
Abstract
Purpose
To describe the clinical features, imaging characteristics, and genetic test results associated with a novel compound heterozygous mutation of the BEST1 gene in two siblings with autosomal recessive bestrophinopathy.
Methods
Two siblings underwent a complete ophthalmic examination, including dilated fundus examination, fundus photography, fundus autofluorescence imaging, spectral-domain optical coherence tomography, fluorescein angiography, electroretinography, and electrooculography. A clinical diagnosis of autosomal recessive bestrophinopathy was established based on ocular examination and multimodal retinal imaging. Subsequently, clinical exome sequencing consisting of a panel of 6670 genes was carried out to confirm the diagnosis and assess genetic alterations in the protein-coding region of the genome of the patients. The identified mutations were tested in the two affected siblings and one of their parents.
Results
Two siblings (a 17-year-old female and a 15-year-old male) presented with reduced visual acuity and bilaterally symmetrical subretinal deposits of hyperautofluorescent materials in the posterior pole, which showed staining in the late phase of fluorescein angiogram. Spectral-domain optical coherence tomography demonstrated hyperreflective subretinal deposits and subretinal fluid accumulation. Both patients shared two mutations in the protein-coding region of the BEST1 gene, c.103G > A, p.(Glu35Lys) and c.313C > A, p.(Arg105Ser) (a novel disease-causing mutation). Sanger sequencing confirmed that the unaffected mother of the proband was carrying p.(Glu35Lys) variant in a heterozygous state.
Conclusions
We have identified and described the phenotype of a novel disease-causing mutation NM_004183.4:c.313C > A, p.(Arg105Ser) in a heterozygous state along with a previously reported mutation NM_004183.4:c.103G > A, p.(Glu35Lys) of the BEST1 gene in two related patients with autosomal recessive bestrophinopathy.
Similar content being viewed by others

Introduction
The BEST1 (alternatively VMD2, RP50, BMD) gene located on chromosome GRCh38 11q12.3 encodes a transmembrane pentameric protein consisting of 585 amino acids with a highly conserved N-terminal region followed by four transmembrane domains (amino acids 1–390) and a carboxy-terminal region (amino acids 391–585) [1]. Structural models of the BEST1 propose the N- and C-termini as being cytosolic with four transmembrane domains (domains 1, 2, 5, and 6) and two cytoplasmic domains (domains 3 and 4) [2, 3]. The protein is predominantly expressed in the basolateral plasma membrane of the retinal pigment epithelium (RPE) and functions as a calcium-activated chloride channel (CaCC) which regulates the flow of chloride and other monovalent anions across cellular membranes in response to intracellular calcium levels [4,5,6,7,8]. Mutation of the BEST1 gene has been associated with lipofuscin accumulating within and beneath the RPE and degeneration of the RPE and the overlying photoreceptors [9]. A wide range of ocular phenotypes resulting from mutations in the BEST1 gene have been described and are collectively termed bestrophinopathies. [10, 11] Autosomal recessive bestrophinopathy (ARB) may result from a total absence (null phenotype) of functional BEST1 protein in the RPE, [12, 13] improper localization to the cell membrane with intact anion channel activity, [14] or lack of the anion channel activity [15].
Schatz et al., in 2006, first described a variant of Best macular dystrophy in two members of a Swedish family presenting with reduced vision, multifocal retinal deposits, and intraretinal cystic changes, harboring biallelic mutations in the BEST1 gene. [16] In 2008, Burgess et al. coined the term autosomal recessive bestrophinopathy (ARB) and identified it as the third distinct phenotype resulting from mutations in the BEST1 gene. [12] Other described phenotypes associated with pathogenic variants of the BEST1 gene include Best vitelliform macular dystrophy (BVMD) [17, 18], adult vitelliform macular dystrophy (AVMD), autosomal dominant vitreoretinochoroidopathy (ADVIRC), [19, 20] autosomal dominant microcornea, rod-cone dystrophy, early-onset cataract, and posterior staphyloma (MRCS) [11], rod-cone dystrophy and retinitis pigmentosa [11, 21]. In contrast to other phenotypes of bestrophinopathies that result from dominant mutations, ARB is associated with recessive biallelic mutations in the BEST1 gene [12, 22, 23].
Patients with ARB typically present in the first two decades of life but may remain asymptomatic as late as the fifth decade [12, 15, 24]. The clinical features of ARB include a gradual and progressive visual loss, hyperopia, predominantly peri-macular sub-retinal yellowish deposits of lipofuscin, seen as hyperautofluorescent areas, accumulation of subretinal and/or intraretinal fluid, absence of light peak in electrooculography, normal or reduced electroretinogram, and sometimes associated with shallow anterior chambers and reduced axial length predisposing the affected patients to angle-closure glaucoma. [12, 25, 26] Full-field electroretinography is typically normal early on in the disease and shows abnormal results from late childhood or adolescence, indicating generalized rod and cone dysfunction. In addition, pattern electroretinography evidence of macular dysfunction is also seen. [12] This article presents the results of clinical evaluation, multimodal imaging, electrophysiological tests, and genetic investigations of two siblings with ARB.
Methods
Clinical investigation
Clinical investigations in patients included a detailed history and physical examination, slit-lamp biomicroscopy, indirect ophthalmoscopy, fundus photography, fundus autofluorescence imaging (FAF), optical coherence tomography (OCT), fluorescein angiography (FA), full-field electroretinography (ERG), and electrooculography (EOG). The ERG and EOG were performed per the guidelines of the International Society for Clinical Electrophysiology of Vision (www.iscev.org).
Genetic analysis
Whole exome sequencing
DNA isolation, exome library preparation, and sequencing
DNA was isolated from the patient’s whole blood sample using QIAamp DNA Blood Mini Kit (QIAGEN, CA, US) and subjected to targeted gene capture using MedGenome Clinical Exome (Ver. 4) which captures a panel of 6670 protein-coding genes. The libraries thus generated were sequenced to mean coverage of > 80-100X on the Illumina HiSeq 4000 sequencing platform (Illumina, CA, US). 100% of the protein-coding region of the BEST1 gene was covered.
Variant calling and annotation
The Genome Analysis Toolkit (GATK) best practices framework was followed to identify the variants in the sample using Sentieon (v201808.07). The sequencing reads were aligned to the human reference genome (GRCh38.p13) using the Sentieon aligner. Sentieon’s version of GATK (IndelRealigner) was used to perform local realignment in regions containing potential indels. Sentieon’s version of GATK Toolkit – BaseRecalibrator was used to recalibrate the quality scores of all the reads [27]. Sentieon DNASeq (v201808.07) HaplotypeCaller was used to identify variants. Gene annotation of the variants was performed using the VEP program against the Ensembl release 99 human gene model [28, 29]. In addition to SNVs and small Indels, copy number variants (CNVs) were detected from targeted sequence data using the ExomeDepth (v1.1.10) method [30]. Clinically relevant mutations were annotated using published variants in literature and a set of disease databases—ClinVar [31], OMIM [32] (updated on 11th May 2020), GWAS [33], HGMD (v2020.2) [34], and SwissVar [35].
Variant filtering and analysis
To identify candidate variants, we selected the variations if their minor allele frequencies are less than 0.05 in 1000 Genome Project [36], gnomAD [37], dbSNP [38], Exome Variant Server [39], 1000 Japanese Genome [40], and internal Indian population database. The identified variations were classified into pathogenic, likely pathogenic, VUS, likely benign, and benign groups according to the variant interpretation guidelines of the American College of Medical Genetics and Genomics (ACMG) [41]. Furthermore, all nucleotide variants present in BEST1 were reviewed. The genes and corresponding variants that qualified these filtering criteria were investigated to determine their significance and relevance in Bestrophinopathy. 8.65 Gb of raw sequencing data was generated, of which > 88% of raw reads passed the on-target alignment. > 90% of the targeted base qualified the Phred score Q30.
Sanger sequencing
Sanger sequencing was performed in the proband and the mother to validate the variants identified by whole exome sequencing and identify the mutation in the parent. Sanger sequencing was performed with these primers:
F1: 5′-ATCGGTGTCCCTCTCTACCA-3′, R1: 5′-CTATGTGGGCCTATGAGTCTG-3′; F2: 5′-CGTCCTGCCGTTAGCAATG-3′, R2: 5′-CACCTTCAGACACCCGACT-3′. The reference sequence NM_004183 of BEST1 was used.
Bioinformatics analysis
The potential functional impact of all the candidate variants was investigated using three programs, including PolyPhen2 (http://genetics.bwh.harvard.edu/pph/, in the public domain), Mutation Taster (http://www.mutationtaster.org/, in the public domain), and SIFT (http://sift.jcvi.org/, in the public domain).
Results
Clinical findings
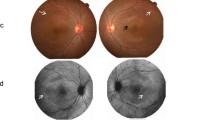
Patient A (proband), a 17-year-old female, reported blurred distance vision in both eyes for three years. Her best-corrected visual acuity was 6/9 in both eyes, which did not change during the follow-up of 1 year. Slit-lamp examination of the anterior segment of both patients was unremarkable. The axial length measured by optical biometry was 21.80 mm and 21.64 mm in the right and the left eye, respectively. Dilated fundus examination revealed bilateral and symmetrical, multifocal subretinal yellowish deposits in the posterior pole and upper nasal region, with peripapillary sparing (Fig. 1A, B). On FAF, the yellowish deposits appeared as hyper-autofluorescent spots surrounding an area of hypo-autofluorescence (Fig. 1C, D). The deposits showed staining in the late phase of the fluorescein angiogram (Fig. 1E, F). On OCT, subretinal fluid and intraretinal hyporeflective spaces (schisis) located predominantly in the outer nuclear layer (ONL) were seen along with elongated photoreceptors and hyperreflective deposits in the subretinal space bilaterally (Fig. 1G, H). The electroretinogram (ERG) was normal, and an absent light peak was noted on EOG.

Clinical features of patient A (proband). A 17-year-old otherwise healthy female presented with blurred distance vision in both eyes, which she first noticed when she was 14 years old. Colour fundus photographs (A, B) show bilateral and symmetrical, multifocal subretinal yellowish deposits in the posterior pole and upper nasal region, with notable peripapillary sparing. The yellowish deposits are hyperautofluorescent on blue light fundus autofluorescence (C, D) and circumscribe areas of hypoautofluorescence. On fluorescein angiography (E, F), the yellowish deposits show diffuse staining in the late phase. Horizontal spectral-domain optical coherence tomography images through the right and left fovea (G, H) show center-involving subretinal fluid and thickening of the ellipsoid zone, with elongation of the photoreceptor outer segments and deposits in the subretinal space. Additionally, intraretinal hyporreflective areas predominantly located in the outer nuclear layer can be seen
Patient B, a 15-year-old male, reported a unilateral decrease in visual acuity and inward deviation of the left eye since the age of four. On ocular examination, he was found to have left esotropia of 15 prism diopters with prescribed correction and 25 prism diopters without the correction for distance. His best-corrected visual acuity was 6/6 in his right eye and 5/60 in his left eye, with an accommodative-convergence over accommodation (AC/A) ratio of 2:1. Slit-lamp examination of the anterior segment was unremarkable. The axial length measured by optical biometry was 21.61 mm and 21.54 mm in the right and the left eye, respectively. Dilated fundus examination revealed two circumscribed areas of bilaterally symmetrical, multifocal subretinal yellowish deposits, one in the posterior pole and the other in the upper nasal region, with peripapillary sparing. (Fig. 2A, B). The yellowish lesions were hyper-autofluorescent surrounding an area of hypo-autofluorescence on FAF (Fig. 2C, D) and showed staining in the late phase of fluorescein angiogram (Fig. 2E, F). On OCT, subretinal fluid (seen as subfoveal hyporeflective space), elongated photoreceptors along with hyperreflective deposits in the subretinal area were observed bilaterally (Fig. 2G, H). The electroretinogram (ERG) was normal, and an absent light peak was noted on EOG.

Clinical features of patient B. A 15-year-old otherwise healthy male presented with a unilateral decrease in visual acuity and inward deviation of the left eye, which was first noticed by his parents when he was four. On colour fundus photographs of the right and the left eye (A, B), two separate areas of bilateral and symmetrical, multifocal subretinal yellowish deposits are seen in the posterior pole and upper nasal region, with notable peripapillary sparing. On blue light fundus autofluorescence (C, D), the yellowish deposits are hyperautofluorescent, and circumscribed areas show hypoautofluorescence. In the late phase of fluorescein angiography (E, F), diffuse staining, seen as diffuse hyperfluorescence of the yellowish deposits is observed. Horizontal spectral-domain optical coherence tomography scans through the fovea (G, H) show center-involving subretinal fluid and thickening of the ellipsoid zone, with elongation of the photoreceptor outer segments and deposits in the subretinal area
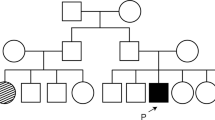
The patients were born of a non-consanguineous marriage from parents of North Indian descent. The parents and unaffected siblings were examined; however, no ocular or systemic abnormalities were observed. (Fig. 3). Both affected siblings were treated with topical carbonic anhydrase inhibitors and followed up for one year. The subfoveal fluid did not improve after one year of treatment. The clinical, imaging, and electrophysiological findings of the affected patients are summarized in Table 1.

Pedigree of the family with two affected members. Patient A, a 17-year-old female (black arrow), and Patient B, a 15-year-old male
Genetic findings
A heterozygous missense mutation, NM_004183.4(BEST1):c.103G > A, was found in exon 2 of the BEST1 gene in both patients (chr11: g.61951909G > A; c.103G > A) and was further validated by Sanger sequencing (Fig. 4A). It resulted in the amino acid substitution of Glutamic acid (negatively charged) for Lysine (positively charged) at codon 35, NM_004183.4:p.(Glu35Lys) (Table 2). The variant is classified as likely pathogenic in the ClinVar database [31] and lies in the RFP-TM, chloride channel domain of the bestrophin protein. The NM_004183.4:p.(Glu35Lys) variant has not been reported in the 1000 genomes [36] and gnomAD databases [37] (accessed 30th January 2022). The in-silico predictions according to PolyPhen-2 is to be probably damaging; and deleterious according to SIFT and MutationTaster2. The reference codon is evolutionarily conserved in mammals.

A Sequence chromatogram and alignment to the reference sequence showing the variant in exon 2 of the BEST1 gene (chr11:g.61951909G > A; c.103G > A; p.Glu35Lys) detected in heterozygous condition in the proband and the unaffected mother. B The variant in exon 4 of the BEST1 gene (chr11:g.61955783C > A; c.313C > A), was detected in the proband but not in the unaffected mother. The reference sequence NM_004183 of BEST1 was used
Another heterozygous missense mutation, NM_004183.4(BEST1):c.313C > A, was found in exon 4 of the BEST1 gene in both patients (chr11:g.61955783C > A; c.313C > A), and was further validated by Sanger sequencing (Fig. 4B). It resulted in the amino acid substitution of Arginine (positively charged) for Serine (uncharged) at codon 105, NM_004183.4:p.(Arg105Ser) (Table 2). The variant lies in the RFP-TM, chloride channel domain of the bestrophin protein. The NM_004183.4:p.(Arg105Ser) variant has not been reported in the 1000 genomes databases [36] and has a minor allele frequency of 0.0007% in the gnomAD database (accessed 30th January 2022) [37]. The in-silico predictions of the variant according to PolyPhen-2 is to be probably damaging, and deleterious according to SIFT and MutationTaster2. The reference codon is evolutionarily conserved in mammals. This mutation has not been previously reported in patients with ARB or VMD. The results of next-gen sequencing are summarized in Table 2. The IGV depicting the distribution of alternate allele and wild type allele for both variations is shown in Supplementary figure S 1A and S 1B.
We analyzed the mother for both variants through Sanger sequencing to clarify whether the two mutations were located on separate BEST1 alleles. The NM_004183.4:p.(Glu35Lys) was detected in a heterozygous condition along with the wild type in the unaffected mother of the proband (Fig. 4A, B).
Discussion
This report analyzed the clinical and imaging characteristics along with the genetic test results of two siblings with ARB. The diagnosis of ARB was established based on clinical observation and multimodal retinal imaging and further confirmed by whole exome sequencing and Sanger sequencing. Patient A demonstrated good central acuity, as seen in other patients with ARB in the first and second decade of life [42]. We noted that the sibling (patient B) had poor visual acuity in one eye due to amblyopia resulting from uncorrected esotropia. In addition, both the patients had short axial lengths in both eye without any abnormal iridocorneal anatomic features or shallow anterior chamber depth. Reduced axial length predisposes patients to angle-closure glaucoma, potentially leading to a further visual decline [42].
The whole exome sequencing revealed a likely compound heterozygous mutations in the BEST1 gene shared by both siblings that likely led to ARB. The variants were validated by Sanger sequencing. One of the alleles carried a missense mutation in exon 2 NM_004183.4(BEST1):c.103G > A, which resulted in the amino acid substitution from negatively-charged Glutamic acid to positively-charged Lysine at the 35th amino acid residue, NM_004183.4:p.(Glu35Lys). This variant was detected in the unaffected mother in a heterozygous state along with the wild type using Sanger sequencing. The variant has been submitted to ClinVar (accession number- RCV000356527) and has previously been reported by Tian et al. and Habibi et al., albeit in a homozygous state [43, 44]. To our knowledge, ours is the first study to report this variant in a compound heterozygous state.
Another variation was observed to be a transversion in exon 4 NM_004183.4(BEST1):c.313C > A, which resulted in the amino acid substitution from positively-charged Arginine to uncharged Serine at 105th amino acid residue, NM_004183.4:p.(Arg105Ser) (Table 2). To our knowledge, this mutation has not been reported previously in patients with either ARB or VMD. This variant is predicted to be pathogenic. The NM_004183.4(BEST1):c.313C > A (p.Arg105Ser) has been submitted to ClinVar previously (accession number- RCV002025371.1) [31]. Two other disease-causing mutations affecting the same codon, p.Arg105Gly in patients with BMD, and p.Arg105Cys in a 69-year-old patient with age-related macular degeneration, have been reported [45, 46]. Interestingly, the p.Arg105Gly mutation resulted in additional extramacular multifocal deposits similar to ARB in three patients with BVMD [45].
Notably, the mutations discovered in this study are localized to the N-terminal region (Fig. 5A-C). The mutation NM_004183.4:p.(Glu35Lys) localizes to the first transmembrane domain, while the NM_004183.4:p.(Arg105Ser) mutation alters an amino acid in the cytoplasmic region distal to the second transmembrane domain (Fig. 5B). The amino acids at these positions are conserved among mammals (Fig. 5D). Among the roughly 335 mutations reported in BEST1 thus far, only about 40 compound heterozygous and homozygous mutations are associated with ARB. [26, 34]

A Schematic representation of BEST1 (NM_004183.4) transcript. Grey boxes represent exons, and lines connecting them represent introns. Green boxes represent the translation start site, while red boxes represent the translation termination site. B Topological representation of BEST1 (Milenkovic et al.) representing mutation sites denoted by red circles. C Generated structural model of BEST1 wild type, E35K, and R105S. D Multiple sequence alignment of BEST1 from different species
Although the detailed pathophysiology that leads to the disease is poorly understood, most of the characterized BEST1 mutations alter the electrophysiological properties of the calcium-activated chloride channel (CaCC), which is thought to be determined by the N-terminus portion of BEST1, affecting the flow to chloride across the RPE [1, 5]. Crystallographic studies of the wild type and mutated proteins suggest that BEST1 variants alter the cytoplasmic pore structure, which affects the permeability of anions or anion-cation selectivity, leading to lipofuscin accumulation and degeneration of the RPE (Fig. 5C) [47].
What this study adds
This study expands the genetic spectrum of the BEST1 variants associated with an ARB phenotype by reporting a novel variant p.(Arg105Ser), found in compound heterozygosity with another clinically significant variant in two affected siblings. Furthermore, the reported variant p.(Arg105Ser) variant appears to contribute to the ARB phenotype as the other variant alone did not cause any disease in the carrier (unaffected mother).
Limitations
Although it does not affect the diagnosis, the genetic testing of the unaffected father and other unaffected siblings would have been ideal but could not be carried out.
Availability of data and materials
All data generated or analyzed during this study are included in this published article. Accession number for the variants: SRR20990872.
Abbreviations
- AC:
-
Anterior Chamber
- ARB:
-
Autosomal recessive bestrophinopathy
- BCVA:
-
Best-corrected visual acuity
- BMD/BVMD:
-
Best macular dystrophy/Best vitelliform macular dystrophy
- EOG:
-
Electroretinogram
- ERG:
-
Electrooculogram
- FA:
-
Fluorescein angiography
- FAF:
-
Fundus autofluorescence
- GWAS:
-
Genome-wide association studies
- HGMD:
-
Human Gene Mutation Database
- NGS:
-
Next-gen sequencing
- OCT:
-
Optical coherence tomography
- OMIM:
-
Online Mendelian Inheritance in Man
- RPE:
-
Retinal pigment epithelium
- SIFT:
-
Sorting Intolerant From Tolerant
- SRF:
-
Subretinal fluid
- VMD:
-
Vitelliform macular dystrophy
- VUS:
-
Variant of uncertain significance
References
Xiao Q, Prussia A, Yu K, Cui YY, Hartzell HC. Regulation of bestrophin Cl channels by calcium: role of the C terminus. J Gen Physiol. 2008;132(6):681–92.
Milenkovic VM, Rivera A, Horling F, Weber BHF. Insertion and topology of normal and mutant bestrophin-1 in the endoplasmic reticulum membrane. J Biol Chem. 2007;282(2):1313–21.
Moskova-Doumanova V, Pankov R, Lalchev Z, Doumanov J. Best1 Shot Through the Eye—Structure, Functions and Clinical Implications of Bestrophin-1 Protein. Biotechnol Biotechnol Equip. 2013;27(1):3457–64.
Marmorstein AD, Marmorstein LY, Rayborn M, Wang X, Hollyfield JG, Petrukhin K. Bestrophin, the product of the Best vitelliform macular dystrophy gene (VMD2), localizes to the basolateral plasma membrane of the retinal pigment epithelium. PNAS. 2000;97(23):12758–63.
Kane Dickson V, Pedi L, Long SB. Structure and insights into the function of a Ca(2+)-activated Cl(-) channel. Nature. 2014;516(7530):213–8.
Sun H, Tsunenari T, Yau KW, Nathans J. The vitelliform macular dystrophy protein defines a new family of chloride channels. Proc Natl Acad Sci U S A. 2002;99(6):4008–13.
Tsunenari T, Sun H, Williams J, Cahill H, Smallwood P, Yau KW, et al. Structure-function analysis of the bestrophin family of anion channels. J Biol Chem. 2003;278(42):41114–25.
Yang T, Liu Q, Kloss B, Bruni R, Kalathur RC, Guo Y, et al. Structure and selectivity in bestrophin ion channels. Science. 2014;346(6207):355–9.
O’Gorman S, Flaherty WA, Fishman GA, Berson EL. Histopathologic findings in Best’s vitelliform macular dystrophy. Arch Ophthalmol. 1988;106(9):1261–8.
Johnson AA, Guziewicz KE, Lee CJ, Kalathur RC, Pulido JS, Marmorstein LY, et al. Bestrophin 1 and retinal disease. Prog Retin Eye Res. 2017;58:45–69.
Boon CJF, Klevering BJ, Leroy BP, Hoyng CB, Keunen JEE, den Hollander AI. The spectrum of ocular phenotypes caused by mutations in the BEST1 gene. Prog Retin Eye Res. 2009;28(3):187–205.
Burgess R, Millar ID, Leroy BP, Urquhart JE, Fearon IM, De Baere E, et al. Biallelic Mutation of BEST1 Causes a Distinct Retinopathy in Humans. Am J Hum Genet. 2008;82(1):19–31.
Davidson AE, Millar ID, Burgess-Mullan R, Maher GJ, Urquhart JE, Brown PD, et al. Functional Characterization of Bestrophin-1 Missense Mutations Associated with Autosomal Recessive Bestrophinopathy. Invest Ophthalmol Vis Sci. 2011;52(6):3730–6.
Johnson AA, Bachman LA, Gilles BJ, Cross SD, Stelzig KE, Resch ZT, et al. Autosomal Recessive Bestrophinopathy Is Not Associated With the Loss of Bestrophin-1 Anion Channel Function in a Patient With a Novel BEST1 Mutation. Invest Ophthalmol Vis Sci. 2015;56(8):4619–30.
Lee CS, Jun I, Choi SI, Lee JH, Lee MG, Lee SC, et al. A Novel BEST1 Mutation in Autosomal Recessive Bestrophinopathy. Invest Ophthalmol Vis Sci. 2015;56(13):8141–50.
Schatz P, Klar J, Andréasson S, Ponjavic V, Dahl N. Variant Phenotype of Best Vitelliform Macular Dystrophy Associated with Compound Heterozygous Mutations in VMD2. Ophthalmic Genet. 2006;27(2):51–6.
Marquardt A, Stöhr H, Passmore LA, Krämer F, Rivera A, Weber BH. Mutations in a novel gene, VMD2, encoding a protein of unknown properties cause juvenile-onset vitelliform macular dystrophy (Best’s disease). Hum Mol Genet. 1998;7(9):1517–25.
Petrukhin K, Koisti MJ, Bakall B, Li W, Xie G, Marknell T, et al. Identification of the gene responsible for Best macular dystrophy. Nat Genet. 1998;19(3):241–7.
Yardley J, Leroy BP, Hart-Holden N, Lafaut BA, Loeys B, Messiaen LM, et al. Mutations of VMD2 splicing regulators cause nanophthalmos and autosomal dominant vitreoretinochoroidopathy (ADVIRC). Invest Ophthalmol Vis Sci. 2004;45(10):3683–9.
Burgess R, MacLaren RE, Davidson AE, Urquhart JE, Holder GE, Robson AG, et al. ADVIRC is caused by distinct mutations in BEST1 that alter pre-mRNA splicing. J Med Genet. 2009;46(9):620–5.
Davidson AE, Millar ID, Urquhart JE, Burgess-Mullan R, Shweikh Y, Parry N, et al. Missense mutations in a retinal pigment epithelium protein, bestrophin-1, cause retinitis pigmentosa. Am J Hum Genet. 2009;85(5):581–92.
Boon CJF, Klevering BJ, den Hollander AI, Zonneveld MN, Theelen T, Cremers FPM, et al. Clinical and genetic heterogeneity in multifocal vitelliform dystrophy. Arch Ophthalmol. 2007;125(8):1100–6.
Boon CJF, Theelen T, Hoefsloot EH, van Schooneveld MJ, Keunen JEE, Cremers FPM, et al. Clinical and molecular genetic analysis of best vitelliform macular dystrophy. Retina. 2009;29(6):835–47.
Borman AD, Davidson AE, O’Sullivan J, Thompson DA, Robson AG, De Baere E, et al. Childhood-onset autosomal recessive bestrophinopathy. Arch Ophthalmol. 2011;129(8):1088–93.
Crowley C, Paterson R, Lamey T, McLaren T, De Roach J, Chelva E, et al. Autosomal recessive bestrophinopathy associated with angle-closure glaucoma. Doc Ophthalmol. 2014;129(1):57–63.
Kinnick TR, Mullins RF, Dev S, Leys M, Mackey DA, Kay CN, et al. Autosomal recessive vitelliform macular dystrophy in a large cohort of vitelliform macular dystrophy patients. Retina. 2011;31(3):581–95.
Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26(5):589–95.
McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26(16):2069–70.
Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J, et al. Ensembl 2018. Nucleic Acids Res. 2018;46(D1):D754–61.
Plagnol V, Curtis J, Epstein M, Mok KY, Stebbings E, Grigoriadou S, et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics. 2012;28(21):2747–54.
Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44(D1):D862-868.
Amberger J, Bocchini CA, Scott AF, Hamosh A. McKusick’s Online Mendelian Inheritance in Man (OMIM®). Nucleic Acids Res. 2009;37(Database issue):D793-6.
Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42(Database issue):D1001-1006.
Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, et al. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet. 2017;136(6):665–77.
Mottaz A, David FPA, Veuthey AL, Yip YL. Easy retrieval of single amino-acid polymorphisms and phenotype information using SwissVar. Bioinformatics. 2010;26(6):851–2.
1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68–74.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–43.
Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29(1):308–11.
Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA (https://www.evs.gs.washington.edu/EVS/).
Nagasaki M, Yasuda J, Katsuoka F, Nariai N, Kojima K, Kawai Y, et al. Rare variant discovery by deep whole-genome sequencing of 1,070 Japanese individuals. Nat Commun. 2015;6:8018.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Casalino G, Khan KN, Armengol M, Wright G, Pontikos N, Georgiou M, et al. Autosomal Recessive Bestrophinopathy: Clinical Features, Natural History, and Genetic Findings in Preparation for Clinical Trials. Ophthalmology. 2021;128(5):706–18.
Habibi I, Falfoul Y, Todorova MG, Wyrsch S, Vaclavik V, Helfenstein M, et al. Clinical and Genetic Findings of Autosomal Recessive Bestrophinopathy (ARB). Genes. 2019;10(12):953.
Tian L, Sun T, Xu K, Zhang X, Peng X, Li Y. Screening of BEST1 Gene in a Chinese Cohort With Best Vitelliform Macular Dystrophy or Autosomal Recessive Bestrophinopathy. Invest Ophthalmol Vis Sci. 2017;58(9):3366–75.
Glavač D, Jarc-Vidmar M, Vrabec K, Ravnik-Glavač M, Fakin A, Hawlina M. Clinical and genetic heterogeneity in Slovenian patients with BEST disease. Acta Ophthalmol. 2016;94(8):e786–94.
Lotery AJ, Munier FL, Fishman GA, Weleber RG, Jacobson SG, Affatigato LM, et al. Allelic variation in the VMD2 gene in best disease and age-related macular degeneration. Invest Ophthalmol Vis Sci. 2000;41(6):1291–6.
Vaisey G, Miller AN, Long SB. Distinct regions that control ion selectivity and calcium-dependent activation in the bestrophin ion channel. Proc Natl Acad Sci U S A. 2016;113(47):E7399–408.
Acknowledgements
MedGenome Labs Limited, Bengaluru, India for the assistance in the genetic workup of the study participants. Dr. Shaik Mohammed Zakir and Dr. Abdul Waris, Institute of Ophthalmology, Jawaharlal Nehru Medical College, Aligarh, India for helping to acquire fundus images and patient care.
Funding
None.
Author information
Authors and Affiliations
Contributions
OIH contributed to the patient’s care, design and implementation of the research, IRB approval, clinical workup, and follow-up of the subjects, investigations, genetic testing of siblings and the parent, analysis of the results, draft preparation, acquiring images, preparing Figs. 1– 5, editing, submission, and revision of the manuscript. AC contributed towards the genetic analysis of the mother and proband, preparing Fig. 4, supplementary Fig. 1 AB, submission of genetic data, and revision of the manuscript; FN analyzed genetic test results and prepared the topological BEST1 protein model in Fig. 5; OA edited the genetic section of the manuscript, analyzed genetic test results and prepared Fig. 5; JPM reviewed, edited, and revised the manuscript and wrote the captions of Figs. 1 and 2, and provided inputs as a consultant for clinical insight into the disease; TA was involved in the genetic testing of the unaffected mother and revision of the manuscript. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Jawaharlal Nehru Medical College Institutional Ethics Committee. It was performed per institutional ethics guidelines per the tenets of the Declaration of Helsinki. Written informed consent for the study was obtained from the parents. The assent of the minors was obtained prior to the study.
Consent for publication
Not Applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
{kind=link}
Cite this article
Haque, O.I., Chandrasekaran, A., Nabi, F. et al. A novel compound heterozygous BEST1 gene mutation in two siblings causing autosomal recessive bestrophinopathy. BMC Ophthalmol 22, 493 (2022). https://doi.org/10.1186/s12886-022-02703-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12886-022-02703-5