
Abstract
Background
Gitelman syndrome (GS) is an autosomal recessive disorder and mild variant of classic Bartter syndrome. The latter is caused by defects in the genes CLCNKB and/or CLCNKA (chloride voltage-gated channel Ka and Kb). Patients with GS usually have loss-of-function mutations in SLC12A3. No patient has been reported with compound heterozygous mutations in these genes. We report a girl with GS with a paternally inherited heterozygous mutation in SLC12A3, and maternally inherited heterozygous variants in both CLCNKB and CLCNKA.
Case presentation
In this report, we reported a female patient (8 y and 10 mo) who had growth retardation (111.8 cm, − 1.62 standard deviation height for age) and normal blood pressure, with persistent hypokalemia, hypomagnesemia, hypocalciuria, hypochloremic alkalosis, and elevated levels of plasma renin and aldosterone. Her younger brother, father, and paternal grandmother all had histories of mild low levels of plasma potassium (3.0–3.5 mmol/L), which were rectified by potassium-rich foods. The genomic DNA of the patient, younger brother, parents, and grandparents were screened for gene variations and pedigree analysis using trio whole exome sequencing (WES). The candidate variants were validated by Sanger sequencing. Protein-protein interaction analysis utilized the following databases: Biogrid, MINT, HPRD, STRING, IntAct, iRefIndex, and ppiTrim. The trio WES screening showed that the patient has paternally inherited SLC12A3 p.N359K, and maternally inherited CLCNKB p.L94I. The paternal grandmother and younger brother are both carriers of SLC12A3 p.N359K. According to the STRING database, SLC12A3 and CLCNKB proteins may interact or coexpress with proteins associated with GS.
Conclusions
Based on clinical phenotypes, genetic evidence of the pedigree, and previous reported studies, this case of GS indicates a digenetic inheritance of SLC12A3 and CLCNKB that resulted in renal tubular dysfunction perhaps, due to a genetic double-hit mechanism. The putative pathogenicity of the CLCNKB p.L94I variant requires confirmation.
Similar content being viewed by others


Background
Gitelman syndrome (GS, or familial hypokalemia-hypomagnesemia; Mendelian inheritance in man [MIM] #263800) is an autosomal recessive renal tubular salt-wasting disorder that is often characterized by potassium and magnesium depletion. Most patients with GS have loss-of-function mutations in SLC12A3, which encodes the thiazide-sensitive sodium-chloride cotransporter (NCC) [1, 2]. Some specific cases have putative mutations in chloride voltage-gated channel Kb (CLCNKB) [3,4,5] (ORPHA:358).
Clinical features of GS include transient periods of muscle weakness and tetany, abdominal pains, and chondrocalcinosis [1]. Asian patients with GS seem to have a greater tendency to hypokalemic paralysis than do patients of other ethnicities (approximately 6%) [6,7,8]. According to a study based on two unrelated Chinese families, female GS patients may have profound hypomagnesemia without neuromuscular symptoms [9].
GS is referred to as a mild variant of classic Bartter syndrome (or renal tubular normotensive hypokalemic alkalosis with hypercalciuria; MIM #607364), which is caused by defects in genes encoding chloride channel components, including CLCNKB, chloride voltage-gated channel Ka (CLCNKA), or both. Thus, previous studies focused on mutations in SLC12A3, CLCNKB, CLCNKA, and other genes associated with renal tubular functions.
Prior to the present, there has been no report of a case of GS associated with compound heterozygous mutations in the above genes. We report a girl aged 8 years and 10 months with GS, who has a paternally inherited heterozygous mutation in SLC12A3, and maternally inherited heterozygous variants in both CLCNKB and CLCNKA. This case may aid understanding of the phenotypic characteristics of the inactivated NCC and CLCNKA and CLCNKB in GS.
Case presentation
The girl (aged 8 years and 10 months) was a full term baby delivered by planned natural birth. At age six years and seven months, she was found with growth retardation (111.8 cm, − 1.62 standard deviation [SD] height for age). She once presented at the local hospital with acute synovitis of the hip. Hypokalemia (2.93 mmol/L, reference: 3.5–5.3 mmol/L) for 2 months was incidentally discovered, but a further diagnosis was not made for the cause of hypokalemia. She had salt craving, thirst, polydipsia, and polyuria. She also complained of intermittent self-remitting tetany that occurred after prolonged activity and usually lasted for a few seconds. She denied other detrimental symptoms such as repeated episodes of fatigue, palsy, dizziness, hypotonia, palpitations, or blurred vision. After supplementation of oral potassium chloride 375 mg/day, her plasma potassium level reached 3.09 mmol/L. She still experienced recurring episodes of hand cramps, and for this reason was admitted to our hospital.
This girl was the first child of a non-consanguineous Chinese couple. She was not on any medications such as laxatives or diuretics, which could lead to a similar symptom. Her paternal grandmother had received a diagnosis of hypokalemia many years previously, at which time the plasma potassium level reached normal after a few days of spironolactone treatment. The girl’s father and younger brother also had been found with slight hypokalemia, and their plasma potassium levels reached normal after taking potassium-rich foods. Other pedigree members were all asymptomatic, and physical examinations were unremarkable.
On admission, the girl was alert, with no sign of anemia. Her vital signs were a temperature of 37.3 °C, pulse rate 92 beats/min, 20 breaths/min, normal blood pressure of 95/64 mmHg, and height 126 cm (− 1.39 SD for age). Her muscle tone and strength, and deep tendon reflexes were normal. The results of other physical examinations were negative.
Laboratory tests showed hypokalemia (3.33 mmol/L), hypomagnesemia (0.49 mmol/L, reference: 0.7–1.1 mmol/L), slight metabolic alkalosis (pH 7.43 and plasma bicarbonate 27.4 mmol/L, reference: 22–27 mmol/L), and hypocalciuria (0.01 mmol/L, reference: 0.48–5.87 mmol/L). The plasma aldosterone (216 pg/mL, reference: 59.5–173.9 pg/mL) and renin activity (13.43 ng/mL/h, reference: 0.15–2.33 ng/mL/h) were elevated on admission. Her thyroid function was normal and electrocardiogram revealed a normal sinus rhythm (95 beats/min) with normal QT interval. Abdominal ultrasound showed no renal stones or nephrocalcinosis.
The parents provided written informed consent prior to genetic testing. The genomic DNA of the patient proband, and her younger brother, parents, and grandparents were screened for gene variations and pedigree analysis using trio whole exome sequencing (WES). Briefly, the DNA was sheared and then hybridized with xGen Exome Research Panel v1.0 probe sequence capture array of IDT (Integrated Device Technology, USA) to enrich the exonic region. The exome libraries were first tested for enrichment by qPCR and for size distribution and concentration using an Agilent Bioanalyzer 2100. Exon-enriched DNA was sequenced on an Illumina HiSeq X Ten (Illumina, USA) platform in accordance with the manufacturer’s instructions. Raw image files were processed by BCL to FASTQ (Illumina) for base calling and generating the raw data. Low-quality variations of the quality score < 20 (Q20) were filtered out. The sequencing reads were aligned to the NCBI human reference genome (hg19) using Burrows-Wheeler Aligner (BWA) software. SAMtools and Pindel software were used to call single-nucleotide variants and indels of the reads. The minor allele frequency (MAF) was annotated using databases dbSNP, 1000 Genomes MAF (Chinese), ExAC, Genome Aggregation Database (gnomAD), and an in-house MAF database. Synonymous substitutions or single-nucleotide variants s with MAF higher than 5% were filtered out. Transcriptions and translations of nonsynonymous variants were predicted using SIFT (Sorting Intolerant from Tolerant; http://sift.jcvi.org), PROVEAN (Protein Variation Effect Analyzer; http://provean.jcvi.org), and PolyPhen-2 (Polymorphism Phenotyping v2; http://genetics.bwh.harvard.edu/pph2/).
The candidate variants were validated by Sanger sequencing and the pathogenicity of variants were annotated according to the American College of Medical Genetics and Genomics (ACMG) standards and guidelines. The UniProt database was searched for protein chain profiles of SLC12A3 (ID P55017) and CLCNKB (ID P51801). Databases of known protein-protein interactions were used, including BioGRID, MINT, HPRD, STRING, and IntAct; iRefIndex data and its trimmed edition, ppiTrim, were both used to perform the interaction search.
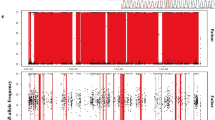
Using trio WES, a paternally inherited SLC12A3 p.N359K (NM_000339) and a maternally inherited CLCNKB p.L94I (NM_000085) were identified, and the variants were confirmed by Sanger sequencing (Fig. 1). The patient’s younger brother and grandmother were the carriers of SLC12A3 p.N359K. No SLC12A3 or CLCNKB variants were observed in other pedigree members (Fig. 2).

Sanger sequencing of candidate variants. (a) A paternally inherited heterozygous mutation was detected in SLC12A3 p.N359K. In addition, maternally inherited heterozygous mutations were detected in (b) CLCNKA c.1054–22(IVS11)delG and (c) CLCNKB p.L94I

Pedigree chart of the proband’s family. The arrow indicates the proband. Men are indicated by squares, and women by circles. III1, the proband presents typical clinical manifestations and laboratory results of GS. I1, II1, and III2 present slight hypokalemia
The SLC12A3 p.N359K is a pathogenic variant according to the ACMG guidelines, with a low MAF < 0.005, and was previously documented pathogenicity [10]. According to the PM1 rating of the ACMG (i.e., located in a mutational hot spot and/or critical and well-establishedfunctional domain [active site of an enzyme] without benign variation), CLCNKB p.L94I is moderate evidence of pathogenicity. The functional predictions are conflicted, with M-CAP and mutation taster scores 0.1415 and 0.9956, respectively. This suggests that the variant is pathogenic. Yet, results of PROVEAN, SIFT, PolyPhen-2 HVAR, and PolyPhen-2 HDIV showed that the variant is tolerated. However, further research in the UniProt database revealed that CLCNKB (id P51801) p.94 is the first residue located at the second helix in the trans-membrane domain. All variants in SLC12A3, CLCNKB, CLCNKA, and the GS-related HNF1B genes were analyzed, and no other potential pathogenic variants were identified.
Using STRING tools, the protein-protein interaction analysis showed that SLC12A3 and CLCNKB interact or coexpress in the human Bartter syndrome protein interaction network (Fig. 3).

Protein-protein interactions in the Bartter syndrome network, according to STRING. (a) Protein-protein coexpressions. The gradient red bar showing the reliability rating. (b) Scores for evidence of the proteins included in the Bartter syndrome network
Discussion and conclusions
The diagnosis of GS was established on clinical rather than genetic grounds, potentially creating confusion with the related disorders classic Bartter syndrome and Bartter syndrome type III. Yet, phenotypic variability has also been documented in genetically confirmed GS patients, including those with identical SLC12A3 mutations. A GS-like phenotype, with hypomagnesemia and hypocalciuria, has also been associated with pathogenic variants in the CLCNKB gene [8, 9]. The phenotype caused by CLCNKB mutations may vary, from the various types of Bartter syndrome to GS. Thus, a differential diagnosis is necessary.
As recommended by the latest KDIGO (Kidney Disease: Improving Global Outcomes) conference report, the next-generation sequencing panels for differential diagnosis of GS should include, at least, SLC12A3, CLCNKB, and HNF1B (hepatocyte nuclear factor-1-beta) [11, 12]. In the present case, a further genetic screening was conducted to exclude deletion or duplication in the SLC12A3 allele (as previous reports have demonstrated [13]). Heterozygous variants in each of SLC12A3, CLCNKA, and CLCNKB, were confirmed.
According to previous studies, some using multiplex ligation-dependent probe amplification (MLPA) for screening, 15–20% of clinical GS patients have only one defect SLC12A3 allele [11, 13]. For these findings, discovering pathogenic variants in other genes is recommended [11]. SLC12A3 p.N359K was previously reported and pathogenicity was demonstrated by Qin et al [10]
The pathogenicity of CLCNKB p.L94I (rs201876924) has not been proved. The ACMG’s PM1 rating for CLCNKB p.L94I seems consistent with the function predictions. However, considering that CLCNKB is a membrane protein and the p.94 is at a trans-membrane domain, the deviation between the function prediction of the trans-membrane domain and other domains should be evaluated. Proving the pathogenicity of CLCNKB p.L94I needs further study.
The other suspect gene is CLCNKA. In the present case, a novel variant was found, a single base deletion that locates deep in intron 11 (c.1054–22 del G) of CLCNKA. Its pathogenicity was excluded using multiple prediction applications.
In the present case, there was no suspected variant of other GS-related or Bartter syndrome-related genes. In addition, no genotype-phenotype associations have been documented in patients with one heterozygous CLCNKB variant [14], which in our patient was inherited from the mother without such a condition. Thus, a digenetic inheritance (DI) may be involved in our case, since a defect in one allele of SLC12A3 or CLCNKB would not cause the phenotypes.
In the present case, the clinical phenotypes of the paternal carriers indicated that heterozygous SLC12A3 p.N359K may be responsible for transient and mild hypokalemia. However, this cannot explain the significant hypokalemia, hypomagnesemia, hypocalciuria, hypochloremic alkalosis, and elevated levels of plasma renin and aldosterone of the patient. On the other hand, compound heterozygous pathogenic variants in CLCNKA and CLCNKB can cause GS, which is considered a canonical pattern of DI. Interestingly, there was no sign of renal tubular dysfunction, and using bioinformatics, no potential pathogenic variant in CLCNKA was identified in the mother carrying the variants (Fig. 2). This helps to exclude the pathogenicity of CLCNKA c.1054–22 (IVS11) delG.
In summary, we identified a pathogenic variant in SLC12A3, a potential pathogenic variant CLCNKB p.L94I, and a likely benign variant c.1054–22 (IVS11) delG in CLCNKA. Since GS has been previously associated with several genes, we speculate that this case may represent a double hit pattern of DI.
Genetic linkage analysis is a reliable method of diagnosis, but has been relatively unsuccessful in identifying genes underlying human DI. In most reports of human DI, knowledge of candidate genes and protein-protein interactions have been used. Because finding ideal DI pedigrees is difficult, most studies base DI conclusions on a few patients without pedigree evidence. Schäffer [15] reviewed and evaluated three methods to identify DI genes, reporting that protein-protein interaction is important evidence. Protein-protein interaction analysis may be best for high-throughput data of variants and rare cases, even without a pedigree.
The coexpression and functional synergy of the proteins SLC12A3 and CLCNKB may be explained based on clinical phenotypes and physiological mechanisms. The results of the present case show that the coexpression of SLC12A3 and CLCNKB is stronger than the coexpression of SLC12A3 and CLCNKA (Fig. 3A), which agrees with the consensus [11]. The phenotype of patients with a CLCNKB defect usually overlaps with the GS phenotype, for example, hypokalemia and hypocalciuria [5, 16]. This may be explained by the localization of CLCNKB in the distal convoluted tubule. Interestingly, gender may influence the regulation of SLC12A3 function, based on a study from China [9] in which the hypokalemia of females with GS was less severe compared with the male family members. The authors speculated that this may be a positive effect of estrogen on SLC12A3 or other modifying genes [9].
In this patient, GS was determined based on typical clinical data and the mutations SLC12A3 p.N359K and CLCNKB p.L94I. We speculate that a digenetic mechanism led to the final phenotype. However, more evidence will be needed to confirm the pathogenicity of CLCNKB p.L94I, and further studies are particularly needed to explore the underlying molecular mechanisms.
Change history
10 September 2019
Following publication of the original article [1], the editor informed that error was found.
Abbreviations
- ACMG:
-
American college of medical genetics and genomics
- DI:
-
digenetic inheritance
- GS:
-
Gitelman syndrome
- MAF:
-
minor allele frequency
- MIM:
-
Mendelian inheritance in man
References
Glaudemans B, Yntema HG, San-Cristobal P, Schoots J, Pfundt R, Kamsteeg EJ, et al. Novel NCC mutants and functional analysis in a new cohort of patients with Gitelman syndrome. Eur J Hum Genet. 2012;20(3):263–70.
Mastroianni N, Bettinelli A, Bianchetti M, Colussi G, De Fusco M, Sereni F, et al. Novel molecular variants of the Na-cl cotransporter gene are responsible for Gitelman syndrome. Am J Hum Genet. 1996;59(5):1019–26.
Konrad M, Vollmer M, Lemmink HH, Van den Heuvel LP, Jeck N, Vargas-Poussou R, et al. Mutations in the chloride channel gene CLCNKB as a cause of classic Bartter syndrome. J Am Soc Nephrol. 2000;11(8):1449–59.
Tavira B, Gomez J, Santos F, Gil H, Alvarez V, Coto E. A labor- and cost-effective non-optical semiconductor (ion torrent) next-generation sequencing of the SLC12A3 and CLCNKA/B genes in Gitelman's syndrome patients. J Hum Genet. 2014;59(7):376–80.
Zelikovic I, Szargel R, Hawash A, Labay V, Hatib I, Cohen N, et al. A novel mutation in the chloride channel gene, CLCNKB, as a cause of Gitelman and Bartter syndromes. Kidney Int. 2003;63(1):24–32.
Cheng NL, Kao MC, Hsu YD, Lin SH. Novel thiazide-sensitive Na-cl cotransporter mutation in a Chinese patient with Gitelman's syndrome presenting as hypokalaemic paralysis. Nephrol Dial Transplant. 2003;18(5):1005–8.
Tajima T, Kobayashi Y, Abe S, Takahashi M, Konno M, Nakae J, et al. Two novel mutations of thiazide-sensitive Na-cl cotrans porter (TSC) gene in two sporadic Japanese patients with Gitelman syndrome. Endocr J. 2002;49(1):91–6.
Cruz DN, Shaer AJ, Bia MJ, Lifton RP, Simon DB. Gitelman's syndrome revisited: an evaluation of symptoms and health-related quality of life. Kidney Int. 2001;59(2):710–7.
Lin SH, Cheng NL, Hsu YJ, Halperin ML. Intrafamilial phenotype variability in patients with Gitelman syndrome having the same mutations in their thiazide-sensitive sodium/chloride cotransporter. Am J Kidney Dis. 2004;43(2):304–12.
Qin L, Shao L, Ren H, Wang W, Pan X, Zhang W. Identification of five novel variants in the thiazide-sensitive NaCl co-transporter gene in Chinese patients with Gitelman syndrome. Nephrology (Carlton). 2009;14(1):52–8.
Blanchard A, Bockenhauer D, Bolignano D, Calò LA, Cosyns E, Devuyst O, et al. Gitelman syndrome: consensus and guidance from a kidney disease: improving global outcomes (KDIGO) controversies conference. Kidney Int. 2017;91(1):24–33.
Verhave JC, Bech AP, Wetzels JF, Nijenhuis T. Hepatocyte nuclear factor 1beta-associated kidney disease: more than renal cysts and diabetes [J]. J Am Soc Nephrol. 2016;27(2):345–53.
Vargas-Poussou R, Dahan K, Kahila D, Venisse A, Riveira-Munoz E, Debaix H, et al. Spectrum of mutations in Gitelman syndrome. J Am Soc Nephrol. 2011;22(4):693–703.
Jeck N, Konrad M, Peters M, Weber S, Bonzel KE, Seyberth HW. Mutations in the chloride channel gene, CLCNKB, leading to a mixed Bartter-Gitelman phenotype. Pediatr Res. 2000;48(6):754–8.
Schaffer AA. Digenic inheritance in medical genetics. J Med Genet. 2013;50(10):641–52.
Fukuyama S, Hiramatsu M, Akagi M, Higa M, Ohta T. Novel mutations of the chloride channel kb gene in two Japanese patients clinically diagnosed as Bartter syndrome with hypocalciuria. J Clin Endocrinol Metab. 2004;89(11):5847–50.
Acknowledgements
Not applicable.
Funding
Not applicable.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
YMK reviewed the case records and wrote the manuscript. KX performed the genetic examinations, and analyzed the molecular data. KY analyzed and interpreted the patient data and reviewed the initial manuscript. ZJF provided the clinical description, and designed the investigation’s structure. WYG performed the genetic examinations, and revise the manuscript. LL was involved in the patient’s treatment and management. CLW reviewed and contributed to the final published version. All authors read and approved the final version of manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The Research Ethics Committee of First Affiliated Hospital, College of Medicine, Zhejiang University approved the study. Written informed consent was obtained from the parents.
Consent for publication
The patient and both of her parents provided written informed consent for publication.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Kong, Y., Xu, K., Yuan, K. et al. Digenetic inheritance of SLC12A3 and CLCNKB genes in a Chinese girl with Gitelman syndrome. BMC Pediatr 19, 114 (2019). https://doi.org/10.1186/s12887-019-1498-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12887-019-1498-3