
Abstract
Background
Primary orbital melanoma is a rare disease and can occasionally develop from a pre-existing neoplasm of the blue naevus family of melanocytic lesions.
Case presentation
Herein we report a rare case of primary orbital melanoma arising from an unusual atypical diffuse (plaque-like) blue naevus/melanocytosis. A 27 year old man presented with mild pain and swelling of the left eye. Magnetic Resonance Imaging revealed a left lateral episcleral orbital mass and an incisional biopsy confirmed the diagnosis of malignant melanoma. Skin-sparing total left orbital exenteration was performed. Histopathological examination of the exenteration specimen revealed a primary orbital melanoma arising in a pre-existing blue naevus like melanocytosis.
We demonstrate the evidence for histological progression, characterise the molecular profile of this tumour and discuss the related literature.
Conclusions
This case emphasises the importance of a meticulous clinicopathological correlation in recognising such a tumour as a primary orbital melanoma rather than a metastasis, which is managed differently.
Similar content being viewed by others


Background
Primary orbital malignant melanoma is a rare tumour. While it can develop de novo, a subset of primary orbital melanomas can arise from a pre-existing melanocytic proliferation either in the form of diffuse melanocytosis or a melanocytic naevus, particularly of the blue naevus family [1,2,3]. Blue melanocytic naevi, as well as diffuse mucocutaneous melanocytosis (naevus of Ota), are seen not infrequently in the orbital and conjunctival region; however, melanoma arising from such lesions is exceedingly rare and only a handful of cases have been reported [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18]. Herein we describe the clinicopathological and molecular characteristics of a melanoma arising in an area of unusual and atypical diffuse blue naevus-like melanocytosis and include a review of the relevant literature.
Case presentation
A 27-year-old Caucasian man presented to the ophthalmology outpatient clinic with mild pain and swelling of his left eye. He had initially presented 18 months previously to the emergency department with an episode of left eye urticaria and spontaneous periorbital bruising, which responded well to topical antihistamine eye drops and lubricants. In addition, for the preceding 6 months the individual had noticed a dark flat lesion on the temporal aspect of his left eye conjunctiva, which was causing a pressure sensation. He had no other significant past medical, surgical or medication history.
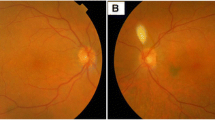
On examination, an area of grey/blue discoloration associated with swelling was evident at the lateral aspect of left bulbar conjunctiva (Fig. 1a). Visual acuity was normal, the individual had a full range of extraocular movements and there was no proptosis. Dilated fundus examination was normal. An orbital Computed Tomography (CT) scan revealed a 29 mm extraconal left orbital mass involving the lacrimal gland and lateral rectus muscle. Magnetic Resonance Imaging (MRI) of the brain and orbits showed a left lateral episcleral orbital tumour, with extension to the lateral rectus muscle, displacing the lacrimal gland (Fig. 1b). Positron Emission Tomography (PET)/CT scan showed no evidence of an 18-fluorodeoxyglucose (FDG)-avid regional or distant metastasis.

a Clinical photo showing an area of blue/grey discoloration and swelling (yellow arrow) involving the temporal bulbar conjunctiva, b MRI images from left to right - T1 coronal, T1 coronal with fat saturation and intravenous Gadolinium contrast, T2 Short Tau Inversion Recovery (STIR) axial and Diffusion Apparent Diffusion Coefficient (ADC) highlighting an intrinsically generally T1 hyper-intense, T2 hypo-intense contrast enhancing lateral retro-septal mass (yellow arrows) which exhibits restriction of water movement on diffusion imaging. This is positioned inferior to the lacrimal gland and lateral to the left globe which itself is mildly distorted by the lesion
A biopsy of the lesion located within the lateral rectus muscle was performed via a conjunctival approach. The histopathologic examination showed parts of a malignant melanocytic tumour characterised by compacted sheet-like to fascicular growth of spindled to epithelioid melanocytes showing enlarged pleomorphic nuclei, inconspicuous nucleoli, and moderate eosinophilic cytoplasm. Mitotic figures were easily identified. The tumour was identified as invading the underlying skeletal muscle tissue (Fig. 2 a-c). In addition, at the periphery of this biopsy, there was a distinctly different zone characterised by a component of less atypical melanocytes forming short fascicles and nests of bland appearing oval to spindle to dendritic cells with slightly elongated nuclei, no nucleoli and scant to moderate amount of cytoplasm with focal pigmentation (Fig. 2d). The latter component was suggestive of a pre-existing melanocytic proliferation with histopathological features of a blue naevus family of melanocytic tumours. By immunohistochemistry, the lesional cells demonstrated diffuse positive staining with melanocytic markers including SOX10, Melan-A, S100 and HMB-45. Ki67 proliferation index was estimated to be 5%. There was an abnormal loss of P16 throughout the tumour. BAP1 was retained in tumour cell nuclei.

a Low power view of a trans-conjunctival incisional biopsy shows portions of a cellular and compacted tumour (H&E, original magnification × 20), b invading into the skeletal muscle tissue with an adjacent less cellular lesion on the right side of this microscopic section (yellow rectangle) (H&E, original magnification × 40), c the main tumour is composed of densely packed round to oval melanocytes with enlarged pleomorphic nuclei and focal melanin pigmentation (brown pigment in the image), (H&E, original magnification × 200) (d) the pre-existing lesion shows features of a blue naevus with loose aggregates of oval to spindle melanocytes with patchy melanin pigmentation (H&E, original magnification × 100)
Molecular testing using Next-Generation sequencing (TruSight 26 gene panel, Illumina, USA) demonstrated a somatic GNAQ mutation [c.626A > T, p.(Gln209Leu)], with wild type BRAF and NRAS. Array comparative genomic hybridisation (aCGH; 60 K array, Agilent, Australia) showed whole chromosome 6, 7, 8 and 20 gains. Fluorescence in situ hybridisation (FISH) testing including Melanoma Four-Colour FISH probe (Vysis, Abbott Molecular, USA) was abnormal, showing absolute and relative gain of RREB1.
The overall immunomorphological, molecular and clinical/imaging findings were in keeping with a “primary orbital malignant melanoma arising from a pre-existing blue naevus-like melanocytic proliferation”.
The patient subsequently underwent lid skin-sparing total left orbital exenteration. Histopathological examination of the specimen confirmed a malignant melanoma arising in a pre-existing blue naevus-like diffuse melanocytosis. The latter was found to circumferentially involve the sub-epithelial soft tissue of the bulbar conjunctiva as well as episcleral soft tissue toward the temporal aspect of the orbit. The pre-existing lesion was histopathologically characterised by an interrupted rim of clusters and nests of spindle to epithelioid melanocytes with patchy pigmentation, mild to moderate cytologic atypia and low-level mitotic activity (Fig. 3a-b). There was an abrupt transition to an expansile tumour (melanoma) showing compacted sheet-like to fascicular growth of atypical plump melanocytes with frequent mitoses (up to 3–4 mitoses per square millimeter), an area of necrosis and extensive infiltration of the lateral rectus muscle. The immunohistochemical profile of the tumour was identical to the prior incisional biopsy (Fig. 3c-d). The lacrimal gland was intact. The internal orbital structures, including the anterior and posterior uvea as well as the optic nerve, were intact with no evidence of a melanocytic proliferation in the iris, ciliary body or choroid. The histopathological findings in the exenteration specimen confirmed the initial diagnosis of malignant melanoma arising in a pre-existing atypical diffuse plaque-like blue naevus/melanocytosis.

a A microscopic section from the exenteration specimen illustrates parts of the eyelid tissue (left, red arrow) and dense fibrous sclera (top, black arrow) with an episcleral/subconjunctival pre-existing melanocytic proliferation (yellow box) giving rise to a densely cellular malignant melanoma (right) (H&E, original magnification × 10), b A large area of tumour necrosis (yellow box) is seen at the border between the melanoma (right) and the pre-existing lesion (left) (H&E, original magnification × 30). c Microscopic high power view of the pre-existing lesion shows a nested growth of round to oval melanocytes with slightly enlarged nuclei, occasional inconspicuous nucleoli and moderate abundant eosinophilic cytoplasm with occasional mitoses (yellow circle), (H&E, original magnification × 400) (d) the malignant melanoma is characterized by compacted fascicular and sheet like growth of highly atypical and mitotically active melanocytes (H&E, original magnification × 200)
The patient declined post-operative radiotherapy and opted for close surveillance with regular PET and MRI scans every 3 months. As of 26 months’ follow-up, no recurrence or metastasis has been detected thus far.
Discussion and conclusion
The blue naevus family of melanocytic neoplasms, including several variably termed melanocytoses such as naevus of Ota, naevus of Ito and Mongolian spot, is a heterogenous group of melanocytic proliferations occurring in the skin and soft tissue, meninges, and mucosa. Such lesions, including oculodermal melanocytosis also known as “naevus of Ota”, can develop in the conjunctiva, eyelid, sclera, and peri-orbital tissue. The exact incidence is not clear; however, blue naevi encompassed 1.5% of pigmented and 0.2% of all conjunctival lesions in a large series of 2455 adult patients reported by Grossniklaus and colleagues [19]. Clinically, these neoplasms manifest as variably pigmented, sometimes very dark/blue lesions, which can be flat or slightly raised. Deep-seated lesions located in the subcutis or peri-orbital soft tissue can be clinically undetectable. Histologically, blue naevi and melanocytoses are characterised by variably dense proliferation of elongated to dendritic melanocytes with thin bland spindle nuclei, often with cytoplasmic melanin pigmentation, within a usually sclerotic stroma with different proportions of intermixed melanophages. Histologic variations including cellular and atypical cellular blue naevi have been described, typically presenting as bulbous to dumbbell-shaped proliferation of fascicles of compacted spindled to ovoid melanocytes with a mild degree of cytologic atypia and limited mitotic activity (≤ 2 mitoses/square millimeter) [20]. At the molecular level, blue naevi and mucocutaneous melanocytoses, similar to uveal melanocytic neoplasms, harbour somatic mutations in genes encoding G protein, including GNAQ and GNA11 in most cases, with a minority showing alterations in PLCB4 or CYCLTR2 genes as an initiating genomic event [21].
While most melanocytic neoplasms of the blue naevus family follow a benign clinical course, a stepwise malignant transformation due to accumulation of additional genomic alterations can occur rarely. Such a transformation has been histologically documented by transition from overtly benign-appearing foci to atypical areas characterised by increased cellularity, cytologic atypia and occasional mitoses to areas showing a frankly malignant appearance with compacted sheet-like and fascicular growth of melanocytes, with significant cytologic atypia, high mitotic rate (> 2 mitoses/square millimeter) and necrosis [13, 20], as observed in the current case. However, a transition such as this is not always observed, particularly in advanced tumours where the melanoma has completely overrun the pre-existing lesion or developed de novo. At the molecular level, these melanomas often show additional genomic alterations including bi-allelic inactivation of BRCA-associated protein 1 (BAP-1) gene or mutations in the SF3B1 gene [21]. In cytogenetic studies, multiple gains and losses of chromosomes, specifically losses of chromosome 3, have often been reported in melanomas associated with blue naevus [20].
The biologic behaviour of histologically malignant tumours of blue naevus origin (variably termed as melanoma associated with blue naevus, melanoma ex-blue naevus, malignant blue naevus and blue naevus-like melanoma) is comparable with conventional melanoma in which local recurrence, metastatic spread and death from the disease are frequently reported [22]. In a previous study, Martin et al. demonstrated no difference in patient survival and overall clinical behaviour of blue naevus-like melanomas compared with conventional melanomas matched for patient age and sex, Breslow thickness, Clark level, ulceration and anatomical location [23]. In addition, it has been shown that tumour thickness — measured as Breslow thickness, or in the absence of epidermis, the largest dimension of tumour — is the only clinicopathological factor significantly correlating with both shorter recurrence-free patient survival and reduced time to distant metastasis. In contrast, other previously established prognostic factors for conventional melanoma, such as patient age and gender, ulceration, tumour necrosis, lymphovascular invasion and mitotic activity, do not correlate with the outcome in this subset of melanomas [15, 23].
Identifying true cases of melanoma arising in blue melanocytic proliferations in the literature is difficult due to the confusion surrounding terminology and variable inclusion of different entities by different authors. The first report of orbital melanoma arising in association with a blue melanocytic proliferation dates back to 1954 when Dorsey and Montgomery reported two cases of malignant melanoma arising from congenital naevus of Ota, one in a 24-year-old woman who died from metastatic disease 3 years later and the other one in a 16-year-old boy [4]. Following on from this, in 1965, Jay presented a clinicopathologically well-documented case of a 66-year-old woman with progressive right eye proptosis due to a retroocular mass on a background of oculodermal melanocytosis and multiple blue naevi in the subcutaneous tissue of the eyelid [5].
Including our patient, in the English literature to date 27 well-documented cases of primary orbital melanoma arising from the blue naevus family of melanocytic neoplasms have been reported (Table 1). This cohort appears to be mainly composed of patients of Caucasian ethnicity, with an average age of 40 years (median 36, range 9–70 years) with no significant gender predilection (F/M = 1.1). The lesions developed in both periorbital skin and deep periorbital tissues, including the intraconal space, with an average size of 20.7 mm (range 8–30 mm). The clinical presentation ranged from an asymptomatic or slightly painful enlarging mass to progressive proptosis, diplopia, and loss of vision. Histologically, the most common pre-existing lesion was a “cellular blue naevus”, seen in 55.5% of patients, sometimes combined with other proliferations of blue naevus family. A naevus of Ota was present in 8 (⁓ 30%) cases and a component of common blue naevus was seen in 11 (⁓ 41%) cases. The melanoma component in these cases has been reported as a nodular expansion of variably pigmented tightly packed atypical melanocytes, often with mixed spindle and epithelioid morphology and significant cytologic atypia with enlarged pleomorphic nuclei and prominent nucleoli. Increased mitotic activity and necrosis were also common findings in these melanomas [2, 5,6,7, 9, 10, 13,14,15] .
While pre-existing lesions in all of the previously reported cases of orbital blue naevus-like melanoma could be easily categorised into the currently known subtypes of blue melanocytic proliferations, the precursor lesion in our case was extraordinary due to the peculiar histologic features including a diffuse circumferential and multifocal architecture and mild cytologic atypia and mitotic activity of constituent melanocytes, albeit with an abrupt transition to a frankly malignant tumour. The diffuse nature of this lesion is akin to what has been previously described as “plaque-like blue naevus” in the skin [24, 25]; however, the atypical cytology and mitotic activity is unusual. Therefore, we opted to classify the lesion descriptively as an “atypical plaque-like blue naevus/melanocytosis” in an orbital location. We are not aware of a similar lesion being described in association with a blue naevus-like melanoma in the orbit.
The molecular profile of these tumours has not been well elucidated and comprehensive molecular testing was available in only two cases, one of which (the current case) showed a GNAQ mutation. Molecular or immunohistochemical data on BRAF V600E was available in 5 cases, all of which were wild type [13, 17]. Loss of BAP-1 protein by immunohistochemistry, indicating bi-allelic inactivation of BAP-1 gene, has been shown in two cases [17]. Although very limited, as expected, the molecular data available suggest that the molecular profile of these tumours is similar to that of uveal melanoma and cutaneous melanoma associated with blue naevus, with frequent initial somatic mutations in G protein-encoding genes followed by bi-allelic inactivation of BAP-1 through different mechanisms including chromosomal loss as well as somatic or germline mutations in BAP-1 gene and possibly epigenomic means in some cases [26].
Follow-up was reported for 20 patients, with an average follow-up time of ~ 38 months. While 4 patients developed local recurrences [2, 12, 18], metastases were reported in 7 patients [4, 5, 7, 12, 15, 16], with tumour spreading to the liver (3 patients), lymph node, brain, and lung and 5 patients dying from the disease [4, 5, 7, 11, 12]. These data confirm the speculation that primary orbital melanoma associated with blue naevus, similar to such tumours elsewhere and conventional melanoma, is potentially an aggressive disease requiring appropriate local clearance with close follow-up.
This case also emphasises the importance of differentiating primary orbital melanoma from metastatic orbital melanoma or spread from adjacent tissues as treatment options differ; exenteration is the treatment of choice for primary melanomas, but non-surgical treatment is preferred in the case of metastasis. In our case, the presence of a pre-existing lesion in the form of an atypical plaque-like blue naevus/melanocytosis in an episcleral/submucosal conjunctival location and the absence of evidence of a primary melanoma in the uvea or an extra orbital location (based on PET scan) pointed toward a primary orbital tumour. Given the rarity of such cases, the role of systemic therapy is not clear.
In summary, we report a primary orbital melanoma developing in an unusual pre-existing melanocytic proliferation of blue naevus family, which we descriptively categorised as “atypical plaque-like blue naevus/melanocytosis”. This phenomenon is extremely rare and seems to occur due to accumulation of genomic alterations at the molecular level. The biologic nature and clinical behaviour of such melanomas are comparable to conventional melanomas. This case highlights the need to recognise “melanoma ex-blue naevus” as a primary orbital tumour that mandates careful histologic examination to look for a pre-existing lesion, with comprehensive clinicopathological correlation to avoid misdiagnosing such melanomas as a metastatic tumour, which is managed differently.
Availability of data and materials
All data generated or analysed during this study are included in this published article.
Abbreviations
- FISH:
-
Fluorescence in situ hybridization
- MRI:
-
Magnetic Resonance Imaging
- CT:
-
Computed Tomography
- PET:
-
Positron Emission Tomography
References
Demirci H, Shields CL, Shields JA, Eagle RC Jr. Malignant melanoma arising from unusual conjunctival blue nevus. Arch Ophthalmol. 2000;118(11):1581–4.
Granter SR, McKee PH, Calonje E, Mihm MC Jr, Busam K. Melanoma associated with blue nevus and melanoma mimicking cellular blue nevus: a Clinicopathologic study of 10 cases on the Spectrum of so-CalledMalignant blue nevus. Am J Surg Pathol. 2001;25(3):316–23.
Mandeville JT, Grove AS, Dadras SS, Zembowicz AM. Primary orbital melanoma associated with an occult episcleral nevus. Arch Ophthalmol. 2004;122(2):287–90.
Dorsey C, Montgomery H. Blue nevus and its distinction from Mongolian spot and the nevus of Ota. J Invest Dermatol. 1954;22(3):225–36.
Jay B. Malignant melanoma of the orbit in a case of oculodermal melanosis. (Naevus of Ota). Br J Ophthalmol. 1965;49(7):359–63 Epub 1965/07/01.
Hagler W, Brown C. Malignant melanoma of the orbit arising in a nevus of Ota. Trans Am Acad Ophthalmol Otolaryngol. 1966;70(5):817–22.
Speakman JS, Phillips MJ. Cellular and malignant blue nevus complicating oculodermal melanosis (nevus of Ota syndrome). Can J Ophthalmol. 1973;8(4):539–47.
Jakobiec FA, Ellsworth R, Tannenbaum M. Primary orbital melanoma. Am J Ophthalmol. 1974;78(1):24–39.
Dutton JJ, Anderson RL, Schelper RL, Purcell JJ, David TT. Orbital malignant melanoma and oculodermal melanocytosis: report of two cases and review of the literature. Ophthalmology. 1984;91(5):497–507.
Löffler K, Witschel H. Primary malignant melanoma of the orbit arising in a cellular blue naevus. Br J Ophthalmol. 1989;73(5):388–93.
Connelly J, Leslie Smith J. Malignant blue nevus. Cancer. 1991;67(10):2653–7.
Gündüz K, Shields JA, Shields CL, Eagle RC Jr. Periorbital cellular blue nevus leading to orbitopalpebral and intracranial melanoma. Ophthalmology. 1998;105(11):2046–50.
Gerami P, Pouryazdanparast P, Vemula S, Bastian BC. Molecular analysis of a case of nevus of ota showing progressive evolution to melanoma with intermediate stages resembling cellular blue nevus. Am J Dermatopathol. 2010;32(3):301–5.
El-Sawy T, Bakhoum MF, Tetzlaff M, et al. Primary orbital melanoma in association with cellular blue nevus. Digit J Ophthalmol. 2014;20(3):35.
Loghavi S, Curry JL, Torres-Cabala CA, et al. Melanoma arising in association with blue nevus: a clinical and pathologic study of 24 cases and comprehensive review of the literature. Mod Pathol. 2014;27(11):1468–78 Epub 2014/04/20.
Hussain A, Sidiropoulos M, Das S, Munoz DG, Nijhawan N. Orbital cellular blue nevus complicated by malignant melanoma. Can J Ophthalmol. 2017;52(3):e111–e3.
Figueira E, Rajak S, McKelvie P, et al. Primary orbital melanoma: a case series and literature review. Orbit. 2018;37(5):352–7.
Odashiro AN, Arthurs B, Pereira PR, Souza Filho JP, Belfort E, Burnier MN Jr. Primary orbital melanoma associated with a blue nevus. Ophthalmic Plast Reconstr Surg. 2005;21(3):247–8.
Grossniklaus HE, Green WR, Luckenbach M, Chan CC. Conjunctival lesions in adults. Cornea. 1987;6(2):78–116.
Hung T, Argenyi Z, Erickson L, et al. Cellular blue nevomelanocytic lesions: analysis of clinical, histological, and outcome data in 37 cases. Am J Dermatopathol. 2016;38(7):499–503.
Griewank KG, Müller H, Jackett LA, et al. SF3B1 and BAP1 mutations in blue nevus-like melanoma. Mod Pathol. 2017;30(7):928–39.
Costa S, Byrne M, Pissaloux D, et al. Melanomas associated with blue nevi or mimicking cellular blue nevi: clinical, pathologic, and molecular study of 11 cases displaying a high frequency of GNA11 mutations, BAP1 expression loss, and a predilection for the scalp. Am J Surg Pathol. 2016;40(3):368–77 Epub 2015/12/10.
Martin RC, Murali R, Scolyer RA, Fitzgerald P, Colman MH, Thompson JF. So-called "malignant blue nevus": a clinicopathologic study of 23 patients. Cancer. 2009;115(13):2949–55 Epub 2009/05/28.
Yan L, Tognetti L, Nami N, et al. Melanoma arising from a plaque-type blue naevus with subcutaneous cellular nodules of the scalp. Clin Exp Dermatol. 2018;43(2):164–7.
Baykal C, Yılmaz Z, Sun G, Büyükbabani N. The spectrum of benign dermal dendritic melanocytic proliferations. J Eur Acad Dermatol Venereol. 2019;33(6):1029–41.
Harbour JW, Onken MD, Roberson ED, et al. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010;330(6009):1410–3.
Acknowledgements
We thank the study participant for granting permission to publish this information and also Dr. Michael Bynevelt for his kind assistance with the interpretation of MRI images.
Funding
There was no funding support for this study.
Author information
Authors and Affiliations
Contributions
TD analysed the clinical case and researched the previous literature. NMA performed the pathology and histology examination. MF and AG were attending ophthalmologists. The first draft of the manuscript was written by TD and NMA. Final editing of manuscript was by TD, MF, AG and NMA. All authors read and approved the final version.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical approval was granted by The Western Australian Department of Health Ethics Committee (#2015/18). The study was performed in accordance with the ethical standards as laid down in the 1964 Declaration of Helsinki. Written informed consent was obtained from the individual participant included in the study.
Consent for publication
Written informed consent was obtained from the participant for publication of this case report and accompanying images. A copy of the consent is available for review by the Editor of this journal.
Competing interests
The authors have no competing interests to declare that are relevant to the content of this article.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Dickens, TA., Franchina, M., Gajdatsy, A. et al. Primary orbital melanoma arising in an atypical diffuse (plaque-like) blue naevus/melanocytosis: a case report and review of literature. BMC Ophthalmol 21, 425 (2021). https://doi.org/10.1186/s12886-021-02176-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12886-021-02176-y