
Abstract
Background
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening disease characterized by some clinical signs (e.g., non-remitting fever, hepatosplenomegaly) and laboratory findings (e.g., cytopenia, increased ferritin level, hypofibrinogenemia, lipid disorders, coagulopathy, and multiple organ failure). Depending on the etiology, HLH is divided into familial (i.e., primary) and acquired (i.e., secondary) forms. Familial HLH (FHL), an autosomal recessive condition, is classified into five subtypes based on underlying genetic defects. The PRF1, STX11, UNC13D, HPLH1, and STXBP2 are the most well-known genes of this type which are related to granule-mediated cytotoxic T and Natural killer (NK) cells. The treatment is based on the HLH-2004 protocol.
Case presentation
The current report presents two cases of HLH with presentations different from each other and previously reported cases. Case 1 was a 15-month-old boy with fever, skin rash, splenomegaly, and bicytopenia, raised triglyceride levels, AST (aspartate transaminase), and ALT (alanine aminotransferase), normal ferritin, and abundant hemophagocytic cell in bone marrow aspiration. He was diagnosed with HLH and received HLH protocol as treatment. The patient had a homozygous intronic mutation; NM_199242: c.2448-13G > A in UNC13D. The associated disease was Familial Hemophagocytic Lymphohistiocytosis 3 (FHL3). Case 2, a 37-day-old female presented with fever, a history of neonatal cholestasis, and huge hepatosplenomegaly. Her whole-exome sequencing report manifested that the patient had the same mutation as case 1. Unfortunately, both patients passed away.
Conclusion
The sequencing of the entire UNC13D gene (coding and non-coding regions) is an applicable and valuable diagnostic procedure for the detection of deep intronic splicing variants and large inversions in patients with atypical manifestations of HLH (such as normal ferritin or triglyceride and cholesterol).
Similar content being viewed by others


Background
Hemophagocytic lymphohistiocytosis (HLH) is an aggressive and life-threatening disease that can be fatal [1]. It is characterized by some clinical signs (e.g., non-remitting fever, hepatosplenomegaly) and laboratory findings (e.g., cytopenia, increased ferritin level, hypofibrinogenemia, lipid disorders, such as hypertriglyceridemia, coagulopathy, and multiple organ failure) [2, 3]. Immune dysregulation in cytotoxic T cells, Natural killer (NK) cells, and histiocytes is considered the leading cause of this disease.
Depending on the etiology, HLH can be divided into familial (i.e., primary) and acquired (i.e., secondary) forms [4,5,6]. Familial HLH (FHL), an autosomal recessive condition, is classified into five subtypes based on underlying genetic defects. The PRF1, STX11, UNC13D, HPLH1, and STXBP2 are the most well-known genes of this type.
Mutations associated with restrained cytotoxic responses of cytotoxic T and NK cells to B cells infected by Epstein-Barr virus (EBV) inducing X-linked lymphoproliferative syndrome (SH2D1A, XIAP) are the third reason. Nevertheless, other primary immunodeficiencies, including combined immunodeficiency, chronic granulomatous disease, autoinflammatory diseases, and antibody deficiencies, have been described to cause HLH [4,5,6,7,8]. Acquired or secondary HLH, also known as macrophage activation syndrome, mainly occurs after severe viral, bacterial, or fungal infections, metabolic disorders, malignancies, and rheumatologic and immune diseases, such as autoimmune or autoinflammatory diseases [4,5,6, 9,10,11].
The treatment is based on HLH-2004 protocol, including chemoimmunotherapy (corticosteroids, etoposide, cyclosporin A, and/or intrathecal methotrexate), supportive therapy, prophylactic antibiotics, and intravenous Ig (IVIG). Hematopoietic stem cell transplantation is recommended in selected patients with refractory and/or relapsed disease after appropriate chemoimmunotherapy [12,13,14]. This treatment is modified regarding the classification and pathophysiology of HLH.
T-cell-directed immunotherapy could be advantageous in patients with primary HLH. In contrast, strong immunosuppression therapy is contraindicated in patients with severe proceeding infections or some primary immunodeficiency diseases other than familial HLH and X-linked lymphoproliferative syndrome [3]. In addition, more T-cell targeting therapies, such as anti-thymocyte globulin, etoposide, and alemtuzumab, can be beneficial to patients with primary HLH due to overactivated T cells [4, 5].
The differentiation between the two mentioned types of HLH is sometimes tricky, particularly in the beginning; moreover, diagnosis at the right time is critical for initiating appropriate and indicated treatment. Therefore, we performed whole-exome sequencing on two separate patients with relatively different symptoms to find the same homozygous intronic UNC13D mutation.
Case presentation
In this study, we present the case of two children with rather different clinical symptoms hospitalized almost at the same time in two different centers affiliated with Tehran University of Medical Sciences. Their ages, symptoms, and signs were different, especially at the beginning; nonetheless, whole-exome sequencing was employed due to the uncertainty surrounding the underlying etiology. Written informed consent was obtained from their parents.
Case 1
The first case was a 15-month-old male infant born to consanguineous parents via normal vaginal delivery. He had normal growth and development until admission due to prolonged fever for 40 days with no specific pattern, vomiting, and maculopapular rash on his face. His physical examination revealed huge splenomegaly (diameter 120 mm) and fever. Liver function tests demonstrated a rise in aspartate aminotransferase (AST) of 88 IU/L and alanine aminotransferase (ALT) of 52 IU/L. His Complete blood count (CBC) showed bicytopenia (white blood cell count of 10.8 thousand/mm3, anemia with hemoglobin of 10.8 g/dl, and thrombocytopenia with a platelet count of 106 *103/μl). Triglyceride (TG) level was initially 425 mg/dl, which increased to 644 mg/dl. The cholesterol level was initially 86 mg/dl, which increased to 182 mg/dl. The c-reactive protein (CRP) was high (73 mg/dl) with a normal erythrocyte sedimentation rate (ESR) at the same time (14 mm/h). Remarkably, the value of serum ferritin was normal throughout all his disease period (150 μg/l).
His abdominal sonography illustrated splenomegaly with a diameter of 120 mm and homogenous echogenicity. Bone marrow aspiration (BMA) performed from the iliac crest appeared normal. He was diagnosed with macrophage activating syndrome in the context of systemic juvenile idiopathic arthritis by a consultant rheumatologist. He received intravenous methylprednisolone pulse therapy (30 mg/kg) and intravenous immune globulin (IVIG). He was discharged from the hospital with oral prednisolone at a dose of 1 mg/kg/day and readmitted with intermittent fever and vomiting after 45 days. He had bicytopenia, increased ALT and CRP during hospitalization, and his ferritin level was 248 μg/l. Antibody assessment against cytomegalovirus (CMV) and Epstein-Barr virus (EBV) displayed no abnormality.
The patient underwent BMA again, and bone marrow revealed abundant hemophagocytic cells. He was treated with cyclosporine A, etoposide, and dexamethasone according to HLH protocol 2004. After 1 month of treatment, CBC turned normal; CRP and TG decreased. Upon the completion of induction therapy with HLH protocol and initiation of maintenance therapy, the patient experienced a disease flare-up and was readmitted with fever and status epilepticus. Laboratory data revealed pancytopenia and increased levels of TG, AST, and ALT again; however, ferritin level was in the normal range, and brain computed tomography (CT) scan showed ventriculomegaly and Mega Cisterna Magna (Table 1).
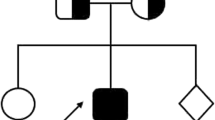
In the last course of hospitalization, the patient passed away because of progressive brain involvement and hydrocephalus, and the whole-exome sequencing (on genomic DNA extracted from peripheral blood cells through an Agilent SureSelect V7 kit and Genome Analyzer HiSeq 4000 (Illumina, USA)) revealed a homozygous intronic UNC13D (NM_199242: c.2448-13G > A), probably affecting splicing. Although Sanger sequencing is not a routine survey for parents, since this mutation is transferred from parents or is a de novo mutation for the assessment of the next child, it was performed and confirmed that his parents were heterozygous carriers for this mutation (Fig. 1). The associated disease was Familial Hemophagocytic Lymphohistiocytosis 3(OMIM 608897) with autosomal recessive inheritance.

Sanger sequencing of c.2448-13G > A mutation in the studied families (Geneious software was used for sanger analysis)
Furthermore, prenatal diagnosis with chorionic villus sampling was implemented in the 12th week of gestational age for the next child of this family and demonstrated that the fetus was only a heterozygous carrier of UNC13D mutation.
Case 2
A 37-day-old female neonate born to consanguineous parents via cesarean section was admitted with fever (Temperature: 39 °C), cough, and a history of neonatal cholestasis. Her physical examination revealed fever and huge hepatosplenomegaly (with spleen preference). She had normal growth before admission despite the previous admission for cholestasis. Her liver function tests resulted in an AST of 33 IU/L that increased to 98 IU/L and an ALT of 21 IU/L that increased to 391 IU/L in her admission process. Her CBC revealed pancytopenia (leukopenia with white blood cell (WBC) of 3.13 thousand/mm3 and ANC of 210, anemia with hemoglobin of 5.1 g/dl, and thrombocytopenia with platelets of 41 *103/μl).
The TG and cholesterol levels were 113 mg/dl and 76 mg/dl, retrospectively. Ferritin level was increased to 2050 μg/l during her hospitalization. The CRP was 28 mg/L, and ESR was in the normal range (16 mm/h). Her abdominal sonography exhibited splenomegaly with a diameter of 144 mm and hepatomegaly with a diameter of 105 mm (both of homogenous echogenicity). The BMA and bone marrow biopsy were performed from the proximal tibia and appeared normal.
Her purified protein derivative (PPD) test turned negative, and a normal TORCH study (assessing antibodies against toxoplasmosis, rubella, CMV, and Herpes simplex virus [HSV]) was detected. The HIV ab was also negative. Metabolic screening tests, including MS/MS, ammonia, lactate, serum amino acid chromatography by HPLC, and urine reducing substance, all were normal. Nevertheless, the evaluation of Galactosemia, Gaucher’s disease, and Niemann-Pick’s disease were not feasible due to the previous history of blood transfusion. Furthermore, fibrinogen was in the normal range.
She was treated with prednisolone (4 mg/kg/day) and IVIG (2 g/kg Stat) as prescribed by a hematologist (with autoimmune thrombocytopenia and anemia diagnosis). Firstly, there was a rapid increase in platelet number and hemoglobin level. Nonetheless, the patient needed multiple blood transfusions and platelets due to sustained bicytopenia. Since the second case did not fulfill the criteria, she did not receive the 2004-HLH treatment protocol as suggested by a consultant rheumatologist. Finally, she died of severe sepsis by abdominal distention and positive blood culture with Pseudomonas aeruginosa. The whole-exome sequencing analysis detected a homozygous c.2448-13G > A UNC13D mutation, the same mutation observed in case 1. Sanger sequencing confirmed that his parents were also the heterozygous carriers of this mutation (data not shown). Sanger sequencing was performed since parents were young and desired to have a healthy child in the future.
Discussion and conclusions
Familial hemophagocytic lymphohistiocytosis (FHL) is a challenging diagnosis due to the absence of any pathognomonic clinical and laboratory signs and symptoms. It is a rare autosomal recessive immune disease caused by the mutations of different genes associated with the formation and function of secretory lysosomes within cytotoxic T lymphocytes and NK cells. FHL type 3 (FHL3) accounts for approximately 30–40% of FHL, and mutation in the UNC13D gene, which encodes Munc13–4 protein, was reported as its underlying cause. This mutation results in defective cytotoxic granule exocytosis, followed by diminished cytotoxic activity of T lymphocytes [11].
In their study, Yoon et al. reported that FHL3 accounts for 89% of FHL cases in Korea, and nearly 20–25% in Japan, suggesting that UNC13D mutations may be responsible for a massive proportion of HLH patients in Asian countries [15]. Functional tests with and/or CTL cells, including perforin detection and degranulation assay quantifying CD107, a surface expression with flow cytometry, can direct genetic analyses [16].
The majority of mutations in the previously reported patients were discovered in deep intronic regions within intron 1 and 9, as well as an inversion in intron 30, advocating the fact that not only coding sequences of the UNC13D gene are essential for the assessment but also introns and non-coding regions should be sequenced and analyzed for the detection of deep intronic splicing variants and large inversions in patients who have failed to show mutations in whole-exome sequencing [17, 18]. The genetic study of both patients revealed a homozygotic intronic c.2448–13 G > A UNC13D mutation previously reported by Alsina et al. [19]. Compound heterozygous mutations reported in the UNC13D gene are frequent in FHL patients [20]. Nonetheless, this case study indicates that homozygous mutations are primarily observed in consanguineous families [17].
This case report confirms the literature claim that more than two-thirds of FHL3 patients suffer from fever and hepatosplenomegaly. Since our first case was presented with status epilepticus, neurological manifestations are also common among HLH patients, according to the literature. Liver failure, lymphadenopathy, consumptive coagulopathy, dermatologist manifestations, edema, and jaundice, like some other manifestations of the disease, were observed in our cases to some extent [18, 21]. Moreover, the laboratory findings of these two cases were in line with the literature (except for ferritin level in case 1), suggesting that neutropenia, thrombocytopenia, lack of NK cell activity, reduced fibrinogen, increased ferritin, and high level of triglycerides should raise doubts about the diagnosis of FHL3 [11].
The FHL3 mostly happens in the first year of life (with a median age of 3 months); nonetheless, the first case was older than 1 year [22]. Ferritin level has been reported high in most previous studies; however, it was interestingly normal in our first case, even during the active phases of the disease [23, 24]. A wide range of bacterial, viral, protozoal, and fungal infections are correlated with HLH [25]. Viral infections are supposed to provoke both primary and secondary types of HLH. Herpesviruses, particularly EBV, are the most prevalent viral infections [26]. Rickettsia [27], Mycobacterium [28], and Histoplasmosis [29] are the most frequent bacterial and fungal infections, retrospectively. Although it could not be justified, the sepsis induced by Pseudomonas aeruginosa could trigger the incidence of HLH in the second patient.
The solitary treatment for familial and severe HLH is bone marrow transplantation [13]. Based on the HLH 2004 protocol, an 8-week monotherapy utilizing dexamethasone, Cyclosporine A, or etoposide is considered the first-line treatment. However, the second case did not receive any treatment due to a failure to fill in the HLH 2004 protocol. Due to the involvement of the central nervous system in readmission, intrathecal methotrexate was administered to our first case with fever and status epilepticus, and bone marrow transplantation was considered the next step to put the disease in remission [30]; however, the neurological involvement did not respond well to the treatment, and after a course of status epilepticus, the patient’s condition got worse, and finally, he passed away.
The FHL, as a challenging diagnosis, may have a wide diversity of clinical manifestations and laboratory findings. Sequencing the entire UNC13D gene (coding and non-coding regions) is an applicable and valuable diagnostic procedure for detecting deep intronic splicing variants and large inversions in patients. Genetic assessment can significantly help in the event of HLH disease with atypical manifestation or situations when primary evaluations cannot detect etiology.
Availability of data and materials
Data of these two patients are available in the Children ‘s Medical Center hospital archive and can be public according to your request and The reported mutations in this study are submitted in ClinVar (Submission ID: SUB12176411).
Abbreviations
- HLH:
-
Hemophagocytic lymphohistiocytosis
- NK:
-
Natural killer
- FHL:
-
Familial HLH
- PRF1:
-
Perforin 1
- STX11:
-
Syntaxin 11
- UNC13D:
-
Unc-13 Homolog D
- HPLH1:
-
Hemophagocytic lymphohistiocytosis 1
- STXBP2:
-
syntaxin binding protein 2
- EBV:
-
Epstein-Barr virus
- IVIG:
-
Intravenous immune globulin
- PID:
-
Primary immunodeficiency
- WES:
-
Whole exome sequencing
- AST:
-
Aspartate transaminase
- ALT:
-
Alanine Transaminase
- CBC:
-
Complete Blood Count
- TG:
-
Triglyceride
- CRP:
-
C-Reactive Protein
- ESR:
-
Erythrocyte Sedimentation Rate
- BMA:
-
Bone Marrow Aspiration
- CMV:
-
Cytomegalovirus
- CT:
-
Computed tomography
- DNA:
-
Deoxyribonucleic Acid
- WBC:
-
White Blood Cells
- ANC:
-
Absolute Neutrophil Count
- BMB:
-
Bone Marrow Biopsy
- PPD:
-
Purified protein derivative
- TORCH:
-
Toxoplasmosis, Others, Rubella, Cytomegalovirus, Herpes Simplex Virus
- HSV:
-
Herpes Simplex Virus
- MS/MS:
-
Tandem mass spectrometry
- HPLC:
-
High-performance liquid chromatography
- FHL3:
-
Familial HLH type 3
- CTL:
-
Cytotoxic T cell
- CD 107:
-
Cluster of differentiation, Cell markers in immunophenotyping
References
Bell R, Brafield A, Barnes N, France N. Familial haemophagocytic reticulosis. Arch Dis Child. 1968;43(231):601.
Giardino G, Veropalumbo C, Ruggiero G, Naddei R, Rubino V, Udhayachandran A, et al. Phenotypic characterization and outcome of paediatric patients affected with haemophagocytic syndrome of unknown genetic cause. Br J Haematol. 2013;162(5):713–7.
Han S-P, Lin Y-F, Weng H-Y, Tsai S-F, Fu L-S. A novel BTK gene mutation in a child with atypical X-linked agammaglobulinemia and recurrent hemophagocytosis: a case report. Front Immunol. 2019;10:1953.
Ammann S, Lehmberg K, Zur Stadt U, Janka G, Rensing-Ehl A, Klemann C, et al. Primary and secondary hemophagocytic lymphohistiocytosis have different patterns of T-cell activation, differentiation, and repertoire. Eur J Immunol. 2017;47(2):364–73.
Bode SF, Ammann S, Al-Herz W, Bataneant M, Dvorak CC, Gehring S, et al. The syndrome of hemophagocytic lymphohistiocytosis in primary immunodeficiencies: implications for differential diagnosis and pathogenesis. Haematologica. 2015;100(7):978.
Faitelson Y, Grunebaum E. Hemophagocytic lymphohistiocytosis and primary immune deficiency disorders. Clin Immunol. 2014;155(1):118–25.
Fleischmann R, Böhmerle W, von Laffert M, Jöhrens K, Mengel A, Hotter B, et al. Adult hemophagocytic lymphohistiocytosis causing multi organ dysfunction in a patient with multiple autoimmune disorders: when the immune system runs amok. Clin Case Rep. 2016;4(2):165.
Sepulveda FE, Maschalidi S, Vosshenrich CA, Garrigue A, Kurowska M, Ménasche G, et al. A novel immunoregulatory role for NK-cell cytotoxicity in protection from HLH-like immunopathology in mice. Blood. 2015;125(9):1427–34.
Ramos-Casals M, Brito-Zerón P, López-Guillermo A, Khamashta MA, Bosch X. Adult haemophagocytic syndrome. Lancet. 2014;383(9927):1503–16.
Lee W-I, Chen S-H, Hung I-J, Yang C-P, Jaing T-H, Chen C-J, et al. Clinical aspects, immunologic assessment, and genetic analysis in Taiwanese children with hemophagocytic lymphohistiocytosis. Pediatr Infect Dis J. 2009;28(1):30–4.
Amirifar P, Ranjouri MR, Abolhassani H, Moeini Shad T, Almasi-Hashiani A, Azizi G, et al. Clinical, immunological and genetic findings in patients with UNC13D deficiency (FHL3): a systematic review. Pediatr Allergy Immunol. 2021;32(1):186–97.
Henter J-I, Samuelsson-Horne A, Arico M, Egeler RM, Gr E, Filipovich AH, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood, the journal of the American society of. Hematology. 2002;100(7):2367–73.
Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124–31.
Seo JJ. Hematopoietic cell transplantation for hemophagocytic lymphohistiocytosis: recent advances and controversies. Blood Res. 2015;50(3):131–9.
Yoon HS, Kim H-J, Yoo K-H, Sung K-W, Koo H-H, Kang HJ, et al. UNC13D is the predominant causative gene with recurrent splicing mutations in Korean patients with familial hemophagocytic lymphohistiocytosis. Haematologica. 2010;95(4):622.
Bryceson YT, Pende D, Maul-Pavicic A, Gilmour KC, Ufheil H, Vraetz T, et al. A prospective evaluation of degranulation assays in the rapid diagnosis of familial hemophagocytic syndromes. Blood. 2012;119(12):2754–63.
Woods CG, Cox J, Springell K, Hampshire DJ, Mohamed MD, McKibbin M, et al. Quantification of homozygosity in consanguineous individuals with autosomal recessive disease. Am J Hum Genet. 2006;78(5):889–96.
Freeman H, Ramanan A. Review of haemophagocytic lymphohistiocytosis. Arch Dis Child. 2011;96(7):688–93.
Alsina L, Colobran R, De Sevilla M, Catala A, Vinas L, Ricart S, et al. Novel and atypical splicing mutation in a compound heterozygous UNC13D defect presenting in familial Hemophagocytic Lymphohistiocytosis triggered by EBV infection. Clin Immunol. 2014;153(2):292–7.
Chen Y, Wang Z, Cheng Y, Tang Y. Novel mutations in the UNC13D gene carried by a Chinese neonate with hemophagocytic lymphohistiocytosis. Yonsei Med J. 2013;54(4):1053–7.
Yazdani R, Amirifar P, Abolhassani H, Azizi G, Parvaneh N, Rezaei N, et al. UNC13D deficiency associated with epileptic seizures and antibody deficiency: the first case from the Iranian National Registry. J Investig Allergol Clin Immunol. 2019;29(2):160–2.
Feldmann J, Callebaut I, Raposo G, Certain S, Bacq D, Dumont C, et al. Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell. 2003;115(4):461–73.
Huang J, Jiang L, Wu X, Cheng Y, Chen C, Xue H. Familial Hemophagocytic Lymphohistiocytosis type 3: early disease onset and unusual manifestation in sibling cases. Iran J Pediatr. 2019;29(2).
Liao C-H, Lee N-C, Jou S-T, Chiang B-L, Yu H-H. UNC13D mutation presenting as fulminant familial hemophagocytic lymphohistiocytosis. J Microbiol Immunol Infect. 2020;53(6):1039–41.
Fisman DN. Hemophagocytic syndromes and infection. Emerg Infect Dis. 2000;6(6):601.
Henter JI, Ehrnst A, Andersson J, Elinder G. Familial hemophagocytic lymphohistiocytosis and viral infections. Acta Paediatr. 1993;82(4):369–72.
Jin Y, Huang L, Fan H, Lu G, Xu Y, Wu Z. Scrub typhus associated with hemophagocytic lymphohistiocytosis: a report of six pediatric patients. Exp Ther Med. 2016;12(4):2729–34.
Hui Y, Pillinger T, Luqmani A, Cooper N. Haemophagocytic lymphohistiocytosis associated with Mycobacterium tuberculosis infection. BMJ Case Rep. 2015;2015:bcr2014208220.
Untanu RV, Akbar S, Graziano S, Vajpayee N. Histoplasmosis-induced hemophagocytic lymphohistiocytosis in an adult patient: a case report and review of the literature. Case Rep Infect Dis. 2016;2016:1358742.
Zhang J-R, Liang X-L, Jin R, Lu G. HLH-2004 protocol: diagnostic and therapeutic guidelines for childhood hemophagocytic lymphohistiocytosis. Zhongguo Dang Dai Er Ke Za Zhi. 2013;15(8):686–8.
Acknowledgments
We thank the parents for giving us their consent to report the cases.
Funding
No person who participated in accomplishing the case report has been sponsored.
Author information
Authors and Affiliations
Contributions
RM and PS admitted and monitored the patient in the first weeks. RM and PS followed the patients in follow-up. GG and RM drafted the manuscript. HR and MC analyzed WES data and wrote the parts of manuscript related to WES and prepared the sanger analysis and figure. All authors read and approved the final manuscript.
Authors’ information
1- Payman Sadeghi is an assistant professor at Tehran University of Medical Sciences.
2- Golnaz Ghazizadeh Esslami is an assistant professor at Tehran University of Medical Sciences.
3- Hassan Rokni-Zadeh works at Zanjan University of Medical Sciences.
4- Majid Changi-Ashtiani works in Institute for Research in Fundamental Sciences (IPM).
5- Reihaneh Mohsenipour is an associate professor at Tehran University of Medical Sciences.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
An written informed consent was obtained from the individual parents of patients whose identifying information is included in this paper.
The whole research was done under the permission of the Ethics committee of Tehran University of Medical Sciences and also Genetic Testing for parents was done beause of their desire to have next healthy child and it was done under the permission of Ethic committee of Tehran University of Medical Sciences.
Consent for publication
We obtained written informed consent from the parents of two cases for publication of their children’s personal or clinical details and images.
Competing interests
The authors declare that there is no competing interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sadeghi, P., Esslami, G.G., Rokni-Zadeh, H. et al. Familial Hemophagocytic Lymphohistiocytosis secondary to UNC13D mutation: a report of two cases. BMC Pediatr 22, 667 (2022). https://doi.org/10.1186/s12887-022-03746-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12887-022-03746-9