
Abstract
Background
Newborn screening (NBS) for cystic fibrosis (CF) not only identifies infants with a diagnosis of CF, but also those with an uncertain diagnosis of cystic fibrosis (CF), i.e. CF transmembrane conductance regulator (CFTR)-related metabolic syndrome (CRMS) or CF screen positive inconclusive diagnosis (CFSPID). These infants have an uncertain long-term outcome and it is currently unclear around time of diagnosis, which infants are at higher risk of later fulfilling a CF diagnosis. In this study, we hypothesised that immunoreactive trypsinogen (IRT) levels, used in NBS as a marker of pancreatic disease and function, may reflect the degree of CFTR dysfunction in each individual and therefore would help to identify those with CRMS/CSPID who are later at risk for meeting the criteria of CF.
Methods
In this longitudinal, prospective study, infants with CRMS/CFSPID and CF were recruited and followed in 9 CF clinics (Canada and Italy). We compared NBS IRT levels between CF and CRMS/CFSPID, and between children with CRMS/CFSPID→CF and CRMS/CFSPID→CRMS/CFSPID during the period of June 2007 to April 2016.
Results
Ninety eight CRMS/CFSPID and 120 CF subjects were enrolled. During the study period, 14 (14.3%) CRMS/CFSPID subjects fulfilled the diagnostic criteria for CF (CRMS/CFSPID→CF), while the diagnosis remained uncertain (CRMS/CFSPID→ CRMS/CFSPID) in 84 (85.7%) subjects. Significantly higher NBS IRT concentrations (ng/ml) were present in CF than CRMS/CFPSID (median (interquartile range): 143.8 (99.8–206.2) vs. 75.0 (61.0–105.9); P < 0.0001). Infants with CRMS/CFSPID→CF (n = 14) had significantly higher NBS IRT concentrations (ng/ml) than CRMS/CFSPID→ CRMS/CFSPID (n = 83) (median (interquartile range): 108.9 (72.3–126.8) vs. 73.7(60.0–96.0); P = 0.02).
Conclusions
Amongst infants who tested positive on NBS for CF, there is a gradation of elevated NBS IRT concentrations. Infants with CF have higher NBS IRT levels than CRMS/CFPSID, and higher NBS IRT concentrations were present in infants with CRMS/CFSPID→CF than CRMS/CFSPID→ CRMS/CFSPID. NBS IRT concentrations, in concert with other factors, may have the potential to predict the likelihood of CF amongst infants with CRMS/CFSPID.
Similar content being viewed by others


Background
Cystic fibrosis (CF) is a life-limiting, autosomal recessive disease caused by pathogenic mutations in the gene encoding the CF transmembrane conductance regulator (CFTR) protein. With the availability and expansion of newborn screening (NBS) programs for CF, a new subgroup of infants with a positive NBS result, but an inconclusive diagnosis of CF has emerged. These infants, who have an uncertain long-term outcome (CF vs. CFTR-related disorder vs. healthy), have been designated as CF transmembrane conductance regulator (CFTR)-related metabolic syndrome (CRMS) or CF screen positive inconclusive diagnosis (CFSPID) (also known as CRMS/CFSPID in the most recent international joint consensus) [1, 2].
Since the first report of newborns with CF having elevated immunoreactive trypsinogen (IRT) levels [3], IRT measurements have become central to NBS protocols worldwide. Immunoreactive trypsinogen levels reflect severity of pancreatic disease and pancreatic function [4,5,6], which in turn is associated with the severity of the CFTR gene defect. Higher and more rapid decline in IRT levels are associated with more severe CFTR variants (Class I-III), while lower levels are observed with less severe variants (Class IV-VI) [7,8,9]. There are also non-CFTR related factors that can lead to a high neonatal IRT level, including stressful or prolonged labor, Black race, and trisomy 21 [1]. We hypothesised that IRT levels may also reflect the severity of the CFTR gene defect among children with CRMS/CFSPID and would be different between children with CRMS/CFSPID who subsequently met the criteria for CF (CRMS/CFSPID→CF) and those whose diagnosis remained uncertain (CRMS/CFSPID→ CRMS/CFSPID). In this study, we compared IRT measured at NBS to differentiate between CF and CRMS/CFSPID, and between children with CRMS/CFSPID who subsequently met the criteria for CF (CRMS/CFSPID→CF) and those whose diagnosis remained uncertain (CRMS/CFSPID→ CRMS/CFSPID).
Materials and methods
Subjects
A clinical-research protocol was developed to evaluate and follow infants with a diagnosis of CRMS/CFSPID in a prospective, longitudinal manner from 9 CF clinics across 5 provinces in Canada (Ontario, British Columbia, Alberta, Saskatchewan and Nova Scotia) and from Verona, Italy. Interim data for the period between June 2007 and April 2016 are reported here. Some of the subjects have been previously reported in a separate study [10].
As part of NBS protocol, dried blood spots were collected in the first 2 days of life. Analyses for IRT levels from these samples were performed using the AutoDELFIA method (Perkin-Elmer Life Sciences, Boston, USA). Accordingly, an infant was considered to be NBS-positive if: [1] IRT exceeded the site-specific cut-off, in addition to at least one CFTR mutation (and/or raised meconium lactase in Verona, Italy); or, [2] IRT exceeded the 99.9th centile when no mutations were identified. All infants who were identified as NBS-positive were then referred to their local CF centre for sweat testing and genotyping to determine whether they fulfilled the diagnostic criteria for CF (based on sweat chloride ≥60 mmol/L and/or CF-causing mutations on both alleles) [1].
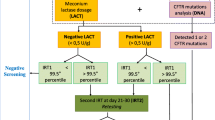
This study was designed intentionally to reflect real-life experiences in the CF clinic following a positive NBS, during which parents/carers are informed as to whether their infant has a diagnosis of CF or CRMS/CFSPID (Fig. 1). For the latter, NBS-positive infants at each centre were defined and entered into the study as CRMS/CFSPID if they had at time of initial diagnosis an indeterminate diagnostic outcome: [1] sweat chloride < 30 mmol/L and 2 CFTR mutations with 0–1 CF-causing CFTR mutations; or [2] sweat chloride 30–59 mmol/L and < 2 CF-causing CFTR mutations [1, 2]. As a comparator group, NBS-positive infants with a clear-cut diagnosis of CF were also enrolled during the same period. Infants who presented with meconium ileus and fulfilled the diagnostic criteria for CF but were negative on NBS (i.e. false negative) were also included as part of the comparator group. Immunoreactive trypsinogen levels obtained at NBS from NBS-positive infants with CRMS/CFSPID and the comparator group of infants with CF were collected for the study.

Flowchart of the study summarising time of initial diagnosis and during the study follow-up period
In our clinical-research protocol, subjects with CRMS/CFSPID were clinically monitored and sweat tested 6 monthly in the first 2 years of life, and annually thereafter. Genotype information was obtained from NBS mutation analyses. Subjects would undergo confirmatory or further genotyping including CFTR gene sequencing, especially for infants identified with less than two CFTR mutations, usually prior to or at time of initial visit/diagnosis. Some of the CRMS/CFSPID subjects were subsequently reclassified as CF during the follow-up period (CRMS/CFSPID→CF) if they at a later time point fulfilled the diagnostic criteria for CF (either repeat sweat chloride levels ≥60 mmol/L or abnormal genotype from reassignment of previously indeterminate CFTR mutation to “CF-causing”) i.e. the participant’s diagnostic label and categorisation/re-categorisation in this prospective study occurred months after the initial diagnosis of CRMS/CFSPID and reflected real life reassignment of diagnosis in clinical practice.
Statistical analysis
Descriptive statistics were presented according to the normality of the data distribution. Continuous data was presented as mean with standard deviation (SD) or median (interquartile range). Comparisons of NBS IRT concentrations were made using the Mann-Whitney test. IBM SPSS Statistics (v 23.0; SPSS Inc., Chicago, Illinois) were used. P-values < 0.05 were considered statistically significant.
Results
Subject characteristics
Out of 218 subjects, 98 CRMS/CFSPID and 120 CF subjects were enrolled. Among the 98 CRMS/CFSPID subjects, 97 subjects had two mutations (no more than one is CF-causing) with intermediate (n = 47) or normal (n = 50) sweat chloride, and 1 subject had one mutation (CF-causing) with intermediate sweat chloride. Mutations were either identified by NBS and/or by confirmatory or further genotyping via the CF clinic prior to or at time of initial visit/diagnosis. During the study period, 14 (14.3%) CRMS/CFSPID subjects fulfilled the diagnostic criteria for CF (CRMS/CFSPID→CF) (Table 1 and Fig. 1), while the diagnosis remained uncertain (CRMS/CFSPID→ CRMS/CFSPID) in 84 (85.7%) subjects. Among the 14 subjects with CRMS/CFSPID→CF, 10 had subsequent elevations of sweat chloride levels ≥60 mmol/L (with 5 based on abnormal sweat test alone and 5 based on both abnormal sweat test and genotype), and 4 based on abnormal genotype alone (Table 1 and Fig. 1). Among the 10 (out of 14) CRMS/CFSPID→CF patients who had abnormal sweat test levels (≥60 mmol/L) on follow-up sweat testing, sweat chloride increased to abnormal levels (mean [SD] of 68.30 [5.60] mmol/L) at a mean (SD) age of 35.97 (21.43) months.
Immunoreactive trypsinogen levels
CRMS/CFSPID vs. CF
Infants with CF had significantly higher NBS IRT concentrations (ng/ml) (median (interquartile range)) than CRMS/CFPSID (143.8 (99.8–206.2) vs. 75.0 (61.0–105.9); P < 0.0001) (Fig. 2).

Comparison of newborn screening IRT levels between children with CF and CRMS/CFSPID
CRMS/CFSPID→CF vs. CRMS/CFSPID→CRMS/CFSPID
Infants with CRMS/CFSPID→CF (n = 14) had significantly higher NBS IRT concentrations (ng/ml) than CRMS/CFSPID→ CRMS/CFSPID (n = 83) (median (interquartile range): 108.9 (72.3–126.8) vs. 73.7(60.0–96.0); P = 0.02) (Fig. 3).

Comparison of newborn screening IRT levels between children with CRMS/CFSPID who subsequently met the criteria for CF (CFSPID→CF) and those whose diagnosis remained uncertain (CFSPID→CFSPID)
Further comparisons of the diagnostic outcomes are shown in Fig. 4 based on initial sweat chloride levels (i.e. done closest to NBS IRT) plotted against NBS IRT concentrations. While still overlapping with the CRMS/CFSPID→ CRMS/CFSPID cohort, infants with CRMS/CFSPID→CF aggregate much closer to the CF cohort compared to the overall CRMS/CFSPID→ CRMS/CFSPID group.

Initial sweat chloride measurements plotted against IRT levels for children with CF (represented by open circle = ○), children with CRMS/CFSPID who subsequently met the criteria for CF (CFSPID→CF; solid triangle = ▲) and those whose diagnosis remained uncertain (CFSPID→CFSPID; open square = □)
Discussion
The outcomes of infants with CRMS/CFSPID remain uncertain. In this prospective longitudinal study of children with CRMS/CFSPID, we identified the potential role for NBS IRT as a biomarker to predict the likelihood of having CF, with significantly different IRT levels between CF and CRMS/CFSPID, and also between CRMS/CFSPID→CF and CRMS/CFSPID→ CRMS/CFSPID.
Our findings have biological plausibility. Among all organ systems affected by CF disease, the exocrine pancreas serves as a phenotypic barometer of CFTR function and correlates well with the underlying genotype [11,12,13]. The pancreatic insufficiency prevalence (PIP) scores, based on the exocrine pancreatic phenotype, have been shown to accurately distinguish the severity of different CFTR mutations [13]. In this study, we speculate that IRT levels reflect the severity of pancreatic disease and thus the severity of the underlying CFTR gene mutation, with higher levels during the first few years of life indicating more extensive pancreatic damage correlating with more severe CFTR dysfunction, akin to higher IRT levels in newborns with CF compared to non-CFs. Our observations are in keeping with the association between trypsinogen levels and the likelihood of having CF disease. However, this relationship is likely complex and non-linear (as observed in Fig. 3). Furthermore, in two separate studies, higher IRT levels were observed in children with two CF-causing mutations compared to those with either one mutation of varying clinical consequence [7] or one non-CF causing mutation [8]. These findings may be highly relevant clinically, as to date, aside from diagnostic tests such as repeat sweat testing, extensive genotyping or other functional CFTR testing, there is no test that predicts or stratifies the risk of a later CF diagnosis following an initial diagnosis of CRMS/CFSPID. In our previous study comparing CRMS/CFSPID→ CRMS/CFSPID and CRMS/CFSPID→CF groups, there were no significant differences in respiratory symptoms and microbiology present [10]. Furthermore, IRT levels are available at the time of diagnosis and thus limiting the delays for risk stratification.
The diagnosis of CF is a dynamic and evolving process among individuals with an indeterminate diagnosis. There is continuous new discovery on the pathogenicity of rare CFTR gene variants due to the ongoing efforts of the CFTR2 project (http://cftr2.org). Furthermore, we have previously demonstrated increases in sweat chloride concentrations over time in a subset of these individuals [10]. In this current study, all 14 subjects with an initial diagnosis of CRMS/CFSPID carried two CFTR variants. Of those, 9 infants were later reassigned a diagnosis of CF (CRMS/CFSPID→CF) based on genotype; 5 of these 9 subjects also had increases in sweat chloride levels above the 60 mmol/L cut-off for CF diagnosis with time while 4 of 9 subjects met the diagnosis of CF by genotyping alone. Thus our experience provides support for comprehensive genotyping in these individuals. While many of these individuals already had their 2nd CFTR mutation identified at time of assessment, these mutations were not considered disease-causing until later in the course of follow-up (with the roll-out of new pathogenic CFTR variants by CFTR2). Since CFTR2 is based on CF patient registries, it is worth noting though that the primary goal of CFTR2 is to identify the most common CF-causing variants rather than the most common non CF-causing variants, which is important in helping distinguish CRMS/CFSPID→ CRMS/CFSPID from CRMS/CFSPID→CF in the long-term. The remaining 5 subjects later fulfilled the diagnosis of CF based on sweat testing alone. As this was a prospective, longitudinal study all these 14 individuals were classified as CRMS/CFSPID and not CF at the time of presentation and recruitment (i.e. as it occurred in real-life clinical practice).
The present study has limitations. Our observations are based on a small sample size, particularly with the CRMS/CFSPID→CF cohort despite the multicentre and multiyear approach of this study. Future efforts would include confirming our observation in another cohort and then either defining a sensitive IRT cut-off or identifying trajectory of longitudinal IRT levels (i.e. levels over time or age) to predict risk of developing CF based on a larger sample size. Due to the multicentre and multinational approach, subjects were identified based on different NBS protocols used. This study also did not account for variability in IRT levels within and across different provinces, and variations in IRT concentration among different races, birth weight and gestational age. However, the phenomenon of identifying CRMS/CFSPID is ubiquitous irrespective of the variations in NBS algorithms and despite this, there were significant differences in IRT levels between the different cohorts of children.
In conclusion, predicting outcomes of children with CRMS/CFSPID remains a challenge, requiring careful monitoring over time. Newborn screening IRT levels are significantly different between children with CRMS/CFSPID who subsequently met the criteria for CF and those whose diagnosis remained uncertain, and may have the potential to predict the likelihood of later fulfilling the diagnostic criteria for CF.
Availability of data and materials
The datasets generated and/or analysed during the current study are not publicly available as the study is ongoing.
Abbreviations
- CF:
-
Cystic fibrosis
- CFSPID:
-
Cystic fibrosis screen positive inconclusive diagnosis
- CFTR:
-
CF transmembrane conductance regulator
- CRMS:
-
CFTR-related metabolic syndrome
- IRT:
-
Immunoreactive trypsinogen
- NBS:
-
Newborn screening
References
Farrell PM, White TB, Ren CL, et al. Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation. J Pediatr. 2017:181S:S4–S15.e1.
Ren CL, Borowitz DS, Gonska T, Howenstine MS, Levy H, Massie J, Milla C, Munck A, Southern KW. Cystic Fibrosis Transmembrane Conductance Regulator-Related Metabolic Syndrome and Cystic Fibrosis Screen Positive, Inconclusive Diagnosis. J Pediatr. 2017:181S:S45–S51.e1.
Crossley JR, Elliott RB, Smith PA. Dried-blood spot screening for cystic fibrosis in the newborn. Lancet. 1979;1:472–4.
Couper RT, Corey M, Durie PR, et al. Longitudinal evaluation of serum trypsinogen measurement in pancreatic-insufficient and pancreatic-sufficient patients with cystic fibrosis. J Pediatr. 1995;127(3):408–13.
Durie PR, Forstner GG, Gaskin KJ, et al. Age-related alterations of immunoreactive pancreatic cationic trypsinogen in sera from cystic fibrosis patients with and without pancreatic insufficiency. Pediatr Res. 1986;20(3):209–13.
Soave D, Miller MR, Keenan K, et al. Evidence for a causal relationship between early exocrine pancreatic disease and cystic fibrosis-related diabetes: a Mendelian randomization study. Diabetes. 2014;63(6):2114–9.
Salinas DB, Sosnay PR, Azen C, Young S, Raraigh KS, Keens TG, Kharrazi M. Benign and deleterious cystic fibrosis transmembrane conductance regulator mutations identified by sequencing in positive cystic fibrosis newborn screen children from California. PLoS One. 2016;11(5):e0155624.
Salinas DB, Sosnay PR, Azen C, Young S, Raraigh KS, Keens TG, Kharrazi M. Benign outcome among positive cystic fibrosis newborn screen children with non-CF-causing variants. J Cyst Fibros. 2015;14(6):714–9.
Sontag MK, Corey M, Hokanson JE, Marshall JA, Sommer SS, Zerbe GO, Accurso FJ. Genetic and physiologic correlates of longitudinal immunoreactive trypsinogen decline in infants with cystic fibrosis identified through newborn screening. J Pediatr. 2006;149(5):650–7.
Ooi CY, Castellani C, Keenan K, Avolio J, Volpi S, Boland M, et al. Inconclusive diagnosis of cystic fibrosis after newborn screening. Pediatrics. 2015;135:e1377–85.
Ahmed N, Corey M, Forstner G, et al. Molecular consequences of cystic fibrosis transmembrane regulator (CFTR) gene mutations in the exocrine pancreas. Gut. 2003;52(8):1159–64.
Ooi CY, Durie PR. Cystic fibrosis transmembrane conductance regulator (CFTR) gene mutations in pancreatitis. J Cyst Fibros. 2012;11(5):355–62.
Ooi CY, Dorfman R, Cipolli M, et al. Type of CFTR mutation determines risk of pancreatitis in patients with cystic fibrosis. Gastroenterology. 2011;140(1):153–61.
Acknowledgements
The authors thank all the doctors, research nurses and coordinators: Louise Taylor, Karen Tam, Julie Avolio (The Hospital for Sick Children, Toronto); Dr. Lenna Morgan (Windsor Regional Hospital, Ontario); Lori Fairservice and Melissa Soles (Alberta Children’s Hospital, Calgary); Shannon Gregory, Amanda Jober and Angela MacDonald (Stollery Children’s Hospital, Edmonton); Adrianna Breen and Natalie Henderson (Hotel Dieu Hospital, Kingston); Monica Dawe, Erin Fleischer and Jennifer Itterman (Children’s Hospital Of Western Ontario, London); Anne Smith (Children’s Hospital Of Eastern Ontario, Ottawa); Caroline Burgess, Amy Schellenberg and Vanessa McMahon (British Columbia’s Children Hospital, Vancouver); Lorna Kosteniuk (Royal University Hospital, Saskatoon); Margaret Surette and Paula Barrett (IWK Health Centre, Halifax); Sonia Volpi, Anna Tamanini, Ciro D’Orazio, Giovanna Amenta, Elisa Calcaterra Nicoletta Vardaro, Ilaria Meneghelli (Azienda Ospedaliera Universitaria Integrata di Verona, Verona, Italy).
Funding
Cystic Fibrosis Canada. The funding body had no role in the design of the study, collection, analysis, interpretation of data, and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
CYO, FR and TG were involved in study conception and design, participant recruitment, data analysis and interpretation, and wrote the draft and final manuscript. RS was involved in the data analysis and interpretation, provided critical review of the manuscript, and approved the final manuscript. CC was involved in study design, participant recruitment, provided critical review of the manuscript, and approved the final manuscript. KK was involved in study coordination, participant recruitment, data collection and database development, provided critical review of the manuscript, and approved the final manuscript. MB, JR, CB, MAC, RvW, SK, AP, DM-C, DH, MS, PZ, JB were involved in participant recruitment, provided critical review of the manuscript, and approved the final manuscript. PRD was involved in study conception and design and participant recruitment. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethics approvals from all participating sites (The Hospital for Sick Children, Toronto; Cystic Fibrosis Centre, Azienda Ospedaliera Universitaria Integrata di Verona, Verona; Children’s Hospital of Eastern Ontario, Ottawa; Alberta Children’s Hospital, Calgary; BC Children’s Hospital, Vancouver; Hotel Dieu Hospital, Kingston; Children’s Hospital Of Western Ontario, London; IWK Health Centre, Halifax; Stollery Children’s Hospital, Edmonton; Royal University Hospital, Saskatoon) were obtained. Informed written consent was obtained from all parents or legal guardians.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ooi, C.Y., Sutherland, R., Castellani, C. et al. Immunoreactive trypsinogen levels in newborn screened infants with an inconclusive diagnosis of cystic fibrosis. BMC Pediatr 19, 369 (2019). https://doi.org/10.1186/s12887-019-1756-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12887-019-1756-4