
Abstract
Background
Perinatal asphyxia and resulting hypoxic-ischemic encephalopathy is a major cause of death and long-term disability in term born neonates. Up to 20,000 infants each year are affected by HIE in Europe and even more in regions with lower level of perinatal care. The only established therapy to improve outcome in these infants is therapeutic hypothermia. Allopurinol is a xanthine oxidase inhibitor that reduces the production of oxygen radicals as superoxide, which contributes to secondary energy failure and apoptosis in neurons and glial cells after reperfusion of hypoxic brain tissue and may further improve outcome if administered in addition to therapeutic hypothermia.
Methods
This study on the effects of ALlopurinol in addition to hypothermia treatment for hypoxic-ischemic Brain Injury on Neurocognitive Outcome (ALBINO), is a European double-blinded randomized placebo-controlled parallel group multicenter trial (Phase III) to evaluate the effect of postnatal allopurinol administered in addition to standard of care (including therapeutic hypothermia if indicated) on the incidence of death and severe neurodevelopmental impairment at 24 months of age in newborns with perinatal hypoxic-ischemic insult and signs of potentially evolving encephalopathy. Allopurinol or placebo will be given in addition to therapeutic hypothermia (where indicated) to infants with a gestational age ≥ 36 weeks and a birth weight ≥ 2500 g, with severe perinatal asphyxia and potentially evolving encephalopathy. The primary endpoint of this study will be death or severe neurodevelopmental impairment versus survival without severe neurodevelopmental impairment at the age of two years. Effects on brain injury by magnetic resonance imaging and cerebral ultrasound, electric brain activity, concentrations of peroxidation products and S100B, will also be studied along with effects on heart function and pharmacokinetics of allopurinol after iv-infusion.
Discussion
This trial will provide data to assess the efficacy and safety of early postnatal allopurinol in term infants with evolving hypoxic-ischemic encephalopathy. If proven efficacious and safe, allopurinol could become part of a neuroprotective pharmacological treatment strategy in addition to therapeutic hypothermia in children with perinatal asphyxia.
Trial registration
NCT03162653, www.ClinicalTrials.gov, May 22, 2017.
Similar content being viewed by others


Background
Neonatal hypoxic-ischemic encephalopathy (HIE) as a result of perinatal asphyxia is a major cause of death and long-term disability in term neonates. About 1–4 per 1000 live births and consequently about 5–20,000 infants per year are affected in Europe [1]. In regions with lower level perinatal care it is even more common. HIE affects about 1 million infants worldwide each year.
Up to now, the only established therapy to improve outcome in infants with HIE is therapeutic hypothermia [2, 3]. However, despite therapeutic hypothermia and modern supportive neonatal intensive care, 45–50% of the infants with moderate or severe HIE (i.e., 2500–10,000 infants per year in Europe) still die or suffer from long-term neurodevelopmental impairment (NDI) such as cerebral palsy (CP), cognitive or behavioral problems [2, 4]. Therefore, additional therapies, including pharmacotherapy, are investigated to further improve the neurodevelopmental outcome of infants with HIE.
One of the potential beneficial pharmacological interventions is allopurinol. Allopurinol is a xanthine oxidase inhibitor, which reduces the production of oxygen radicals, most importantly of superoxide [5]. Superoxide radicals damage mitochondria resulting in secondary energy failure and apoptosis affecting neurons and glial cells after reperfusion of hypoxic brain tissue, this is called reperfusion injury [6, 7]. This reperfusion injury leads to additional brain injury occurring in the hours after birth and may affect much larger areas of brain tissue than the area primarily affected during the sentinel event [7]. Superoxide production, which is reduced by allopurinol, reaches its peak within 30 min after birth and therefore early administration is important to reduce reperfusion injury [8]. Furthermore, allopurinol, especially in higher concentrations, possibly chelates non-protein bound iron and scavenges the hydroxyl free radicals [9, 10]. Allopurinol also prevents adenosine degradation, which is an anti-excitatory neuromodulator [11]. Thereby, allopurinol might reduce reperfusion injury and improve outcome in neonates with HIE.
Several preclinical and three small clinical studies in neonates with HIE suggested a possible neuroprotective effect of allopurinol (recently reviewed in Annink et al. [8]). In the first two studies of van Bel et al. and Benders et al. allopurinol was administered within 4 h after birth. Allopurinol improved neurodevelopmental outcome in infants with moderate HIE, but not in severe HIE [12,13,14]. Gunes et al. administered allopurinol for three days and found improved outcome at one year of age [15]. All three studies were conducted before therapeutic hypothermia became standard of care, so the effect of allopurinol in addition to therapeutic hypothermia has not been investigated yet.
Based on the hypothesis that administration within 4 h after birth was too late to achieve full neuroprotective effect, allopurinol was administered antenatally in case of suspected hypoxia in the antenatal allopurinol trial for reduction of birth asphyxia induced brain damage (ALLO-trial) [16]. In girls, biomarkers as S100B were reduced in the allopurinol group compared to the placebo group. However, there was substantial overtreatment on the one hand and on the other moderately and severely asphyxiated infants were often missed [16].
Consequently, in this study on the effects of ALlopurinol in addition to hypothermia treatment for hypoxic-ischemic Brain Injury on Neurocognitive Outcome (ALBINO), allopurinol will be administered intravenously within 30 (max. 45) minutes after birth to optimize the timing and inhibition of superoxide formation in asphyxiated infants with evolving HIE.
Importantly, in all antenatal and neonatal studies in HIE, no severe side-effects were seen [12, 13, 15,16,17,18,19]. Also, in other neonatal populations, such as preterm infants and infants with congenital cardiac abnormalities, no severe side effects have been reported following (intravenous or oral) administration of allopurinol [20,21,22,23,24]. In the ALLO-trial, 4.5% of the mothers who received allopurinol had an irritation of the perivascular tissue, caused by the high pH of the allopurinol solution, but this was reversible in all cases [16]. In adults, a rare hypersensitivity reaction to allopurinol has been described after daily administration for a median of two to three weeks [25, 26]. An allopurinol sensitivity reaction in neonates has never been reported and is expected to be extremely unlikely.
Methods/design
Trial objectives
The main objective of the ALBINO trial is to evaluate the effect of early postnatal allopurinol administered in addition to standard of care (including therapeutic hypothermia if indicated) on the incidence of death and severe NDI at 24 months of age in newborns with HIE.
Secondly, safety of early postnatal intravenous allopurinol will be evaluated, as well as the pharmacokinetic profile of intravenous allopurinol and the short-term effect of early allopurinol on brain injury assessed by magnetic resonance imaging (MRI) of the brain, cerebral ultrasound, heart function assessed by echocardiography, electro-encephalography (EEG), and biochemical biomarkers.
Trial design
The ALBINO trial is a European double-blinded randomized placebo-controlled parallel group multicenter trial for superiority of allopurinol versus placebo (mannitol) in addition to therapeutic hypothermia where indicated (Phase III). More than 60 hospitals in ten countries will participate in this study, and ALBINO may expand to additional sites in further countries, after appropriate approvals have been obtained from ethics committees and authorities.
Population
Term and near-term infants (≥36 weeks) with severe perinatal asphyxia and potentially evolving encephalopathy can be included in the ALBINO trial:
Inclusion criteria
Infants must meet at least one of the following five criteria of severe perinatal asphyxia: 1) umbilical or postnatal blood gas within 30 min after birth with a pH < 7.0 or 2) with a base deficit ≥16 mmol/l; 3) need for ongoing cardiac massage at/beyond 5 min postnatally; 4) need for adrenalin administration during resuscitation and/or 5) Apgar score ≤ 5 at 10 min after birth.
Further, the infant must meet two out of the following four criteria for potentially evolving encephalopathy to participate in the study: 1) altered state of consciousness (reduced or absent response to stimulation or hyperexcitability); 2) severe muscular hypotonia or hypertonia; 3) absent or insufficient spontaneous respiration (i.e. gasping only) with need for respiratory support at 10 min postnatally and/or 4) abnormal primitive reflexes (absent suck/gag/ corneal/Moro reflex) or abnormal movements (i.e. potential clinical correlates of seizure activity).
Exclusion criteria
Infants will be excluded if the gestational age is below 36 weeks, birth weight is below 2500 g, in the presence of severe congenital malformations or syndromes requiring neonatal surgery or affecting long-term outcome. Furthermore, infants will be excluded if their postnatal age is > 30 min at the end of the screening phase, the neonate is considered “moribund” or “non-viable”, there is a decision of ‘comfort care only’ before study drug administration or if parents decline study participation.
Randomization and allocation concealment
Randomization will be performed with randomization software (Randlist Version 1.2) in blocks of four and stratified per center.
Randomization will be performed by allocation of the next consecutive study medication box (including first and second vial of study medication and two vials with sterile water for reconstitution) to an infant.
Study intervention
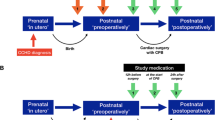
Infants included in the ALBINO trial will receive either allopurinol or placebo (mannitol). Study medication will be administered by intravenous infusion in one or two doses (see Fig. 1). The first dose (20 mg/kg body mass reconstituted in 2.0 ml/kg sterile water for infusion) will be given as soon as possible after birth. The start of infusion of study medication should preferably be within 30 min after birth, but no later than 45 min after birth.

Study interventions in ALBINO
The second dose (10 mg/kg body mass reconstituted in 1.0 ml/kg sterile water for infusion) will be administered 12 h after the first dose. This second dose will only be administered to infants treated with therapeutic hypothermia. Infants who recover quickly and do not qualify for and hence do not undergo hypothermia will not receive a second dose.
Placebo (mannitol) will be given in the same dose, volume and intervals as allopurinol.
Concomitant interventions and medication
Any concomitant medication that is medically necessary for the patients will be allowed in the study, except open-label allopurinol in any dosage and any application mode.
Where indicated according to respective national standards or treatment protocols, hypothermia treatment (whole body cooling to 33.5 °C for 72 h) should be started as soon as possible according to local protocols [3, 27].
Primary outcome
The primary endpoint will be death or severe NDI versus survival without severe NDI at the age of two years. Severe NDI is hereby defined as any of the following: cognitive or language delay defined as a cognitive-composite-score or a language-composite-score on the Bayley Scales of Infant and Toddler Development (3rd edition) < 85 and/or cerebral palsy (CP) according to the Surveillance of Cerebral Palsy in Europe (SCPE) criteria.
Secondary and further outcomes
The primary endpoint will be reconstituted as dichotomized composite secondary endpoint (survival without NDI versus Death or language-composite-score < 85 or cognitive-composite-score < 85 or CP present). Furthermore, the incidences of death and CP and the composite scores derived from the Bayley test (continuous and dichotomized) as well as the Gross Motor Function Classification Score will be analyzed as secondary outcome variables.
Further important secondary outcome parameters are brain injury assessed by MRI of the brain, cerebral ultrasound, amplitude-integrated EEG, full scale multichannel EEG, heart function assessed by echocardiography, concentrations of peroxidation products and S100B which are markers for brain injury in the blood. Furthermore, pharmacokinetics of allopurinol will be investigated in 48 to 52 patients. Finally, the opinions of parents experiencing two different consent procedures will be evaluated.
Parental perspectives
Following study participation, parents will be approached again and asked for their opinion on and satisfaction with the consent procedure to inform future investigators in the field of HIE therapy.
Ethical Considerations
Because allopurinol has to be administered as early as possible after birth to reduce formation of oxygen radicals during reperfusion and because the emergency situation of perinatal asphyxia is very stressful for the parents, the usual procedure of provision of comprehensive oral and written information, time for consideration and full written informed parental consent before study entry is not feasible in the setting of ALBINO. This problem and the various alternative approaches (antenatal consent, short information and oral consent and later full information and written confirmation, waiver of consent for 1st dose and deferred information and consent), have been discussed with external experts on perinatal HIE as well as medical ethicists and a balance between the need to inform the parents and the feasibility of the study was sought in collaboration with the relevant ethics committees in each participating country.
Community Engagement
Information material, such as posters and flyers, that provides short information for parents, will be available in prenatal clinics and delivery areas and will direct interested parents to the study home page (www.albino-study.eu).The homepage will grant access to nationally approved full parent information material. A press release will inform the community around study sites about the ALBINO study.
Parents, who do not want to participate in the ALBINO-trial, will have the option to deny participation even before delivery verbally or on a ‘declaration of intent’-form printed on the flyers informing about the study. This can be completed and kept in the maternal health passport to inform the staff in the delivery room.
Form of Consent
According to the relevant ethics committee’s decisions, either a deferred consent or an initial short oral consent approach will be used for obtaining parental consent.
The deferred consent procedure has previously been used in emergency research in adults and is in compliance with §30 of the Declaration of Helsinki (Fortaleza 2013 [28]). In the case that a child fulfills the inclusion criteria and meets no exclusion criterion, physicians will administer the first dose of study medication in the delivery room without prior consent (i.e., a ‘consent waiver’ was granted for the 1st dose of study medication). Parents will receive detailed information later and will be asked for written informed consent for continued participation in the study (as soon as possible, at the latest before the 2nd dose of study medication if indicated). The deferred consent procedure has been approved in Austria, Belgium, Estonia, Finland and Norway.
In Germany, the Netherlands, Italy, Switzerland and Spain, the ethical committees did not agree on the deferred consent procedure, so in these countries the short oral consent procedure will apply: short oral information (duration < 5 min) on the indication and the potential benefits and risks of the study medication must be provided to at least one parent and oral (or written) consent of this parent must be obtained before the 1st dose of study medication can be administered. Again, both parents will receive more detailed information and will be asked for full written consent as soon as possible and at the latest before the 2nd dose of study medication will be administered (if indicated).
Statistical analysis
Sample size, power and study duration
The primary assessment for efficacy will compare the proportions of infants surviving without severe NDI versus those of infants who died or survived with severe NDI at the age of two years.
Based on the above referenced (preliminary) clinical studies from the pre-therapeutic hypothermia era and clinical studies on hypothermic treatment, it is estimated that the combined incidence of death and severe NDI in the control group will be 37 and 27% in the allopurinol group. Therefore, we calculated with a two-sided X2-test (alpha = 0.05, power 80%) a sample size of 682 infants (341 per treatment group) in which the primary outcome should be ascertained. Assuming a drop-out rate of 10% for loss to follow-up, a total of 760 infants need to be enrolled. And assuming that 10% of the parents will refuse participation after the initial dose of the study drug, 846 infants have to be randomized (see Fig. 2).

Anticipated Trial Flow
We estimate a recruitment of about 35 patients per month in approximately 70 study centers (the recruitment of additional study sites is ongoing) and therefore recruitment will last 24 months.
Data analysis
All statistical analyses will be described in detail in a statistical analysis plan completed before closure of the database.
Monitoring safety
An independent Data Monitoring Committee (DMC) will monitor the participants’ well-being and the overall risk/benefit-ratio of the study. National monitors will monitor the accuracy and completeness of the data and the safety issues such as the presence of serious adverse events.
Regulatory aspects
Trial sponsor
Sponsor of the ALBINO-trial is the University Hospital Tuebingen, Geissweg 3, 72076 Tuebingen, Germany. Contact is available under albino@med.uni-tuebingen.de.
Orphan Drug Designation
The Committee for Orphan Medicinal Products (COMP) has given a positive advice to ACE Pharmaceuticals for the orphan drug designation for allopurinol sodium for treatment of perinatal asphyxia (EU/3/15/1493) and an Orphan Drug Designation has been granted by the European Medicines Agency. The public summary is available at: https://www.ema.europa.eu/documents/ orphan-designation/eu/3/15/1493-public-summary-opinion-orphan-designation-allopurinol-sodium-treatment-perinatal-asphyxia_en.pdf.
Scientific Advice from the European Medicines Agency
In November 2015, ACE Pharmaceuticals has requested Scientific Advice and Protocol Assistance from the European Medicines Agency, including questions specifically related to the study protocol and the intended procedure of deferred consent. Written scientific advice was received in May 2016 and after careful consideration by the Steering Committee, the relevant issues were subsequently incorporated into the study protocol.
Medical ethics committees
At the time of publication, the relevant ethics committees in ten European countries approved the study with either the short oral consent procedure or the deferred consent procedure. Applications for approvals are currently underway in two additional countries.
National Regulatory/Competent Authorities
At the time of publication, eleven European National Regulatory/Competent Authorities approved the study. Application for approval is currently underway in one additional country.
Discussion
ALBINO is a randomized controlled trial investigating the safety and efficacy of allopurinol in (near-) term infants with HIE.
A decision was made for a large phase III trial for efficacy and safety because preliminary clinical data from postnatal and prenatal allopurinol trials already suggested a reduction in brain injury by allopurinol without apparent adverse effects. Another small proof-of-principle or dose seeking study would have added little with respect to safety and clinically relevant outcomes. Survival without NDI was selected as the primary endpoint of this study, because this outcome parameter is most meaningful to the children and their families.
The calculated starting dose was based on previous studies: the doses used in the first studies with allopurinol in neonates undergoing extracorporeal membrane oxygenation and in neonates diagnosed with hypoplastic left heart syndrome (10 and 30 mg/kg birth weight respectively) gave 100% xanthine-oxidase inhibition [21, 23]. Higher concentrations may be needed for the iron chelating and reactive oxygen scavenging effect of allopurinol. Even with higher doses (up to 40 mg/kg birth weight per day for 3 days) no adverse effects were seen, with special attention to skin rashes and leukopenia [15].
A significant beneficial effect of allopurinol in moderately asphyxiated neonates has been found on long-term (4–5 years) neurodevelopmental outcome by Kaandorp et al. (2012), which was a meta-analyses of the study from van Bel et al. (1998) and Benders et al. (2006) [12, 13]. These latter trials administered 2 times 20 mg/kg birth weight allopurinol with 12 h interval. The doses of the ALBINO trial are based on these three studies. However, pharmacokinetics of allopurinol during hypothermia have not yet been determined in neonates with HIE. The second dose in the ALBINO study (only during hypothermia) is adjusted for the hypothermia treatment which may possibly slow-down allopurinol and oxypurinol metabolism and elimination. In the latter case this would lead to higher circulating concentrations of allopurinol and, respectively, oxypurinol.
In previous studies the plasma concentrations of allopurinol were often supra-therapeutic without any side effect [19, 29]. These supra-therapeutic levels seem to be important for the direct scavenging of hydroxyl and free iron by allopurinol. However, to ensure that in addition to therapeutic hypothermia plasma concentrations are not lower than in the earlier clinical trials indicating efficacy, blood sampling for pharmacokinetic analyses will be performed in 48 to 52 infants (in selected centers) recruited during the first year of the study and may lead to adaptation of doses.
Mannitol is used as placebo, since its freeze-dried white powder and the reconstituted solution, have the same visual aspects and volume as the freeze-dried sodium salt of allopurinol and its reconstitution solution (10 ml of a colorless, clear solution in a 20 ml vial). The dosage of mannitol is 50 times lower than the dose of mannitol used for neuroprotection [30], and a normal daily dose of intravenous paracetamol will include more mannitol as supporting agent than the dose administered in ALBINO (i.e. 100 ml solution for injection contains 1000 mg acetaminophen and 3670-3850 mg mannitol [31, 32]. For each single dose of 12.5 mg/kg paracetamol i.v. [33], 45.9–48.1 mg/kg of Mannitol are concomitantly administered).
Inclusion and exclusion criteria were selected to recruit a patient population similar to the TOBY trial of whole body cooling [3], but took into account that the assessment for eligibility has to be done much earlier, i.e., within 30 min after birth.
The ALBINO study group extensively discussed the various ethical implications of need for additional treatment for HIE, need to administer allopurinol very early for best efficacy, need for parental consent to ensure patient autonomy and burden to the parents in the emergency situation of perinatal HIE.
The European Foundation for the Care of Newborn Infants (EFCNI), which is composed of parents, healthcare experts, scientists and politicians, has been asked for advice. The EFCNI endorsed the conduct of the ALBINO trial in a letter of support in September 2016. Because perinatal HIE occurs rarely and unpredictably and because of the need for very early administration of allopurinol, the EFCNI agreed with the approach of deferred consent.
Furthermore, independent ethics experts provided advice. Whereas all experts agreed that regular informed consent by the parents, which includes appropriate time for reflection and further questions is not feasible before administration of the 1st dose of study medication in the context of ALBINO due to the unpredictable emergency situation. Opinions within the group as well as among external experts ranged from ‘deferred consent is unacceptable’ to ‘deferred consent is justified and the better option’, so that the decision was left to the national leading ethics committees in each country.
Currently, we are conducting a survey among parents-to-be and parents of infants with a history of HIE to better understand how parents might feel about deferred versus short oral consent. An additional survey will follow parents of infants enrolled in the ALBINO study to capture their satisfaction with the various approaches and to inform future trials in similar situations.
In conclusion, infants with HIE still suffer from death and long-term NDI despite improved standards of care including therapeutic hypothermia. The neurodevelopmental outcome of infants with HIE should be further improved with additional neuroprotective interventions. The aim of the ALBINO trial is to investigate the neuroprotective effect of very early allopurinol within 45 min after birth aiming to reduce the formation of the toxic superoxide and subsequent secondary energy failure and apoptosis.
Trial status
Protocol version 5: 19. December 2017. Recruitment has started in April 2018 and is expected to be finalized in April 2020. The last patient out (after follow-up) will then be expected in April 2022.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Abbreviations
- ALLO-trial:
-
Antenatal allopurinol trial for reduction of birth asphyxia induced brain damage
- CP:
-
Cerebral Palsy
- COMP:
-
Committee for Orphan Medicinal Products
- DMC:
-
Data Monitoring Committee
- ALBINO:
-
Effect of Allopurinol in addition to hypothermia for hypoxic-ischemic brain injury on neurocognitive outcome
- EEG:
-
Electro-encephalography
- EFCNI:
-
European Foundation for the Care of Newborn Infants
- HIE:
-
Hypoxic-ischemic Encephalopathy
- MRI:
-
Magnetic Resonance Imaging
- NDI:
-
Neurodevelopmental Impairment
- SCPE:
-
Surveillance of Cerebral Palsy in Europe
- TOBY:
-
Total Body Hypothermia for Neonatal Encephalopathy Trial
References
Kurinczuk JJ, White-Koning M, Badawi N. Epidemiology of neonatal encephalopathy and hypoxic-ischaemic encephalopathy. Early Hum Dev. 2010;86(6):329–38.
Azzopardi D, Strohm B, Marlow N, Brocklehurst P, Deierl A, Eddama O, et al. Effects of hypothermia for perinatal asphyxia on childhood outcomes. N Engl J Med. 2014;371(2):140–9.
Azzopardi DV, Strohm B, Edwards AD, Dyet L, Halliday HL, Juszczak E, et al. Moderate hypothermia to treat perinatal asphyxial encephalopathy. N Engl J Med. 2009;361(14):1349–58.
Edwards AD, Brocklehurst P, Gunn AJ, Halliday H, Juszczak E, Levene M, et al. Neurological outcomes at 18 months of age after moderate hypothermia for perinatal hypoxic ischaemic encephalopathy: synthesis and meta-analysis of trial data. BMJ. 2010;340:c363.
Day RO, Graham GG, Hicks M, McLachlan AJ, Stocker SL, Williams KM. Clinical pharmacokinetics and pharmacodynamics of allopurinol and oxypurinol. Clin Pharmacokinet. 2007;46(8):623–44.
McCord JM. Oxygen-derived free radicals in postischemic tissue injury. N Engl J Med. 1985;312(3):159–63.
Bel F, Groenendaal F. Drugs for neuroprotection after birth asphyxia: pharmacologic adjuncts to hypothermia. Semin Perinatol. 2016;40(3):152–9.
Annink KV, Franz AR, Derks JB, Rudiger M, Bel FV, Benders M. Allopurinol: old drug, new indication in neonates? Curr Pharm Des. 2017;23(38):5935–42.
Shadid M, Buonocore G, Groenendaal F, Moison R, Ferrali M, Berger HM, et al. Effect of deferoxamine and allopurinol on non-protein-bound iron concentrations in plasma and cortical brain tissue of newborn lambs following hypoxia-ischemia. Neurosci Lett. 1998;248(1):5–8.
Moorhouse PC, Grootveld M, Halliwell B, Quinlan JG, Gutteridge JM. Allopurinol and oxypurinol are hydroxyl radical scavengers. FEBS Lett. 1987;213(1):23–8.
Marro PJ, Mishra OP, Delivoria-Papadopoulos M. Effect of allopurinol on brain adenosine levels during hypoxia in newborn piglets. Brain Res. 2006;1073-1074:444–50.
Benders MJ, Bos AF, Rademaker CM, Rijken M, Torrance HL, Groenendaal F, et al. Early postnatal allopurinol does not improve short term outcome after severe birth asphyxia. Arch Dis Child Fetal Neonatal Ed. 2006;91(3):F163–5.
Van Bel F, Shadid M, Moison RM, Dorrepaal CA, Fontijn J, Monteiro L, et al. Effect of allopurinol on postasphyxial free radical formation, cerebral hemodynamics, and electrical brain activity. Pediatrics. 1998;101(2):185–93.
Kaandorp JJ, van Bel F, Veen S, Derks JB, Groenendaal F, Rijken M, et al. Long-term neuroprotective effects of allopurinol after moderate perinatal asphyxia: follow-up of two randomised controlled trials. Arch Dis Child Fetal Neonatal Ed. 2012;97(3):F162–6.
Gunes T, Ozturk MA, Koklu E, Kose K, Gunes I. Effect of allopurinol supplementation on nitric oxide levels in asphyxiated newborns. Pediatr Neurol. 2007;36(1):17–24.
Kaandorp JJ, Benders MJ, Schuit E, Rademaker CM, Oudijk MA, Porath MM, et al. Maternal allopurinol administration during suspected fetal hypoxia: a novel neuroprotective intervention? A multicentre randomised placebo controlled trial. Arch Dis Child Fetal Neonatal Ed. 2015;100(3):F216–23.
Torrance HL, Benders MJ, Derks JB, Rademaker CM, Bos AF, Van Den Berg P, et al. Maternal allopurinol during fetal hypoxia lowers cord blood levels of the brain injury marker S-100B. Pediatrics. 2009;124(1):350–7.
Chaudhari T, McGuire W. Allopurinol for preventing mortality and morbidity in newborn infants with suspected hypoxic-ischaemic encephalopathy. Cochrane Database Syst Rev. 2008;(2):Cd006817. https://doi.org/10.1002/14651858.CD006817.pub2
van Kesteren C, Benders MJ, Groenendaal F, van Bel F, Ververs FF, Rademaker CM. Population pharmacokinetics of allopurinol in full-term neonates with perinatal asphyxia. Ther Drug Monit. 2006;28(3):339–44.
Russell GA, Cooke RW. Randomised controlled trial of allopurinol prophylaxis in very preterm infants. Arch Dis Child Fetal Neonatal Ed. 1995;73(1):F27–31.
McGaurn SP, Davis LE, Krawczeniuk MM, Murphy JD, Jacobs ML, Norwood WI, et al. The pharmacokinetics of injectable allopurinol in newborns with the hypoplastic left heart syndrome. Pediatrics. 1994;94(6 Pt 1):820–3.
Clancy RR, McGaurn SA, Goin JE, Hirtz DG, Norwood WI, Gaynor JW, et al. Allopurinol neurocardiac protection trial in infants undergoing heart surgery using deep hypothermic circulatory arrest. Pediatrics. 2001;108(1):61–70.
Marro PJ, Baumgart S, Delivoria-Papadopoulos M, Zirin S, Corcoran L, McGaurn SP, et al. Purine metabolism and inhibition of xanthine oxidase in severely hypoxic neonates going onto extracorporeal membrane oxygenation. Pediatr Res. 1997;41(4 Pt 1):513–20.
Boda D, Nemeth I, Hencz P, Denes K. Effect of allopurinol treatment in premature infants with idiopathic respiratory distress syndrome. Dev Pharmacol Ther. 1984;7(6):357–67.
Ramasamy SN, Korb-Wells CS, Kannangara DR, Smith MW, Wang N, Roberts DM, et al. Allopurinol hypersensitivity: a systematic review of all published cases, 1950-2012. Drug Saf. 2013;36(10):953–80.
Stamp LK, Day RO, Yun J. Allopurinol hypersensitivity: investigating the cause and minimizing the risk. Nat Rev Rheumatol. 2016;12(4):235–42.
Shankaran S, Laptook AR, Ehrenkranz RA, Tyson JE, McDonald SA, Donovan EF, et al. Whole-body hypothermia for neonates with hypoxic-ischemic encephalopathy. N Engl J Med. 2005;353(15):1574–84.
World Medical Association. World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;310(20):2191–4. https://doi.org/10.1001/jama.2013.281053
Kaandorp JJ, van den Broek MP, Benders MJ, Oudijk MA, Porath MM, Bambang Oetomo S, et al. Rapid target allopurinol concentrations in the hypoxic fetus after maternal administration during labour. Arch Dis Child Fetal Neonatal Ed. 2014;99(2):F144–8.
Tavakkoli F. Review of the role of mannitol in the therapy of children. 18th expert committee on the selection and use of essential medicines mannitol review (children); 2011. p. 16.
Cadence Pharmaceuticals, Inc. - OFIRMEV® (acetaminophen) injection - Approval Label (PDF) 11/2010 - https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/022450lbl.pdf (downloaded 2019/01/29).
Fresenius Kabi USA, LLC - acetaminophen injection, for intravenous use - approval label (PDF) 10/2015 - https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/204767s000lbl.pdf (downloaded 2019/01/29).
Mallinckrodt hospital products Inc. - OFIRMEV® (acetaminophen) injection - labeling-package insert (PDF) 04/2018 - https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/022450s011lbl.pdf (downloaded 2019/01/29).
Acknowledgements
The ALBINO consortium is indebted to Silke Mader and Nicole Thiele from the European Foundation for the Care of Newborn Infants (EFCNI) who granted a letter of support for the ALBINO study after careful evaluation of the various arguments.
We would also like to thank the members of the Data Monitoring Committee: Michael Weindling (University of Liverpool), Sandra Juul (University of Washington), Steven Miller (Hospital for Sick Children Toronto), Edwin Spaans (Erasmus University Rotterdam) and Josef Högel (University of Ulm), and the members of the ALBINO External Advisory Board: Seetha Shankaran,(Wayne State University Detroit) and Neil Marlow (University College London).
ALBINO study group:
Coordinating Investigators: Axel R. Franz (University Hospital Tuebingen, Germany; corresponding and senior author) and Mario Rüdiger (University Hospital C.G. Carus - Medizinische Fakultät der TU Dresden, Germany).
Beneficiaries / National Coordinators: Axel R. Franz and Christian F. Poets (Tuebingen, Germany), Mario Rüdiger (Dresden, Germany), Manon Benders and Frank van Bel (Utrecht, the Netherlands), Karel Allegaert and Gunnar Naulaers (Leuven, Belgium), Dirk Bassler (Zurich, Switzerland), Katrin Klebermaß-Schrehof (Vienna, Austria), Maximo Vento (Valencia, Spain), Hercilia Guimarães (Porto, Portugal), Tom Stiris (Oslo, Norway), Luigi Cattarossi (Udine, Italy), Marjo Metsäranta (Helsinki, Finland), Sampsa Vanhatalo (Helsinki, Finland), Jan Mazela (Poznan, Poland), Tuuli Metsvaht (Tartu, Estonia), Cees K.W. van Veldhuizen (Zeewolde, the Netherlands).
Data Management, Biometry, Monitoring, and Study Coordination, all at the Center for Pediatric Clinical Studies, University Hospital Tuebingen: Corinna Engel, Christian A. Maiwald, Gabriele von Oldershausen, Iris Bergmann, Monika Weiss, Caroline J. B. R. Wichera, Andreas Eichhorn, Michael Raubuch, Birgit Schuler.
Industry Partner: Cees K.W. van Veldhuizen, Bas Laméris, Yannique Jacobs, Roselinda van der Vlught-Meijer (all ACE Pharmaceuticals BV, Zeewolde, the Netherlands).
Recruiting Hospitals and Local Principal Investigators:
Austria: Medizinische Universitaet Wien Katrin Klebermaß-Schrehof, Medizinische Universität Graz Gerhard Pichler, Tirol Kliniken - Universitätskliniken Innsbruck Elke Griesmaier, Uniklinikum Salzburg Johannes Brandner.
Belgium: University Hospitals Leuven Gunnar Naulaers, CHU St. Pierre University Hospital Brüssel Marie Tackoen and Ruth Reibel, CHR - Grand Hopital de Charleroi Chantal Lecart, UZ Brussel Filip Cools, AZ Sint-Jan Brugges Luc Cornette, Tivoli, La Louviere Genevieve Malfilatre, CHR Citadelle, Liege Renaud Viellevoye.
Estonia: Tartu University Hospital Tuuli Metsvaht, Tallinn Children’s Hospital Mari-Liis Ilmoja, West Tallinn Central Hospital Pille Saik and Ruth Käär, East Tallinn Central Hospital Pille Andresson.
Finland: Helsinki University Hospital (HUS) Marjo Metsäranta,
Germany: University Hospital Tuebingen Axel R. Franz, Klinikum der J. W. Goethe-Universität Frankfurt am Main Rolf Schloesser, Universitätsklinikum Münster Torsten Ott, Universitätsklinikum C. G. Carus - Medizinische Fakultät der TU Dresden Stefan Winkler, Universitätsklinikum Duesseldorf Thomas Hoehn, Universitätsklinikum der Ruhr-Universität Bochum Norbert Teig, Cnopf’sche Kinderklinik/Klinik Hallerwiese Nürnberg Michael Schroth, Universitätsklinik der Paracelsius Med. Privatklinik, Klinikum Nürnberg Süd Christoph Fusch, Universitätsklinikum Leipzig Ulrich H. Thome, Department of General Pediatrics and Neonatology, Justus-Liebig-University Gießen Harald Ehrhardt.
Italy: Azienda sanitaria universitaria integrata di Udine Luigi Cattarossi and Isabella Mauro, Università degli studi di Padova Eugenio Baraldi, Azienda ospedaliero universitaria Ospedali Riuniti di Ancona Virgilio Carnielli, Fondazione MBBM - Ospedale San Gerardo di Monza Giuseppe Paterlini, Ospedale Evangelico Betania (Naples) Marcello Napolitano, Ospedale Valduce Como Paola Francesca Faldini, ASST FBF-Sacco Ospedale dei Bambini “V.Buzzi” Milano Gianluca Lista, Ospedale di Treviso Gianluca Visintin, ASST-Lariana, Ospedale Sant’anna San Fermo della Battaglia Mario Barbarini and Laura Pagani, Presidio Ospedaliero S.Anna, Città della Salute e della Scienza di Torino Emmanuele Mastretta, Fondazione Policlinico Universitario A. Gemelli IRCCS - Università Cattolica del Sacro Cuore Rome Giovanni Vento, Fondazione IRCCS Cà Granda Ospedale Maggiore Policlinico Milano Monica Fumagalli, Ospedale Maggiore della Carità Novara Marco Binotti.
Netherlands: VU Medical Center Mirjam M. van Weissenbruch, Isala Klinieken Henrica L.M. van Straaten, Universitair Medisch Centrum Utrecht Manon J.N.L. Benders, Kim V. Annink, Frank van Bel, Jeroen Dudink, Jan B. Derks, Diakonessenhuis Utrecht Inge P. de Boer, Meander Medisch Centrum Amersfoort Clemens B. Meijssen, Academic Medical Center Timo R. de Haan, Medisch Spectrum Twente Linda G. van Rooij, St. Antonius Ziekenhuis Jacqueline L. van Hillegersberg and Minouche van Dongen, Elisabeth Tweesteden Ziekenhuis Jos Bruinenberg, Deventer Ziekenhuis A.C.M. Dassel, Maxima Medical Center Veldhoven Koen P. Dijkman, Spaarne Gasthuis Marlies A. van Houten, OLVG Sophie R.D. van der Schoor.
Norway: Oslo Universitetssykehus HF Tom Stiris, Haukeland Univ. Sykehus Bergen Bodil Salvesen, Vestfold Hospital Trust Tonsberg Moritz Schneider and Eirik Nestaas, Akershus University Hospital (AHUS) Lørenskog Britt Nakstad, Sykehuset Innlandet Lillehammer Dag Helge Frøisland.
Poland: Poznan University of Medical Sciences - Department of Neonatology Jan Mazela and Lukas Karpinski, Instytut Centrum Zdrowia Matki Polki Ewa Gulczynska, Wroclaw Medical University Department of Neonatology Barbara Królak-Olejnik, Neonatal and Intensive Care Department Medical University of Warsaw Renata Bokiniec
Portugal: Centro Hospitalar Universitário São João (CHUSJ) Porto Ana I. Vilan, Centro Materno Infantil do Norte (CMIN) Porto Liliana Flores de Pinho, Hospital Pedro Hispano (HPH) Porto Claudia Ferraz, Hospital de Braga (HB) Almerinda Pereira, Hospital Fernando Fonseca (HFF) Amadora (Lisboa) Rosalina Barroso, Hospital Santa Maria - Centro Hospitalar Universitario de Lisboa Norte André Mendes da Graça, Centro Hospitalar Universitário de Lisboa Central (CHULC) Teresa Tomé and Filomena Pinto.
Spain: Hospital Universitario y Politécnico La Fe, Valencia, Maximo Vento and Juan Martínez Rodilla, Complejo Hospitalario Universitario Santiago de Compostela Maria Luz. Couce Pico, Hospital Puerta del Mar Cádiz Simón Lubián, General University Hospital of Alicante Caridad Tapia Collados, Quironsalud Madrid University Hospital Fernando Cabañas, Hospital Sant Joan de Déu Barcelona Marta Camprubí Camprubí, Hospital Virgen de las Nieves Granada José Antonio Hurtado Suazo, Hospital Universitario La Paz Madrid Eva Valverde, Hospital Reina Sofía Córdoba Inés Tofé, Complejo Hospitalario Universitario Vigo José Ramón Fernández Lorenzo, Hospital Clínico San Carlos Madrid José Martinez Orgado, Hospital Vall de Hebrón Barcelona Héctor Boix, Hospital Regional Universitario de Málaga Mercedes Chaffanel, Hospital Virgen del Rocío Sevilla Francisco Jimenez Parrilla, Hospital Gregorio Marañón Madrid Dorotea Blanco, Hospital de Cruces Barakaldo Begoña Loureiro, Hospital 12 de Octubre Madrid Maria Teresa Moral-Pumarega, Hospital Miguel Servet Zaragoza Segundo Rite.
Switzerland: UniversitaetsSpital Zuerich Dirk Bassler, Julia Maletzki and Claudia Knoepfli, Kinderspital Zürich (KiSpi ZH) Cornelia Hagmann, Kantonsspital Winterthur Michael Kleber, Universitäts-Kinderspital beider Basel (UKBB) Sven Schulzke, Kantonsspital Luzern Martin Stocker, Ostschweizer Kinderspital (St.Gallen) André Birkenmaier, Kantonsspital Graubünden (Chur) Thomas Riedel.
Funding
This study is funded under the Horizon 2020 Framework Program of the European Union, call H2020-PHC-2015-two-stage, grant 667224. The European Union/European Commission had no influence on the design of the study, on collection, analysis and interpretation of data and on writing this manuscript.
Publication of this manuscript was supported by Deutsche Forschungsgemeinschaft and the Open Access Publishing Fund of the University of Tuebingen. They had no influence on the design of the study, on collection, analysis and interpretation of data and on writing this manuscript.
Author information
Authors and Affiliations
Consortia
Contributions
CAM and KVA drafted the first version of the manuscript together on behalf of the ALBINO study group (shared first authorship). All other members of the ALBINO study group revised the manuscript, making important contributions and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The ALBINO trial is performed in accordance with the Declaration of Helsinki and the guidelines of Good Clinical Practice (GCP). Written informed consent must be obtained by the parents or legal guardians before full participation in the study (i.e. before administration of the second dose of study medication (if indicated) and before data-entry into the database). Whether oral consent by at least one parent is obtained following short information or an approved waiver of consent is applied before administration of the first dose of study medication, depends on the approvals of the responsible national ethics committees (as detailed elsewhere). At the time of publication, the ALBINO trial is currently taking place in 10 European countries and may expand to other countries, including Poland and Portugal, once ethical approval has been obtained.
Austria: Ethics: Ethikkommission Medizinische Universität Wien, reference no. 1731/2017, approved with deferred consent; Authority: Bundesamt für Sicherheit im Gesundheitswesen, reference no. 10680185 approved conduct.
Belgium: Coordinating ethical committee: Ethical Committee UZ Leuven reference no. S60224, approved with deferred consent; Authority: Federal Agency for Medicines and Health Products Brussels, reference no. FAGG/R&D/MMN approved conduct.
Estonia: Ethics: Research Ethics Committee of the University of Tartu (UT REC), reference no. 272/T-13, approved with deferred consent; Authority: State Agency of Medicines clinical trial, reference no. 17–044 approved conduct.
Finland: Ethics: Naisten, lasten ja psykiatrian eettinen toimikunta, Helsingin ja Uudenmaan sairaanhoitopiiri reference no. HUS/1528/2017 approved with deferred consent; Authority: Finnish Medicines Agency (FIMEA) reference no. 44/ 2017 approved conduct.
Germany: Ethics: Ethics Committee at the University Hospital Tuebingen, reference no. 703/2016AMG1, approved with short oral consent; Authority: Bundesinstitut für Arzneimittel und Medizinprodukte, reference no. 4041912 approved conduct.
Italy: Ethics: COMITATO ETICO UNICO REGIONALE sede operative CENTRO di RIFERIMENTO ONCOLOGICO reference no. 6.1 21/11/2017 - ID 2167 approved with short oral consent; Authority: AIFA- Agenzia Italiana del Farmaco reference no. 97707 approved conduct.
Netherlands: Ethics: The ethical committee of the University Medical Center Utrecht reference no. NL57237.041.16 approved with short oral consent; Authority: Centrale Commissie Mensgebonden Onderzoek (CCMO) reference no. NL57237.041.16 approved conduct.
Norway: Ethics: REK – Regionale komiteer for medisinsk og helsefaglig forskningsetikk reference no. 2017/800 approved with deferred consent; Authority: Norwegian Medicines Agency reference no. 17/04729–11 approved conduct.
Poland: Ethics: to be submitted Authority: to be submitted.
Portugal: Ethics: CEIC - Comissão de Ética para a Investigação Clínica - Waiting for approval; Authority: INFARMED - Autoridade Nacional do Medicamento e Produtos de Saúde, I.P. - approved conduct.
Spain: Ethics: Ethics Committee for Research with Medications at the Hospital Universitario y Politécnico de La Fe. reference no. 2016–000222-19 approved with Short Oral Consent; Authority: Spanish Agency of Medicines reference no.2016–000222-19 approved with Short Oral Consent.
Switzerland: Ethics: Kantonale Ethikkommission Zürich, reference no. 2017/00961, approved with short oral consent; Authority: Swissmedic - Swiss agency for therapeutic products, reference no. 2017DR3135 approved conduct.
Consent for publication
Not applicable.
Competing interests
Y. Jacobs and R. van der Vlught-Meijer are employees of ACE Pharmaceuticals, the company that holds the Dutch marketing authorization registration for Acepurin® (allopurinol 1 g/100 ml) for intravenous application for treatment of gout. C. van Veldhuizen and B. Laméris are the former owners of ACE Pharmaceuticals. All four contributed to the development of the study protocol. All other contributors declare that they do not have competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Maiwald, C.A., Annink, K.V., Rüdiger, M. et al. Effect of allopurinol in addition to hypothermia treatment in neonates for hypoxic-ischemic brain injury on neurocognitive outcome (ALBINO): study protocol of a blinded randomized placebo-controlled parallel group multicenter trial for superiority (phase III). BMC Pediatr 19, 210 (2019). https://doi.org/10.1186/s12887-019-1566-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12887-019-1566-8