
Abstract
Background
Citrin deficiency (CD) is a recessive metabolic disease caused by biallelic pathogenic variants in SLC25A13. Although previous studies have reported ketosis in CD, it was observed at the time of euglycemia or mild hypoglycemia. Blood ketone levels concomitant with symptomatic or severe hypoglycemia in CD have not been a topic of focus despite its importance in identifying the etiology of hypoglycemia and assessing the ability of fatty acid utilization. Herein, we describe a patient with CD who had repeated episodes of hypoglycemia with insufficient ketosis.
Case presentation
A 1-year-old boy with repetitive hypoglycemia was referred to us to investigate its etiology. The fasting load for 13 h led to hypoketotic hypoglycemia, indicating the possibility of partial β-oxidation dysfunction. A genetic test led to the diagnosis of CD. The hypoglycemic episodes disappeared after switching to a medium-chain triglyceride-containing formula.
Conclusions
This case report suggests that symptomatic or severe hypoglycemia in patients with CD could be associated with relatively low levels of ketone bodies, implying that β-oxidation in these patients might possibly be partially disrupted. When encountering a patient with hypoglycemia, clinicians should check blood ketone levels and bear in mind the possibility of CD because excessive intravenous administration of glucose can cause decompensated symptoms in patients with CD as opposed to other disorders presenting with hypoketotic hypoglycemia, such as fatty acid oxidation disorders. Further studies in a large-scale cohort are warranted to confirm our speculation.
Similar content being viewed by others


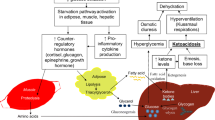
Background
Citrin deficiency (CD) is caused by biallelic pathogenic variants in SLC25A13 (MIM 603859) that encodes citrin, a mitochondrial membrane protein that is mainly expressed in the liver [1, 2]. Citrin protein exchanges aspartic acid with glutamic acid between the mitochondria and cytosol to maintain NADH and NAD+ balance [3]. The NADH/NAD+ ratios increase in the cytosol in patients with CD [4], resulting in NADH/NAD+ imbalance after glycolysis. Individuals with CD have unique preferences for low-carbohydrate and high-fat foods, which are probably associated with energy production balance [5]. CD often markedly causes hypoglycemia during childhood; thus, physicians should consider the possibility of CD in such children. The assessment of ketone body levels during hypoglycemia is crucial for differential diagnosis [6]. A previous study diagnosed this as ketosis based on the results of urine analysis and not those of blood analysis, at the time of the episode of hypoglycemia [7]. Urine ketone tests could overestimate the values owing to abnormal urine concentration and might not explain the appropriate blood ketone status at the time of assessment. However, blood ketone levels concomitant with symptomatic or severe hypoglycemia in patients with CD have not been studied.
Herein, we describe the case of a patient with CD who had repeated episodes of hypoglycemia with insufficient ketosis.
Case presentation
The patient was born at 37 weeks of gestation without adverse perinatal events and neonatal jaundice. His body weight and height at birth were 2530 g (− 0.39 SD) and 47.0 cm (− 0.63 SD), respectively. Newborn screening tests were negative. At the age of 1 year, he started presenting afebrile convulsions on days when he fell asleep without having milk at night. Laboratory examination indicated hypoglycemia (glucose levels, 0.83 [normal, 3.89–6.11] mM). He was transferred to a previous hospital to investigate the cause of the hypoglycemic episode. His height and body weight were 73.1 cm (− 0.7 SD) and 9.6 kg (+ 0.3 SD), respectively. He had a plump face but no hepatosplenomegaly. After 12 h of fasting, blood samples revealed hypoglycemia without ketosis (glucose, 1.61 mM; insulin, 1.2 pmol/L; total ketone bodies, 477 μM; acetoacetic acid, 87 μM; and 3-hydroxybutylate, 390 μM). Laboratory examination revealed mild liver injury and hypertriglyceridemia (AST, 94 [normal, 23–57] U/L; ALT, 66 [normal, 9–38] U/L; and TG, 8.81 [normal, 0.23–1.69] mM). The blood ACTH, cortisol, IGF-1, thyroid hormones, galactose, and amino acid levels were normal. Urine organic acid and serum acylcarnitine analyses with hypoglycemia showed nonspecific results. Glycogen storage disease (GSD) type Ia was suspected on the basis of these laboratory findings. The patient was given a high-glucose-containing formula for GSD to avoid hypoglycemia; however, he still presented repetitive hypoglycemia when fasting over 12 h. In total, three episodes of hypoglycemia with glucose levels below 2.5 mM were observed. At the age of 1 year and 1 month, he was referred to our institution to investigate the cause of repetitive hypoglycemia despite the consumption of a formula for GSD. Given the possibility of fatty acid oxidation disorders (FAODs) or GSDs, we measured sequential glucose and ketone levels in a fasting state as well as glucose loading (Table 1). A venous cannula was inserted on the last day; all feeding was stopped at 9 PM, and blood glucose and 3-hydroxybutyrate levels were checked every 3 h until 12 h or every 1 h after 12 h using portable blood glucose or ketone meters. After 13 h of fasting, the baby boy showed signs of hypoglycemia without severe ketosis [6, 8]. Shortly thereafter, he orally consumed 20 mL/kg of 10% glucose solution enthusiastically. His blood glucose levels increased, but his blood lactate levels remained unchanged before and after glucose loading. Serum acylcarnitine levels were mildly elevated in multiple acylcarnitines, which is sometimes observed in hypercatabolism. Urine organic acid analysis revealed nonketotic dicarboxylic aciduria. At the first outpatient visit after the test, the baby’s mother mentioned his food preferences: He particularly liked soybeans. This led to the suspicion of CD. Genetic testing revealed compound heterozygous variants of SLC25A13 (NM_014251: c.[1019_1177del]; [1813C > T]), both of which are reported as prevalent pathogenic variants [9], leading to a diagnosis of CD. The hypoglycemic episodes and mild liver injury disappeared after switching the high-glucose-containing formula for a medium-chain triglyceride (MCT)-containing formula (AST, 32 U/L and ALT, 16 U/L).
Discussion and conclusions
Here, we described the clinical history of a baby boy with repetitive episodes of hypoglycemia without adequate ketone production. The fasting tolerance test showed that the patient demonstrated hypoglycemia without appropriate ketone body elevation. A genetic test revealed pathogenic SLC25A13 gene variants and led to the diagnosis of CD; the use of MCT-containing formula resolved the hypoglycemic episodes.
We collected serial blood samples during a starvation test, allowing us to exactly determine the ketone body dynamics in response to hypoglycemia. Inappropriately low levels of ketones in the presence of severe hypoglycemia may provide a diagnostic clue for CD. Characterizing the metabolic profile using accessible and convenient laboratory tools could help physicians and patients because CD exhibits nonspecific symptoms and has little diagnostic evidence [10]. Moreover, it is important to recognize that a hypoglycemic state in CD is associated with insufficient ketone levels because relatively high ketone levels have been reported in patients with CD. A previous case of CD reported as ketotic hypoglycemia was determined using a urine sample, while the blood ketone levels might have been relatively low for blood glucose (0.78 mM) because 2+ result of qualitative tests for urine ketones would be approximately 400 μM of acetoacetic acid [7]. The blood ketone levels might be relatively low in the hypoglycemic situation. Moreover, another study describing ketotic hypoglycemia revealed a CD case with hypoglycemic convulsion [11], while blood ketone levels (total ketone body level was 1.8 mM) were certainly elevated but relatively low for the predicted compensatory change (expected total ketone body level was > 5 mM) [8]. This result should be considered as insufficient ketosis with hypoglycemia. Further large-scale cohort studies are required to verify our speculation.
The underlying mechanism of inappropriately low ketone levels with hypoglycemia in patients with CD is unknown; however, this hypoketotic state may be associated with the altered redox states of both cytosol and mitochondria in CD. The fasting test showed that the 3-hydroxybutylate to acetoacetic acid ratio was lower than expected, and that the reduction of 3-hydroxybutylate may be a secondary effect due to the elevation in cytosolic NADH/NAD+. The patient was first suspected of having GSD Ia and prescribed with a high-glucose formula to prevent hypoglycemic episodes. A high glucose level may increase NADH/NAD+ levels in the cytosol and disturb the NADH/NAD+ balance via glycolysis, and the shift in NADH/NAD+ possibly caused hypoglycemia in this patient [12]. The hypoglycemic episodes disappeared after switching from the high-glucose-containing formula to an MCT-containing formula. This could be due to the improvement of NAD+/NADH imbalance by MCT supplementation via an increase in the availability of NAD+ [13]. In addition, the NADH/NAD+ imbalance, which may be exacerbated by high glucose intake, might be associated with mild liver injury long after the infantile period when patients with CD usually present with cholestasis because NADH/NAD imbalance can lead to oxidative stress and damage in the hepatocyte [14].
The partial impairment of β-oxidation might possibly occur and could contribute to hypoketotic condition via peroxisome proliferator-activated receptor (PPAR) dysregulation; this hypothesis might have explained the phenotypic discrepancy between patients with CD and FAODs if these disorders commonly occurred due to errors in β-oxidation. PPARα, a subtype of PPAR, is enriched in the liver. It maintains lipid homeostasis and positively regulates multiple enzymes of β-oxidation [15]. A previous study reported that PPARA mRNA expression was downregulated but PPARD was not downregulated in patients CD [16]. In PPARs, the downregulation of PPARα and not PPARδ or PPARγ is associated with liver steatosis in patients with CD [16]; in contrast, PPARα seemed to be upregulated in patients with FAODs, such as in those with trifunctional protein deficiency and very-long-chain acyl-CoA dehydrogenase deficiency [17, 18]. Ppara knockout mice seemingly did not show any phenotypes; however, they showed hypoketotic hypoglycemia in a fasting condition and could easily affect patients with fatty liver, which is similar to that in CD [19]. Transgenic mice of Ppara and not Ppard or Pparg showed hypertrophic cardiomyopathy, which can result in FAODs [20, 21]. Thus, PPARα dysregulation could be related to pathogenicity in CD. In our patient, urine organic acid analysis revealed nonketotic dicarboxylic aciduria, which is consistent with the partial inhibition of β-oxidation. Moreover, the extremely high free fatty acid levels might result from the partial blockade of β-oxidation or physical and mental burden of the fasting test. MCT might help the hypoketotic state of the patient with CD to overexpress enzymes in β-oxidation [22, 23]. The difference between CD and FAODs may simply be due to the expressed tissues [1, 24]. Additional studies are warranted to confirm the relationship between PPARα dysregulation and phenotypes of CD.
In summary, CD might possibly cause hypoketotic hypoglycemia due to partial impairment of ketogenesis, and physicians should measure blood ketone levels in cases of unexplained hypoglycemia. It is important to consider the possibility of CD during the differential diagnosis of hypoketotic hypoglycemia because high-glucose intake may cause hyperglycemia and exacerbate the metabolic state rather than prevent hypoglycemia.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- CD:
-
Citrin deficiency
- FAOD:
-
Fatty acid oxidation disorder
- GSD:
-
Glycogen storage disease
- MCT:
-
Medium-chain triglyceride
- PPAR:
-
Peroxisome proliferator-activated receptor
References
Kobayashi K, Sinasac DS, Iijima M, et al. The gene mutated in adult-onset type II citrullinaemia encodes a putative mitochondrial carrier protein. Nat Genet. 1999;22(2):159–63. https://doi.org/10.1038/9667.
Ohura T, Kobayashi K, Tazawa Y, et al. Neonatal presentation of adult-onset type II citrullinemia. Hum Genet. 2001;108(2):87–90. https://doi.org/10.1007/s004390000448.
Palmieri L, Pardo B, Lasorsa FM, et al. Citrin and aralar1 are Ca(2+)-stimulated aspartate/glutamate transporters in mitochondria. EMBO J. 2001;20(18):5060–9. https://doi.org/10.1093/emboj/20.18.5060.
Saheki T, Kobayashi K. Mitochondrial aspartate glutamate carrier (citrin) deficiency as the cause of adult-onset type II citrullinemia (CTLN2) and idiopathic neonatal hepatitis (NICCD). J Hum Genet. 2002;47(7):333–41. https://doi.org/10.1007/s100380200046.
Okano Y, Ohura T, Sakamoto O, Inui A. Current treatment for citrin deficiency during NICCD and adaptation/compensation stages: strategy to prevent CTLN2. Mol Genet Metab. 2019;127(3):175–83. https://doi.org/10.1016/j.ymgme.2019.06.004.
Morris AA, Thekekara A, Wilks Z, Clayton PT, Leonard JV, Aynsley-Green A. Evaluation of fasts for investigating hypoglycaemia or suspected metabolic disease. Arch Dis Child. 1996;75(2):115–9. https://doi.org/10.1136/adc.75.2.115.
Hachisu M, Oda Y, Goto M, et al. Citrin deficiency presenting with ketotic hypoglycaemia and hepatomegaly in childhood. Eur J Pediatr. 2005;164(2):109–10. https://doi.org/10.1007/s00431-004-1549-z.
Bonnefont JP, Specola NB, Vassault A, et al. The fasting test in paediatrics: application to the diagnosis of pathological. Eur J Pediatr. 1990;150(2):80–5. https://doi.org/10.1007/bf02072043.
Kikuchi A, Arai-Ichinoi N, Sakamoto O, et al. Simple and rapid genetic testing for citrin deficiency by screening 11 prevalent mutations in SLC25A13. Mol Genet Metab. 2012;105(4):553–8. https://doi.org/10.1016/j.ymgme.2011.12.024.
Song Y-Z, Deng M, Chen F-P, et al. Genotypic and phenotypic features of citrin deficiency: five-year experience in a Chinese pediatric center. Int J Mol Med. 2011;28(1):33–40. https://doi.org/10.3892/ijmm.2011.653.
Otsuka H, Sasai H, Abdelkreem E, et al. Effectiveness of medium-chain triglyceride oil therapy in two Japanese Citrin-deficient siblings: evaluation using Oral glucose tolerance tests. Tohoku J Exp Med. 2016;240(4):323–8. https://doi.org/10.1620/tjem.240.323.
Zhao Y, Yang Y. Real-time and high-throughput analysis of mitochondrial metabolic states in living cells using genetically encoded NAD(+)/NADH sensors. Free Radic Biol Med. 2016;100:43–52. https://doi.org/10.1016/j.freeradbiomed.2016.05.027.
Hayasaka K, Numakura C, Toyota K, et al. Medium-chain triglyceride supplementation under a low-carbohydrate formula is a promising therapy for adult-onset type II citrullinemia. Mol Genet Metab Rep. 2014;1:42–50. https://doi.org/10.1016/j.ymgmr.2013.12.002.
Wu J, Jin Z, Zheng H, Yan L-J. Sources and implications of NADH/NAD(+) redox imbalance in diabetes and its complications. Diabetes Metab Syndr Obes. 2016;9:145–53. https://doi.org/10.2147/DMSO.S106087.
Wang Y, Nakajima T, Gonzalez FJ, Tanaka N. PPARs as metabolic regulators in the liver: lessons from liver-specific PPAR-null mice. Int J Mol Sci. 2020;21(6):2061. https://doi.org/10.3390/ijms21062061.
Komatsu M, Kimura T, Yazaki M, et al. Steatogenesis in adult-onset type II citrullinemia is associated with down-regulation of PPARalpha. Biochim Biophys Acta. 2015;1852(3):473–81. https://doi.org/10.1016/j.bbadis.2014.12.011.
Wakabayashi M, Kamijo Y, Nakajima T, et al. Fatty acid accumulation and resulting PPARα activation in fibroblasts due to trifunctional Protein deficiency. PPAR Res. 2012;2012:371691. https://doi.org/10.1155/2012/371691.
Yang Y, Feng Y, Zhang X, et al. Activation of PPAR<i>α</i> by fatty acid accumulation enhances fatty acid degradation and Sulfatide synthesis. Tohoku J Exp Med. 2016;240(2):113–22. https://doi.org/10.1620/tjem.240.113.
Kersten S, Seydoux J, Peters JM, Gonzalez FJ, Desvergne B, Wahli W. Peroxisome proliferator–activated receptor α mediates the adaptive response to fasting. J Clin Invest. 1999;103(11):1489–98. https://doi.org/10.1172/JCI6223.
Finck BN, Lehman JJ, Leone TC, et al. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J Clin Invest. 2002;109(1):121–30. https://doi.org/10.1172/JCI14080.
Burkart EM, Sambandam N, Han X, et al. Nuclear receptors PPARbeta/delta and PPARalpha direct distinct metabolic regulatory programs in the mouse heart. J Clin Invest. 2007;117(12):3930–9. https://doi.org/10.1172/JCI32578.
Iemitsu M, Shimojo N, Maeda S, et al. The benefit of medium-chain triglyceride therapy on the cardiac function of SHRs is associated with a reversal of metabolic and signaling alterations. Am J Physiol Heart Circ Physiol. 2008;295(1):H136–44. https://doi.org/10.1152/ajpheart.01417.2006.
Liberato MV, Nascimento AS, Ayers SD, et al. Medium chain fatty acids are selective peroxisome proliferator activated receptor (PPAR) γ activators and pan-PPAR partial agonists. Moschetta A, ed. PloS one. 2012;7(5):e36297. https://doi.org/10.1371/journal.pone.0036297.
Uhlén M, Fagerberg L, Hallström BM, et al. Proteomics. Tissue-based map of the human proteome. Science (New York, NY). 2015;347(6220):1260419. https://doi.org/10.1126/science.1260419.
Acknowledgments
Not applicable.
Funding
This work was supported by JSPS KAKENHI Grant Number 20 K16845 (to YW).
Author information
Authors and Affiliations
Contributions
YW conceptualized and designed the study, collected data, drafted the initial manuscript, and reviewed and revised the manuscript. NA-I and OS conceptualized and designed the study, collected data, and reviewed and revised the manuscript. AK and KS interpreted the data and reviewed and revised the manuscript. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was performed in accordance with the Declaration of Helsinki and was approved by the Ethics Committee of Tohoku University Hospital (approval number: 17946). The patient’s parents provided written informed consent.
Consent for publication
Written informed consent was obtained from the parents for the publication of this case report.
Competing interests
The authors have no conflicts of interest to disclose.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wada, Y., Arai-Ichinoi, N., Kikuchi, A. et al. Hypoketotic hypoglycemia in citrin deficiency: a case report. BMC Pediatr 20, 444 (2020). https://doi.org/10.1186/s12887-020-02349-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12887-020-02349-6