe lack in blood, brain, and liver, but high values in intestines, heart, kidneys spleen, pancreas and skeletal muscle [10].
In this patient, elongation and tortuosity of the intracranial blood vessels, numerous wormian bones, flaring of the ribs, and metaphyseal changes in the long bones were observed. The pathogenic mechanism of tortuous blood vessels may be related to impaired elastin and collagen cross-linkages due to the impairment of LOX activity. Normal cross-linking of collagen is thought to be essential for the mineralization of bone tissue [9]. Cerebral and cerebellar atrophy is another typical feature that likely contributes to the combined effects of metabolic impairment, prolonged status epilepticus, lactic acidosis, and mitochondrial failure [11,12,13]. White matter abnormalities vary among individuals, the lesions show vasogenic edema-like features. However, the exact pathogenic mechanism is uncertain and may be related to altered blood-brain barriers due to impaired copper metabolism [14]. Typically, the bilateral but asymmetric involvement of the basal ganglia results in cytotoxic edema, which may contribute to mitochondrial dysfunction in the respiratory chain [15].
In addition to these common imaging presentations, Manara et al. [15] reported that subdural collections could be detected on MRI. Matthias K. Bernhard et al. [16] reported a 4-month-old child demonstrated dural sinus widening on cerebral angiography. Grange et al. [17] reported a case showed aneurysmal dilatation of the right internal jugular vein, along with dilatation of the superior vena cava, the odd vein, and the innominate vein on magnetic resonance venography (MRV). The chest computed tomography (CT) and computer tomography pulmonary angiography (CTPA) revealed panlobular emphysema and multiple pulmonary alveoli in both lungs, along with pulmonary hypoplasia and tortuous dilatation of the pulmonary arteries. The autopsy findings of this child revealed multiple polyps in the stomach and sigmoid colon. Kim and Zaffanello et al. [18, 19] reported that the most common complicating urologic abnormality in MD is multiple diverticula of the bladder, the others include bladder stones, neurogenic bladder, vesicoureteral reflux, ureteral effusion, renal scarring and cryptorchidism. Peng et al. [20] reported a 4-year-old boy presented with a spontaneous retroperitoneal hematoma. Osteoporosis and fracture of the long bones of the extremities without clear history of accidental trauma have been reported [21, 22]. Hill et al. [23] reported a clinical trial that included 35 children with Menkes’ disease and found a combined C2 posterior arch defect in approximately 11%.
The differential diagnosis of MD can vary among different body systems according to imaging features. Wilson’s disease, another copper metabolism disorder, may be considered in cases involving the basal ganglia, thalamus, and brainstem [24]. However, it typically exhibits less extensive white matter involvement and cerebral and intracranial arterial tortuosity. The other copper metabolism diseases, such as Huppke-Brendel Syndrome, MEDNIK syndrome and CCS deficiency, should be considered. Huppke-Brendel Syndrome presents with hypomyelination, cerebellar hypoplasia, and wide subarachnoid spaces, without basal ganglia, intracranial vessels and extensive white matter involvement. Moreover, the wormian bones have not been reported. MEDNIK syndrome (mental retardation, enteropathy, deafness, peripheral neuropathy, ichthyosis, keratodermia) mainly shows cerebral atrophy and bilateral symmetrical hyperintensities in caudate nuclei and putamina at T2-weighted images and severe osteoporosis on X-ray. These are similar to that seen in Wilson’s disease. CCS deficiency displays progressive brain atrophy, hypomyelination and symmetrical lesions of the thalami, but absence of vascular abnormalities. Occipital Horn Syndrome, known as the milder form of MD, has similar hair and connective tissue abnormalities. The main distinction is radiographic finding of the peculiar occipital horns, which are symmetric exostoses extend from the occipital bone [25,26,27]. Moyamoya disease (MMD), characterized by progressive stenosis, occlusion, and twisted vascular anastomoses in the circle of Willis, is a potential differential diagnosis in children with excessive tortuosity of intracranial vessels, but it differs from Menkes disease in terms of the pattern of vascular abnormalities [28]. Metabolic bone diseases, such as rickets, scurvy, and osteogenesis imperfecta, can be considered; however, numerous wormian bones and progressive neurodegenerative changes are not common features in these disorders.
There is currently no curative treatment for the patients of Menkes disease. Symptomatic and supportive measures remain the mainstay management strategies, such as seizure management, feeding or gastrostomy tube placement to enhance caloric intake, antibiotic prophylaxis to prevent infection and surgery for bladder diverticula [3, 8]. The researchers suggest that subcutaneous copper histidine injections at early age (usually in the neonatal period) have been shown effectively to improve neurodevelopment outcomes, decrease seizures frequency and severity, and increase survival [29, 30]. Recently, adeno-associated viral gene therapy in combination with copper and a membrane traversing drug like elesclomol or elesclomol-Cu2+ hold promise for the treatment [31, 32].
Even though the typical physical examination signs of the patients are distinctive, early diagnosis might be difficult for clinicians due to the rarity. The increased ratios of dopamine to norepinephrine, as well as dihydroxyphenylacetic acid to dihydroxyphenylglycol, are a promising test for neonatal diagnosis of Menkes disease [3]. Meanwhile, radiologists can provide more diagnostic ideas based on the featured imaging findings, such as numerous wormian bones, cerebral and cerebellar atrophy, tortuous intracerebral vessels with parenchymal abnormalities. Due to the multisystem involvement of the disease, differential diagnosis might be difficult for radiologists. Systemic and comprehensive evaluation of radiological findings, detailed physical examination and biochemical testing are important for early diagnosis.
Data availability
No datasets were generated or analysed during the current study.
Abbreviations
- MD:
-
Menkes Disease
- MRI:
-
Magnetic Resonance Imaging
- MRA:
-
Magnetic Resonance Angiography
- CSF:
-
Cerebrospinal fluid
- T1WI:
-
T1-weighted image
- T2WI:
-
T2-weighted image
- T2-FLAIR:
-
T2-fluid-attenuated inversion recovery
- ACMG:
-
American College of Medical Genetics and Genomics classification
- PAM:
-
Peptidylαamidating monooxygenase
- LOX:
-
Lysyl oxidase
- DβH:
-
Dopamine-β hydroxylases
- COX:
-
Cytochrome c oxidase
- MRV:
-
Magnetic resonance venography
- CT:
-
Computer tomography
- CTPA:
-
Computer tomography pulmonary angiography
- MMD:
-
Moyamoya disease
References
J H Menkes Malter, G K Steigleder. A sex-linked recessive disorder with retardation of growth, peculiar hair, and focal cerebral and cerebellar degeneration. Pediatrics. 1962;29:764–79.
Kevin Fung KF, Kwong YY, Mak WS, Elaine Kan YL. Case 280: Trichopoliodystrophy. Radiology. 2020;296:463–7.
Kaler SG, Goldstein DS, Godwin SC, Sato S. Neonatal diagnosis and treatment of Menkes Disease. N Engl J Med. 2008;358:605–14.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Sci. 2015;17:405–24.
Maung MT, Carlson A, Olea-Flores M, Elkhadragy L, Schachtschneider KM, Navarro‐Tito N, et al. The molecular and cellular basis of copper dysregulation and its relationship with human pathologies. FASEB j. 2021;35(9):e21810.
Zlatic S, Comstra HS, Gokhale A, Petris MJ, Faundez V. Molecular basis of neurodegeneration and neurodevelopmental defects in Menkes disease. Neurobiol Dis. 2015;81:154–61.
Tümer Z. An overview and update of ATP7A mutations leading to Menkes Disease and Occipital Horn Syndrome. Hum Mutat. 2013;34:417–29.
Kaler SG. ATP7A-related copper transport diseases—emerging concepts and future trends. Nat Rev Neurol. 2011;7:15–29.
Horn N, Tümer Z. Menkes Disease and the Occipital Horn Syndrome. In: Royce PM, Steinmann B, editors. Connective tissue and its heritable disorders. Hoboken, NJ, USA: John Wiley & Sons, Inc.; 2002. pp. 651–85.
Kaler SG. Inborn errors of copper metabolism. Handbook of clinical neurology. Elsevier; 2013. pp. 1745–54.
Bindu PS, Taly AB, Kothari S, Christopher R, Gayathri N, Sinha S, et al. Electro-clinical features and magnetic resonance imaging correlates in Menkes disease. Brain Develop. 2013;35:398–405.
Rennert J, Doelken R, Doelken M, Doerfler A. Menkes disease: MRI appearance of a rare neurodegenerative disorder. J Pediatr Neurol. 2015;07:317–20.
Leventer RJ, Kornberg AJ, Phelan EM, Kean MJ. Early magnetic resonance imaging findings in Menkes’ Disease. J Child Neurol. 1997;12:222–4.
Manara R, D’Agata L, Rocco MC, Cusmai R, Freri E, Pinelli L, et al. Neuroimaging changes in Menkes Disease, Part 1. AJNR Am J Neuroradiol. 2017;38:1850–7.
Manara R, Rocco MC, D’agata L, Cusmai R, Freri E, Giordano L, et al. Neuroimaging changes in Menkes Disease, Part 2. AJNR Am J Neuroradiol. 2017;38:1858–65.
Bernhard M, Merkenschlager A, Mayer T, Muntau A, Pfluger T. The spectrum of neuroradiological features in Menkes disease: Widening of the cerebral venous sinuses. J Pediatr Neuroradiol. 2015;01:121–5.
Grange DK, Kaler SG, Albers GM, Petterchak JA, Thorpe CM, deMello DE. Severe bilateral panlobular emphysema and pulmonary arterial hypoplasia: unusual manifestations of Menkes disease. Am J Med Genet. 2005;139A:151–5.
Kim MY, Kim JH, Cho MH, Choi YH, Kim SH, Im YJ, et al. Urological problems in patients with Menkes Disease. J Korean Med Sci. 2019;34:e4.
Zaffanello M, Maffeis C, Fanos V, Franchini M, Zamboni G. Urological complications and copper replacement therapy in childhood Menkes syndrome. Acta Paediatr. 2006;95:785–90.
Peng C-H, Hsu C-H, Wang N-L, Lee H-C, Lin S-P, Chan W-T, et al. Spontaneous retroperitoneal hemorrhage in Menkes disease: a rare case report. Medicine. 2018;97:e9869.
Droms RJ, Rork JF, McLean R, Martin M, Belazarian L, Wiss K. Menkes Disease mimicking child abuse. Pediatr Dermatol. 2017;34.
Adams PC, Strand RD, Bresnan MJ, Lucky AW. Kinky hair syndrome: serial study of radiological findings with emphasis on the similarity to the battered child syndrome. Radiology. 1974;112:401–7.
Hill SC, Dwyer AJ, Kaler SG. Cervical spine anomalies in Menkes disease: a radiologic finding potentially confused with child abuse. Pediatr Radiol. 2012;42:1301–4.
Bandmann O, Weiss KH, Kaler SG. Wilson’s disease and other neurological copper disorders. Lancet Neurol. 2015;14:103–13.
Huppke P, Brendel C, Kalscheuer V, Korenke GC, Marquardt I, Freisinger P, et al. Mutations in SLC33A1 cause a Lethal autosomal-recessive disorder with congenital cataracts, hearing loss, and low serum copper and Ceruloplasmin. Am J Hum Genet. 2012;90:61–8.
Martinelli D, Travaglini L, Drouin CA, Ceballos-Picot I, Rizza T, Bertini E, et al. MEDNIK syndrome: a novel defect of copper metabolism treatable by zinc acetate therapy. Brain. 2013;136:872–81.
Huppke P, Brendel C, Korenke GC, Marquardt I, Donsante A, Yi L, et al. Molecular and biochemical characterization of a unique mutation in CCS, the human copper chaperone to superoxide dismutase. Hum Mutat. 2012;33:1207–15.
Ihara M, Yamamoto Y, Hattori Y, Liu W, Kobayashi H, Ishiyama H, et al. Moyamoya disease: diagnosis and interventions. Lancet Neurol. 2022;21:747–58.
Kaler SG, DiStasio AT. ATP7A-Related Copper Transport Disorders.
Vairo FPe, Chwal BC, Perini S, Ferreira MAP, de Freitas Lopes AC, Saute JAM. A systematic review and evidence-based guideline for diagnosis and treatment of Menkes disease. Mol Genet Metab. 2019;126:6–13.
Haddad MR, Choi E-Y, Zerfas PM, Yi L, Martinelli D, Sullivan P, et al. Cerebrospinal Fluid-Directed rAAV9-rsATP7A plus subcutaneous copper histidinate advance survival and outcomes in a Menkes Disease Mouse Model. Mol Therapy - Methods Clin Dev. 2018;10:165–78.
Guthrie LM, Soma S, Yuan S, Silva A, Zulkifli M, Snavely TC, et al. Elesclomol alleviates Menkes pathology and mortality by escorting Cu to cuproenzymes in mice. Science. 2020;368:620–5.
Acknowledgements
The authors would like to thank the patient and his family for their consent to publish this report.
Funding
This study was funded by Sichuan Science and Technology Program and Natural Science Foundation of Sichuan Province (2022NSFSC0830).
Author information
Authors and Affiliations
Contributions
Juncheng Zhu collected the patient’s clinical information, reviewed the literature and drafted the initial manuscript. Yi Liao designed the study and revised the manuscript. Fenglin Jia helped to review the article. Xuesheng Li and Xinmao Ma provided imaging data. Haibo Qu provided critical editing and revision to the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Approval was obtained from the ethics committee of Sichuan University. The procedures used in this study adhere to the tenets of the Declaration of Helsinki.
Consent for publication
Written informed consent for publication of this case report and accompanying images was obtained from the parents.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhu, J., Liao, Y., Li, X. et al. Brain and the whole-body bone imaging appearances in Menkes disease: a case report and literature review. BMC Pediatr 24, 411 (2024). https://doi.org/10.1186/s12887-024-04885-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12887-024-04885-x