
Abstract
Background
To explore the molecular genetic cause of a four-generation autosomal dominant retinitis pigmentosa family in China.
Methods
Targeted region sequencing was performed to detect the potential mutation, and Sanger sequencing was used to validate the mutation. Multiple sequence alignment from different species was performed by CLUSTALW. The structures of wild-type and the mutant RHO were modeled by Swiss-Model Server and shown using a PyMOL Molecular Graphic system.
Results
A novel heterozygous nonsense mutation (c.1015 A > T, p.Lys339Ter, p.K339X) within RHO, which cosegregated with retinitis pigmentosa phenotype was detected in this family. Bioinformatics analysis showed the mutation was located in a highly conserved region, and the mutation was predicted to be pathogenic.
Conclusions
We identified a novel heterozygous nonsense mutation of RHO gene in a Chinese family with retinitis pigmentosa by target region sequencing and our bioinformatics analysis indicated that the mutation is pathogenic. Our results can broaden the spectrum of RHO gene mutation and enrich the phenotype-genotype correlation of retinitis pigmentosa.
Similar content being viewed by others


Background
Retinitis pigmentosa (RP) is a heterogeneous group of inherited progressive retinal degenerative conditions and a leading cause of irreversible blindness. Typical fundus changes of RP include attenuated arterioles, waxy pallor of the optic disc, and characteristic perivascular and midperipheral bone-spicule pigmentation, which is resulted from melanin-containing vesicles accumulation in the retinal vascular layer and around the Muller cells [1].
RP affects 1 in 3500 individuals worldwide and there are several inherited patterns of RP, including autosomal dominant (15-20% cases), autosomal recessive (20-25% cases), X-linked (10-15% cases) or even mitochondrial inheritance [2]. To date, more than 89 genes have been reported to cause non-syndromic RP [3], with RP-GTPase regulator gene (RPGR; 10-20% of cases) and rhodopsin gene (RHO; 8-10% of cases) being the most commonly involved genes [4,5,6,7,8].
In this study, by target region sequencing, we identified a novel RHO nonsense mutation (c.1015 A > T, p.Lys339Ter, p.K339X) in a Chinese RP family.
Methods
Patients
This study was approved by the medical ethics committee of Tianjin Medical University Eye Hospital. A four-generation Chinese family suffered from RP was recruited and written informed consent was obtained. A 5 ml venous blood sample of each enrolled participant was collected from antecubital vein puncture, drawn into an ethylenediamine tetraacetic acid (EDTA) sample tube and stored in − 20 °C refrigerator for further analysis.
Target region capture and sequencing
The target region capture and next-generation sequencing (NGS) procedure was described previously [9]. Briefly, genomic DNA was firstly extracted according to the manufacturer’s standard procedure (MagPure Buffy Coat DNA Midi KF Kit, Magen, China). After that, the genomic DNA of the proband was sequenced with PE100 + 100 on MGISEQ-2000. Finally, the targeted sequences were captured using the NimbleGen SeqCap EZ Choice XL Library 24 Reaction 150217_HG19_CllnE_EZ_HX1 chip (Roche, Madison, USA) containing 156 genes related to retinal diseases (Table 1).
Data analysis
To detect the potential variants in the family, we performed bioinformatics processing and data analysis after receiving the primary sequencing data. We used previously published filtering criteria to generate “clean reads” for further analysis [10]. The “clean reads” (with a length of 90 bp) derived from targeted sequencing and filtering were then aligned to the human genome reference (hg19) using the BWA (Burrows Wheeler Aligner) Multi-Vision software package [11]. After alignment, the output files were used to perform sequencing coverage and depth analysis of the target region, single-nucleotide variants (SNVs) and INDEL calling. We used GATK software [12] to detect SNVs and indels. All SNVs and indels were filtered and estimated via multiple databases, including NCBI dbSNP, HapMap, 1000 human genome dataset and database of 100 Chinese healthy adults. The Human Gene Mutation Database (HGMD, http://www.hgmd.cf.ac.uk/ac/index.php) was used to screen mutations reported in published studies.
Sanger verification
All mutations and potential pathogenic variants were validated using conventional Sanger sequencing methods. Segregation analysis was performed in all available family members.
Protein model construction
Multiple sequence alignment from different species was performed by CLUSTALW (https://www.genome.jp/tools-bin/clustalw). In addition, the structures of homomeric wild-type and the mutant RHO were modeled by Swiss-Model Server (https://swissmodel.expasy.org) and shown using a PyMOL Molecular Graphic system, using the solved structure of rhodopsin coded by RHO gene as template (Protein Data Bank No. 5W0P).
Results
Clinical evaluation
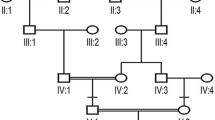
Two patients (III:2, III:3) and three normal individuals (III:4, III:5, IV:2) from this family were enrolled in this study (Fig. 1). The proband was a sixty-year-old man (III:2) and was diagnosed with RP when he was 17 years old. The patient suffered from night blindness when he was young and accepted cataract surgery 20 years ago. On presentation, the visual acuity of both eyes was light perception. The axial length was 25.99 mm and 25.56 mm, the central corneal thickness was 586 μm and 595 μm, the flat K was 42.56D and 43.27 D, the steep K was 43.80 D and 43.79 D, the intraocular pressure was 20.1 mmHg and 18.6 mmHg for the right eye and left eye, respectively. Under slit-lamp microscopy, the cornea was clear, the anterior chamber was deep and quiet, the pupil was sluggish and the intraocular lens was central and clear. Fundoscopic examination revealed attenuated arterioles, waxy pallor of the optic disc, atrophy of the choroid and diffuse bone-spicule pigmentation in both eyes (Fig. 2). The sister of the proband (III:3) also suffered from night blindness and was diagnosed with RP when she was 15 years old and she had similar fundus changes with the proband. There was no consanguineous marriage in this family.

Pedigree map of the family. The arrow indicates the proband. Squares and circles symbolize males and females, respectively. Black and white denotes affected and unaffected individuals, respectively

Fundus photographs of the proband. Note the attenuated arterioles, waxy pallor of the optic disc, atrophy of the choroid and diffuse bone-spicule pigmentation in both eyes
Mutation identification in RHO
Targeted region sequencing containing 156 retinal diseases-related genes of the proband (III:2) revealed a transversion in exon 5 (c.1015A > T) of RHO, which was not reported previously. The variant causes the replacement of a wild-type Lysine with stop codon at codon 339 (p.Lys339Ter; Fig. 3A). The variant can be detected in all affected patients enrolled in this study (III:2, III:3) and was not found in the unaffected family member (III:4, III:5, IV:2) by further Sanger sequencing. The variant co-segregated with the disease in family members and was not found in NCBI dbSNP, HapMap, 1000 human genome dataset and database of 100 Chinese healthy adults, suggesting the variant may be the pathogenic mutation in this family.

Sanger sequencing of RHO and bioinformatics analysis of the mutation. A Sanger sequencing of RHO detected a c.1015A > T transversion in affected patients which caused the replacement of a wild-type Lysine with stop codon at codon 339. B Multiple-sequence alignments of RHO in various species showed codon 339 was located within a highly conserved region
Protein model construction
Multiple-sequence alignments of RHO in various species showed codon 339 was located within a highly conserved region (Fig. 3B). Structure model of rhodopsin-arrestin complex showed normally, arrestin ‘reads’ phosphorylation codes of rhodopsin through code-sensing pockets in the C-terminus, while p.K339X mutation of RHO is predicted to produce a truncated protein and interfere with arrestin recruitment (Fig. 4).

Structure model of rhodopsin-arrestin complex. Left: C-terminus of rhodopsin was shown in sticks, and the phosphorylation codes (D330, E332, T336, S338, E341) were highlighted. Arrestin was shown in vacuum electrostatics, blue area represents positive charge while red area represents negative charge. Normally, arrestin ‘reads’ phosphorylation codes of rhodopsin through code-sensing pockets. Right: K339X mutation of RHO produced a truncated protein and interfered the arrestin recruitment
Discussions
In this study, we identified a RHO nonsense mutation (c.1015 A > T, p.Lys339Ter, p.K339X) in a Chinese RP family by target region sequencing and Sanger verification, which co-segregated with the disease in family members. This mutation appeared to be novel, which is neither reported in literature nor registered in HGMD. Our bioinformatics analysis showed codon 339 was located within a highly conserved region in various species and structure model showed the mutation could interfere arrestin recruitment, indicating this mutation was pathogenic and responsible for RP in this family.
RHO, located on chromosome 3q21-25, contains five exons. RHO was the first photoreceptor specific gene found to be mutated in autosomal dominant RP (adRP) and is the most common gene implicated in adRP. RHO gene accounts for 25-30% of adRP in Americans [13, 14], and the rate of RHO mutations in Asians is lower than in Americans [15,16,17,18]. The RHO gene encodes a 348-amino acids rod-specific protein rhodopsin, which is a typical G-protein-coupled receptor [19]. Rhodopsin localizes to the outer segment of rods and contains extracelluar, transmembrane, and cytoplasmic domains. RHO mutations often cause protein misfolding and retention in the endoplasmic reticulum, leading to cellular stress and eventual cell death [7].
Since the first p.P23H mutation in the RHO gene was discovered in 1990, more than 230 different mutations have been reported to be associated with RP in different ethnic populations, most of which are point mutations (HGMD). Based on their biochemical and cellular properties, these mutations can be grouped into class I or class II [20]. Class I mutations, located predominantly in the C-terminus of the protein, can fold normally, but are not correctly transported to the outer segment. Class II mutations, which usually occur in the intradiscal and transmembrane domains of the protein, are not folded correctly and are retained in the endoplasmic reticulum. Clinically, class I mutations lead to severe diffuse abnormal rod function early in life, while in class II mutations, the disease progresses slowly and the rods have nearly normal function even in adult patients.
Up to now, a total of 10 nonsense mutations have been reported in RHO (HGMD). It is suggested that the heterozygous truncating variants at the downstream of K296 could result in adRP [21]. The underlying mechanism for this may be that a truncated protein resulted from a heterozygous truncating variant after K296 could escape nonsense-mediated mRNA decay (NMD) and, thus, produce harmful truncated proteins. In contrast, the truncating variants before K296 are very close to benign variants, because the abnormal products from the truncating variants at the upstream of K296 will be cleared by the NMD process being triggered [21]. This is consistent with our results. The nonsense heterozygous mutation p.K339X detected in our study, located at the downstream of K296, led to adRP with severe phenotype.
It is recently reported that rhodopsin C-terminal tail served as an interface for arrestin recruitment, and the β strand (K339-T342) is one of the anchor points for rhodopsin-arrestin interaction [22]. Several mutations (Ala333Val; Thr340Met; Glu341Lys; Glu341Ter; Thr342Met; Ser343Asn; Ser343Cys; Gln344Pro; Gln344Arg; Gln344Ter; Val345Met; Val345Leu; Val345Gly; Ala346Pro; Pro347Thr; Pro347Gln; Pro347Arg; Pro347Leu; Pro347Ala; Pro347Ser; Pro347Cys; Ter349Gln; Ter349Glu, etc) have been reported in the C-terminal tail of rhodopsin (HGMD), indicating the C-terminus is a hotspot of mutation. The mutation detected in our study (p.K339X) is the first mutation found in codon 339, highlighting its role to maintain the normal function of rhodopsin. The mutation can interfere the arrestin recruitment and block the downstream signaling transmission, as shown in Fig. 4. As a class I mutation, p.K339X is expected to fold correctly but affect the post-Golgi trafficking of rhodopsin and impair its normal targeting to the photoreceptor outer segment [20], and lead to RP eventually.
Conclusions
In conclusion, by target region sequencing, we identified a novel RHO nonsense mutation in a Chinese RP family. Our results can broaden the spectrum of RHO gene mutation, enrich the phenotype-genotype correlation of RP, and provide target for potential gene therapy in the future.
Availability of data and materials
All data generated or analysed during this study are included in this published article.
Abbreviations
- RP:
-
Retinitis pigmentosa
- RPGR:
-
RP-GTPase regulator gene
- RHO:
-
Rhodopsin gene
- EDTA:
-
Ethylenediamine tetraacetic acid
- NGS:
-
Next-generation sequencing
- SNVs:
-
Single-nucleotide variants
- HGMD:
-
Human Gene Mutation Database
- adRP:
-
Autosomal dominant RP
- NMD:
-
Nonsense-mediated mRNA decay
References
Kalloniatis M, Fletcher EL. Retinitis pigmentosa: understanding the clinical presentation, mechanisms and treatment options. Clin Exp Optom. 2004;87:65–80.
Wang DY, Chan WM, Tam PO, et al. Genetic markers for retinitis pigmentosa. Hong Kong Med J. 2005;11:281–8.
Wang J, Xu D, Zhu T, et al. Identification of two novel RHO mutations in Chinese retinitis pigmentosa patients. Exp Eye Res. 2019;188:107726.
Breuer DK, Yashar BM, Filippova E, et al. A comprehensive mutation analysis of RP2 and RPGR in a north American cohort of families with X-linked retinitis pigmentosa. Am J Hum Genet. 2002;70:1545–54.
Briscoe AD, Gaur C, Kumar S. The spectrum of human rhodopsin disease mutations through the lens of interspecific variation. Gene. 2004;332:107–18.
Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–809.
Wright AF, Chakarova CF, Abd El-Aziz MM, Bhattacharya SS. Photoreceptor degeneration: genetic and mechanistic dissection of a complex trait. Nat Rev Genet. 2010;11:273–84.
Foote KG, Wong JJ, Boehm AE, et al. Comparing cone structure and function in RHO- and RPGR-associated retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2020;61:42.
Li D, Xu C, Huang D, et al. Identification and functional analysis of a novel missense mutation in GJA8, p.Ala69Thr. BMC Ophthalmol. 2020;20:461.
Wei X, Ju X, Yi X, et al. Identification of sequence variants in genetic disease causing genes using targeted next-generation sequencing. PLoS One. 2011;6:e29500.
Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25:1754–60.
McKenna A, Hanna M, Banks E, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
Dryja TP, McGee TL, Hahn LB, et al. Mutations within the rhodopsin gene in patients with autosomal dominant retinitis pigmentosa. N Engl J Med. 1990;323:1302–7.
Sohocki MM, Daiger SP, Bowne SJ, et al. Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Hum Mutat. 2001;17:42–51.
Kim KJ, Kim C, Bok J, et al. Spectrum of rhodopsin mutations in Korean patients with retinitis pigmentosa. Mol Vis. 2011;17:844–53.
Li S, Xiao X, Wang P, Guo X, Zhang Q. Mutation spectrum and frequency of the RHO gene in 248 Chinese families with retinitis pigmentosa. Biochem Biophys Res Commun. 2010;401:42–7.
Sullivan LS, Bowne SJ, Birch DG, et al. Prevalence of disease-causing mutations in families with autosomal dominant retinitis pigmentosa: a screen of known genes in 200 families. Invest Ophthalmol Vis Sci. 2006;47:3052–64.
Yang G, Xie S, Feng N, Yuan Z, Zhang M, Zhao J. Spectrum of rhodopsin gene mutations in Chinese patients with retinitis pigmentosa. Mol Vis. 2014;20:1132–6.
Rosenfeld PJ, Cowley GS, McGee TL, Sandberg MA, Berson EL, Dryja TP. A null mutation in the rhodopsin gene causes rod photoreceptor dysfunction and autosomal recessive retinitis pigmentosa. Nat Genet. 1992;1:209–13.
Mendes HF, van der Spuy J, Chapple JP, Cheetham ME. Mechanisms of cell death in rhodopsin retinitis pigmentosa: implications for therapy. Trends Mol Med. 2005;11:177–85.
Luo H, Xiao X, Li S, Sun W, Yi Z, Wang P, et al. Spectrum-frequency and genotype-phenotype analysis of rhodopsin variants. Exp Eye Res. 2021;203:108405.
Zhou XE, He Y, de Waal PW, et al. Identification of phosphorylation codes for arrestin recruitment by G protein-coupled receptors. Cell. 2017;170:457–469.e13.
Acknowledgements
Not applicable.
Funding
Supported by The Science&Technology Development Fund of Tianjin Education Commission for Higher Education (2017KJ216), Tianjin Clinical Key Discipline Project (TJLCZDXKQ023) and Open Project of Tianjin Key Laboratory of Retinal Functions and Diseases (2020tjswmq003). The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Author information
Authors and Affiliations
Contributions
WL and JJ designed and supervised the study; HH performed the bioinformatics analysis; WL and RG drafted the manuscript; WL, RG and HH analyzed the data. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the medical ethics committee of Tianjin Medical University Eye Hospital. A four-generation Chinese family was recruited and written informed consent was obtained.
Consent for publication
Consent for publication has been obtained from the patients enrolled in this study.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, W., Guo, R., Hao, H. et al. Identification of a novel RHO heterozygous nonsense mutation in a Chinese family with autosomal dominant retinitis pigmentosa. BMC Ophthalmol 21, 360 (2021). https://doi.org/10.1186/s12886-021-02125-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12886-021-02125-9