
Abstract
Background
Mutations in the COL2A1 gene cause type II collagenopathies characterized by skeletal dysplasia with a wide spectrum of phenotypic severity. Most COL2A1 mutations located in the triple-helical region, and the glycine to bulky amino acid substitutions (e.g., glycine to serine) in the Gly-X-Y repeat were identified frequently. However, the same COL2A1 mutations are associated with different phenotypes and the genotype-phenotype relationship is still poorly understood. Therefore, the studies of more patients about the recurrent mutations in COL2A1 will be needed for further research to provide more comprehensive clinical and genetic data. In this paper, we report a rare recurrent c.G1636A (p.G546S) mutation in COL2A1 associated with different metaphyseal changes in a Chinese family.
Case presentation
The proband (III-3) was the second child of the family with skeletal dysplasia. She was 2 years and 3 months old with disproportional short stature, short neck, pectus carinatum, genu varum, bilateral pes planus, and obvious waddling gait. Notably, she displayed severe metaphyseal lesions, especially typical “dappling” and “corner fracture” appearance, whereas no particular metaphyseal involvement was detected in the proband’s mother (II-3) and elder sister (III-2) in the family. We identified a heterozygous mutation (c.1636G > A) in COL2A1 in the three patients, causing the substitution of glycine to serine in codon 546. Although the same mutation has been reported in two previous studies, the phenotypes of the previous patients were different from those of our patients, and the characteristic “dappling” and “corner fracture” metaphyseal abnormalities were not reported previously.
Conclusions
In this study, we identified a c.G1636A (p.G546S) mutation in the COL2A1 associated with different metaphyseal changes, which was never reported in the literature. Our findings revealed a different causative amino acid substitution (glycine to serine) associated with the “dappling” and “corner fracture” metaphyseal abnormalities, and may provide a useful reference for evaluating the phenotypic spectrum and variability of type II collagenopathies.
Similar content being viewed by others


Background
The type II collagen gene (COL2A1, MIM #108300) encodes the alpha 1(II) chain of procollagen type II, which is crucial for constructing functional collagen. Mutations in this gene cause type II collagenopathies, which are skeletal dysplasias with a wide spectrum of phenotypic severity [1]. The most severe phenotypes include achondrogenesis type II and hypochondrogenesis, which are associated with neonatal death [2]; the intermediately severe phenotypes, such as spondyloepiphyseal dysplasia congenita (SEDC) [3] and spondyloepimetaphyseal dysplasia (SEMD), Strudwick type [4], are associated with disproportionately short stature, abnormal epiphyses, scoliosis, and/or ocular conditions; and the mildest phenotypes, such as osteoarthritis [5] and stickler syndrome type I [6] manifesting only in late childhood or adulthood, and present as isolated joint or ocular disease.
According to the Leiden Open Variation Database (LOVD, http://databases.lovd.nl/shared /genes/COL2A1), 455 variations in COL2A1 have been reported (updated on March 24, 2016). Due to the rarity of recurrent mutations, no mutational hot spots have been identified. Type II collagen is a homotrimer composed of three alpha1 (II) chains. Each alpha 1 (II) chain contains a triple-helical structure formed by a characteristic Gly-X-Y repeat sequence. The X and Y position of the Gly-X-Y repeat are occupied by proline and hydroxyproline residues, respectively [7]. Most COL2A1 mutations are located in the triple-helical region, and glycine to bulky amino acid substitutions (e.g., glycine to serine) in the Gly-X-Y repeat have been identified frequently [8], however, the same COL2A1 mutation may cause different phenotypes and the genotype- phenotype relationship is still poorly understood. In this study, we identified a recurrent c.G1636A (p.G546S) COL2A1 mutation in a Chinese family. The clinical phenotypes of three affected family members were described. This mutation is associated with a specific spondyloepimetaphyseal dysplasia characterized by “dappling” and “corner fracture” metaphyseal abnormalities in one of the three family members with skeletal dysplasia, which was never reported in the previous literature.
Case presentation
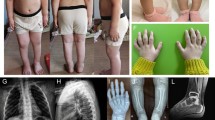
The pedigree of the patients is shown in Fig. 1a. The proband (III-3) was the second child in the family with skeletal dysplasia. She was born at 40+3 weeks of gestation by cesarean. Her birth length and weight were reported to be 46.0 cm (<3rd centile) and 2700 g (3rd–10th centile), respectively. She was brought to the Department of Pediatrics at the age of 2 years and 3 months for disproportional short stature. Her height was 66.5 cm (<3rd centile); her weight was 8.0 kg (<3rd centile); and her head circumference was 48.2 cm (50th–75th centile). Other physical examination findings included short neck, pectus carinatum, genu varum, bilateral pes planus, and an obvious waddling gait (Fig. 1b). Her early motor development was slightly delayed, while her intellectual development was normal. In contrast, the proband’s elder sister (III-2) displayed milder symptoms: she was born at 40+5 weeks of gestation by cesarean. Her birth length and weight were reported to be 48.0 cm (10th–25th centile) and 2800 g (10th–25th centile), respectively. She was brought to our clinic at the age of 8 years and 7 months. Her height was 108.5 cm (<3rd centile), and her weight was 21.0 kg (3rd–10th centile). Besides short stature, no remarkable abnormalities were found in the physical examination (Fig. 1c). The proband’s mother (II-3) was 33 years old when she received the physical examination. Her height was 128.5 cm (<3rd centile), and her weight was 35.2 kg (<3rd centile). Similar to her first child, no remarkable abnormalities were found except for the short stature. Unfortunately, she did not consent to taking pictures of her profile. None of the three patients displayed ocular defects, hearing impairment, inguinal hernia, or cleft palate.

Pedigree and pictures of the patients. a Pedigree of the patients. b Pictures of patient III-3. c Pictures of patient III-2
Radiographic examinations were performed on the three patients (Figs. 2 and 3). In general, the skeletal defects of patients II-3 and III-2 were milder than those of patient III-3. For patients II-3 and III-2, the major affected structures were the spine and epiphyses, whereas in patient III-3, skeletal defects were found in the spine, epiphyses, and pelvis. Notably, patient III-3 displayed severe metaphyseal lesions, especially a typical “dappling” and “corner fracture” appearance. In contrast, no particular metaphyseal involvement was detected in patients II-3 and III-2.

Radiographic findings of patient III-3. a Radiographic findings of the spine of patient III-3. The patient displayed platyspondyly (C3–C7), defects on the edge of the anterior vertebral bodies (L3–L5), and a slight shift of the vertebral axis. In addition, ovoid vertebral bodies, which are indicators of dysplasia, were observed in the CT images of the cervical spine. b Radiographic findings of the long bones of patient III-3. Bilateral humeri, ulnae, radii, femurs, and tibiofibulas were shortened. Bilateral femoral heads and necks, as well as the femoral head epiphyses and distal humeral epiphyses, were absent. The epiphyses of the upper humeri and distal tibias were dysplastic. The metaphyses of the proximal femurs and proximal humeri displayed a “dappling” appearance, resulting from the irregular intermingling of radiolucencies and radiodensities. The metaphyses in the proximal tibias were flared and irregular. Notably, “corner fracture” phenomena were observed in the right proximal humerus and bilateral femurs (arrows). c Radiographic findings of the pelvis of patient III-3. An irregular acetabular roof was observed

Radiographic findings of patients III-2 and II-3. a Radiographic findings of the spine of patient III-2. The patient displayed platyspondyly (C3–C6) and defective anterior vertebral bodies (T7–T12, particularly at the lower edge). b Radiographic findings of the long bones of patient III-2. Dysplasia was detected in the bilateral femoral heads and distal tibial epiphysis. No particular changes were found in the metaphyses. c Radiographic findings of the pelvis of patient III-2. No particular abnormalities were found in the pelvis. d Radiographic findings of the spine of patient II-3. Os odontoideum and atlantoaxial subluxation were observed in the CT scan of the cervical spine. Other findings included multiple Schmorl’s nodes (T5–T12), platyspondyly (C5–C6, T6–T9), lumbar lordosis, and a marked increase in the lumbosacral angle. e Radiographic findings of the long bones of patient II-3. Dysplasia was found in the bilateral femoral heads and distal tibial epiphysis. No particular changes were found in the metaphyses. f Radiographic findings of the pelvis of patient II-3. No particular abnormalities were found in the pelvis
Written informed consent was obtained from the patients (or guardian) and their family members for conducting the genetic tests and publishing the research data. The study protocol was approved by the ethics committee of Xiamen Maternal and Child Health Hospital. We collected peripheral blood samples from three generations of the patients’ family (Fig. 1a). Genomic DNA was extracted from 200 μL of blood using the Super/HF16 plus DNA Extraction System (MagCore, Xiamen, China) according to the manufacturer’s protocol. DNA samples from the three patients (II-3, III-2, and III-3) were analyzed by commercial whole exome sequencing (WES; Sinopath, Beijing, China). A guanine to adenosine change at position 1636 of the coding sequence of the COL2A1 gene (c.G1636A), leading to a corresponding glycine to serine change in the protein sequence (p.G546S), was detected in all of three patients by WES. The mutation was confirmed by Sanger sequencing. Related family members were also examined for this mutation by Sanger sequencing. The sequencing results revealed that the mutation found in patient II-3 was a de novo mutant because it was absent in the genomes of her parents (I-1 and I-2). In addition, a total of 15,116 variants were unique in the exome of patient III-3 compared to in patients II-3 and III-2, including three heterozygous variants in COL2A1 (Table 1).
Eventually, based on previous studies and the current classification of skeletal dysplasia [9,10,11,12,13], patient III-3 was diagnosed with a variant of SEMD, Strudwick type, and patients II-3 and III-2 were diagnosed with mild SEDC.
Discussion
In the differential diagnosis, the “dappling” metaphyseal appearance, which results from irregular ossification, is characteristic of SEMD, Strudwick type (MIM #184250), while the “corner fracture” metaphyseal appearance, which was considered as an extra ossification center, is characteristic of spondylometaphyseal dysplasia, corner fracture type (MIM #184255). Thus far, four publications have reported a phenotype similar to that of patient III-3, with a combination of “dappling” and “corner fracture” metaphyseal abnormalities, and COL2A1 mutations were also detected in the patient in those studies [9,10,11,12]. Including our patient, a total of six patients of different gender and race have been described who display similar phenotypes, including the characteristic “dappling” and “corner fracture” metaphyseal abnormalities, disproportional short stature, relatively mild abnormalities in the spine with platyspondyly, shortened long bones with relatively normal small tubular bones in the hands and feet, dysplasia of the femoral heads and necks, hip dysplasia, and genu varum/valgum (Table 2). According to previous studies and the current classification of skeletal dysplasia [9,10,11,12,13], this distinct phenotype was classified as a variant of SEMD, Strudwick type. An interesting finding from these studies is that most COL2A1 mutations associated with the “dappling” and “corner fracture” metaphyseal abnormalities were glycine to arginine substitutions (in four of six patients), which suggests a potential molecular mechanism. Although more patients are needed to delineate a possible molecular mechanism, our patient reveals a different causative amino acid substitution (glycine to serine), which expands the mutational spectrum of this specific phenotype. We anticipate more patients will be discovered, which will further delineate and decipher this specific variant of SEMD, Strudwick type.
Currently, the genotype-phenotype correlations in type II collagenopathies cannot be clarified for several reasons [14, 15]. First, there are no mutational hot spots, and most mutations are unique. Second, there is a wide range of phenotypic variation among patients, even in individuals who share the same mutation. Moreover, age-dependent transitions and/or other unidentified factors could also complicate the clinical phenotypes. However, the study of recurrent COL2A1 mutations provides an opportunity to gain insight into the phenotypic spectrum and variability of individual mutations or mutation groups, which could facilitate a more precise prognosis early in life, thus improving individualized medical care and patients’ quality of life.
Recurrent COL2A1 mutations have been reported in several studies; some mutations displayed similar phenotypes, while others displayed distinct phenotypes [8, 15,16,17,18,19,20]. For example, Silveira et al. reported clinical and radiological follow-up of six unrelated patients with a R989C mutation that was associated with a severe SEDC phenotype, which was consistent with the phenotypes of twelve other R989C mutation cases [18]. In contrast, three patients with a G504S mutation showed mild SEDC, SEDT, and severe SEDC phenotypes [8, 15]. Likewise, a G513S mutation in a 4-year-old was associated with mild SEDC, but was also associated with a lethal form of SEDC that resulted in neonatal death [15, 19]. Based on previous limited data, unlike glycine to non-serine substitutions, glycine to serine substitutions produced variable effects, with both inter- and intra-familial phenotypic variation [8, 15].
Two previous reports of the c.G1636A (p.G546S) mutation were found in the online database. Xu et al. reported the c.G1636A mutation in a familial case of SEDC [20]. Unlike our patients, the major skeletal abnormalities in Xu et al.’s patients were concordant among affected family members and included dysplasia of the femoral heads and necks, abnormal acetabular roofs, moderate or mild scoliosis, and thoracic hyperkyphosis. Most of these skeletal abnormalities were not found in our patients, except for dysplasia of the femoral heads and necks and abnormal acetabular roofs, which were observed in patient III-3. In addition, marked metaphyseal abnormalities were noted in one of our patients (III-3), which was distinct from the phenotypes of Xu et al.’s patients. Kaissi et al. reported another patient of a c.G1636A mutation in a patient in Germany [21]. As the authors stated in the English abstract, the patient was characterized by short stature associated with acetabulo femoral dysplasia, spinal osteochondritis (Scheuermann’s disease), and mild thoracic kyphosis. According to the limited phenotypic information, the skeletal abnormalities in this patient were similar to those observed in Xu et al.’s patients. Therefore, in agreement with the previous findings for glycine to serine substitutions [8, 15], in this study, patients with the G546S mutation show inter- and intra-familial phenotypic variation. Due to the small number of patients with insufficient genetic information and the complicated genotype-phenotype correlation, the reason why the same COL2A1 mutation causes different phenotypes is still unclear. A reasonable hypothesis is that in addition to the causative COL2A1 mutation in a critical domain, other genetic, epigenetic, and environmental factors can be attributed to inter- and intra-familial phenotypic variation by influencing the microenvironments within the collagen domains or complex interactions with other proteins [22]. In our WES data, numerous variants were found to be unique in the exome of patient III-3 compared to in the other two patients, particularly two variants in COL2A1: one was a benign c.2854 C > A (p.P952T) located outside the triple helix repeat domain while the other was a c.4317 + 43G > A variation located in the intron region (Table 1). These data provide potential candidates for gaining insight into the phenotypic spectrum and variability of type II collagenopathies. However, the contribution of these genetic variations should be further investigated in a larger number of clinical samples and functional studies using genetic animal models. The use of genome-wide strategies, e.g., genome-wide association study, whole genome/exome sequencing, and whole genome bisulfate sequencing, with large cohorts of patients may reveal the basis of the indefinite genotype-phenotype correlation of COL2A1.
Conclusion
Our case reported a recurrent c.G1636A (p.G546S) mutation of COL2A1 in a Chinese family with skeletal dysplasia. Specific spondyloepimetaphyseal dysplasia characterized by “dappling” and “corner fracture” metaphyseal abnormalities was observed in one of the three family members. Our finding revealed a different causative amino acid substitution (glycine to serine) associated with the “dappling” and “corner fracture” metaphyseal abnormalities, and may provide a useful reference for evaluating the phenotypic spectrum and variability of type II collagenopathies.
Abbreviations
- SEDC:
-
Spondyloepiphyseal dysplasia congenital
- SEMD:
-
Spondyloepimetaphyseal dysplasia
- WES:
-
Whole exome sequencing
References
Spranger J, Winterpacht A, Zabel B. The type II collagenopathies: a spectrum of chondrodysplasias. Eur J Pediatr. 1994;153(2):56–65.
Bogaert R, Tiller GE, Weis MA, Gruber HE, Rimoin DL, Cohn DH, Eyre DR. An amino acid substitution (Gly853-->Glu) in the collagen alpha 1(II) chain produces hypochondrogenesis. J Biol Chem. 1992;267(31):22522–6.
Anderson IJ, Goldberg RB, Marion RW, Upholt WB, Tsipouras P. Spondyloepiphyseal dysplasia congenita: genetic linkage to type II collagen (COL2AI). Am J Hum Genet. 1990;46(5):896–901.
Tiller GE, Polumbo PA, Weis MA, Bogaert R, Lachman RS, Cohn DH, Rimoin DL, Eyre DR. Dominant mutations in the type II collagen gene, COL2A1, produce spondyloepimetaphyseal dysplasia, Strudwick type. Nat Genet. 1995;11(1):87–9.
Beighton P, Christy G, Learmonth ID. Namaqualand hip dysplasia: an autosomal dominant entity. Am J Med Genet. 1984;19(1):161–9.
Stickler GB, Belau PG, Farrell FJ, Jones JD, Pugh DG, Steinberg AG, Ward LE. Hereditary progressive Arthro-Ophthalmopathy. Mayo Clin Proc. 1965;40:433–55.
Prockop DJ, Kivirikko KI. Collagens: molecular biology, diseases, and potentials for therapy. Annu Rev Biochem. 1995;64:403–34.
Terhal PA, Nievelstein RJ, Verver EJ, Topsakal V, van Dommelen P, Hoornaert K, Le Merrer M, Zankl A, Simon ME, Smithson SF, et al. A study of the clinical and radiological features in a cohort of 93 patients with a COL2A1 mutation causing spondyloepiphyseal dysplasia congenita or a related phenotype. Am J Med Genet A. 2015;167A(3):461–75.
Kaitila I, Korkko J, Marttinen E, Ala-Kokko L. Phenotypic expressions of a Gly 154Arg mutation in type II collagen in two unrelated patients with spondyloepimetaphyseal dysplasia (SEMD). Am J Med Genet. 1996;63(1):111–22.
Matsubayashi S, Ikema M, Ninomiya Y, Yamaguchi K, Ikegawa S, Nishimura G. COL2A1 mutation in Spondylometaphyseal dysplasia Algerian type. Mol Syndromol. 2013;4(3):148–51.
Vikkula M, Ritvaniemi P, Vuorio AF, Kaitila I, Ala-Kokko L, Peltonen L. A mutation in the amino-terminal end of the triple helix of type II collagen causing severe osteochondrodysplasia. Genomics. 1993;16(1):282–5.
Walter K, Tansek M, Tobias ES, Ikegawa S, Coucke P, Hyland J, Mortier G, Iwaya T, Nishimura G, Superti-Furga A, et al. COL2A1-related skeletal dysplasias with predominant metaphyseal involvement. Am J Med Genet A. 2007;143A(2):161–7.
Warman ML, Cormier-Daire V, Hall C, Krakow D, Lachman R, LeMerrer M, Mortier G, Mundlos S, Nishimura G, Rimoin DL, et al. Nosology and classification of genetic skeletal disorders: 2010 revision. Am J Med Genet A. 2011;155A(5):943–68.
Barat-Houari M, Dumont B, Fabre A, Them FT, Alembik Y, Alessandri JL, Amiel J, Audebert S, Baumann-Morel C, Blanchet P, et al. The expanding spectrum of COL2A1 gene variants IN 136 patients with a skeletal dysplasia phenotype. Eur J Hum Genet. 2016;24(7):992–1000.
Nishimura G, Haga N, Kitoh H, Tanaka Y, Sonoda T, Kitamura M, Shirahama S, Itoh T, Nakashima E, Ohashi H, et al. The phenotypic spectrum of COL2A1 mutations. Hum Mutat. 2005;26(1):36–43.
Chung BH, Luk HM, Lo IF, Lam ST, Li RH. A second report of p.Pro986Leu variant in COL2A1-phenotypic overlap with SEDC and other forms of type II collagenopathies. Am J Med Genet A. 2013;161A(4):918–20.
Hoornaert KP, Dewinter C, Vereecke I, Beemer FA, Courtens W, Fryer A, Fryssira H, Lees M, Mullner-Eidenbock A, Rimoin DL, et al. The phenotypic spectrum in patients with arginine to cysteine mutations in the COL2A1 gene. J Med Genet. 2006;43(5):406–13.
Silveira KC, Bonadia LC, Superti-Furga A, Bertola DR, Jorge AA, Cavalcanti DP. Six additional cases of SEDC due to the same and recurrent R989C mutation in the COL2A1 gene--the clinical and radiological follow-up. Am J Med Genet A. 2015;167A(4):894–901.
Mortier GR, Weis M, Nuytinck L, King LM, Wilkin DJ, De Paepe A, Lachman RS, Rimoin DL, Eyre DR, Cohn DH. Report of five novel and one recurrent COL2A1 mutations with analysis of genotype-phenotype correlation in patients with a lethal type II collagen disorder. J Med Genet. 2000;37(4):263–71.
Xu L, Qiu X, Zhu Z, Yi L, Qiu Y. A novel mutation in COL2A1 leading to spondyloepiphyseal dysplasia congenita in a three-generation family. Eur Spine J. 2014;23(Suppl 2):271–7.
Al Kaissi A, Laccone F, Karner C, Ganger R, Klaushofer K, Grill F. Hip dysplasia and spinal osteochondritis (Scheuermann's disease) in a girl with type II manifesting collagenopathy. Orthopade. 2013;42(11):963–8.
Deng H, Huang X, Yuan L. Molecular genetics of the COL2A1-related disorders. Mutat Res Rev Mutat Res. 2016;768:1–13.
Acknowledgments
We thank the family for their cooperation.
Funding
This work was supported by the Natural Science Foundation for Distinguished Young Scholars of Fujian Province (project no. 2015D012), Natural Science Foundation of Fujian Province (project no. 2014D003), the Medical Innovation Foundation of Fujian Province (project no. 2014-CXB-46), and the Science and Technology Project of Xiamen City (project no. 3502Z20164029).
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Author information
Authors and Affiliations
Contributions
JC cared for the patient, collected samples, and drafted the manuscript. QG designed the study, analyzed the sequencing results, and revised the manuscript.GL advised the study and critically read the manuscript. XM analyzed the radiographic results. YZ advised the study and critically read the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was performed in accordance with the Declaration of Helsinki, after written informed consent obtained from the participants or legal guardians, and approved by the Human Research Ethics Committee of Xiamen Maternal and Child Health Hospital (KY-2016002).
Consent for publication
Written informed consent was obtained from the participants or legal guardians for publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor of BMC Pediatrics.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Chen, J., Ma, X., Zhou, Y. et al. Recurrent c.G1636A (p.G546S) mutation of COL2A1 in a Chinese family with skeletal dysplasia and different metaphyseal changes: a case report. BMC Pediatr 17, 175 (2017). https://doi.org/10.1186/s12887-017-0930-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12887-017-0930-9