
Abstract
Background
Aponermin, a circularly permuted tumor necrosis factor-related apoptosis-inducing ligand, is a potential death receptor 4/5-targeted antitumour candidate. Previous phase 1/2 studies have demonstrated the efficacy of aponermin in patients with relapsed or refractory multiple myeloma (RRMM). To confirm the superiority of aponermin plus thalidomide and dexamethasone (aponermin group) over placebo plus thalidomide and dexamethasone (placebo group) in RRMM, a randomized, double-blinded, placebo controlled phase 3 trial was performed.
Methods
Four hundred seventeen patients with RRMM who had previously received at least two regimens were randomly assigned (2:1) to receive aponermin, thalidomide, and dexamethasone or placebo, thalidomide, and dexamethasone. The primary endpoint was progression-free survival (PFS). Key secondary endpoints included overall survival (OS) and overall response rate (ORR).
Results
A total of 415 patients received at least one dose of trial treatment (276 vs. 139). The median PFS was 5.5 months in the aponermin group and 3.1 months in the placebo group (hazard ratio, 0.62; 95% confidence interval [CI], 0.49–0.78; P < 0.001). The median OS was 22.4 months for the aponermin group and 16.4 months for the placebo group (hazard ratio, 0.70; 95% CI, 0.55–0.89; P = 0.003). Significantly higher rates of ORR (30.4% vs. 13.7%, P < 0.001) and very good partial response or better (14.1% vs. 2.2%, P < 0.0001) were achieved in the aponermin group than in the placebo group. Treatment with aponermin caused hepatotoxicity in some patients, as indicated by the elevated alanine transaminase, aspartate transaminase, or lactate dehydrogenase levels (52.2% vs. 24.5%, 51.1% vs. 19.4% and 44.9% vs. 21.6%, respectively), mostly grade 1/2, transient and reversible. The main grade 3/4 adverse events included neutropenia, pneumonia and hyperglycemia. The incidence of serious adverse events was similar between the two groups (40.6% vs. 37.4%). There was no evidence that aponermin leads to hematological toxicity, nephrotoxicity, cardiotoxicity, or secondary tumors.
Conclusions
Aponermin plus thalidomide and dexamethasone significantly improved PFS, OS and ORR with manageable side effects in RRMM patients who had received at least two prior therapies. These results support the use of aponermin, thalidomide, and dexamethasone as a treatment option for RRMM patients.
Trial registration
The trial was registered at http://www.chictr.org.cn as ChiCTR-IPR-15006024, 17/11/2014.
Similar content being viewed by others


Introduction
Although in the past 20 years, several new drugs have been approved, multiple myeloma (MM) remains an incurable hematological malignancy, and almost all patients eventually become drug-resistant [1,2,3,4]. New drugs are critically needed.
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) induces apoptosis selectively by activating death receptor 4 or 5 (DR4/5) in a wide range of cancers while sparing normal cells [5,6,7]. Several recombinant TRAIL-fusion proteins and multimeric anti-DR5 agonist antibody are in clinical trials for cancers [8,9,10,11,12,13].
Aponermin is a recombinant circularly permuted human TRAIL (CPT), by connecting the amino end and the carboxy end of the native TRAIL fragment (amino acid 121–281) with the linker (Gly-Gly-Gly-Gly-Gly) and breaking the TRAIL fragment at the site of amino acid 135 to create new amino and carboxy termini. The primary sequence of the aponermin protein was reordered, while its secondary structure and activity were retained. Aponermin is a more stable homotrimer and has shown a higher affinity for DR4/5, more potent antitumor activity and longer half-life than native TRAIL [14,15,16,17].
In the phase 1b study in patients with relapsed or refractory multiple myeloma (RRMM) [18], aponermin monotherapy was well tolerated with doses ranging 5–15 mg/kg. An overall response rate (ORR) of 18.5% was achieved across dose ranges. In the phase 2 study of aponermin combined with thalidomide in RRMM patients [19], a higher ORR (22.0% vs. 16.7%) and more cases of complete response (CR) or near CR (12.2% vs. 0) were observed compared to the aforementioned phase 1b results [18] at the same dose level, even though the patients were more heavily pretreated in the phase 2 trial.
Thalidomide combined with dexamethasone (TD regimen) has been approved for the treatment of MM in 2006. Although TD regimen is no longer widely used in developed countries with the approval of novel drugs, it is still a good option in low- and middle-income countries due to its accessibility and affordability [20, 21]. Preclinical studies in xenografted nude mice of human multiple myeloma showed that the antitumor effect of aponermin combined with TD was significantly better than that of aponermin alone or TD alone (P < 0.05) (data not published). In a randomized, open-labelled phase 2 trial [22], a prolonged progression-free survival (PFS) (6.7 months) was observed in the aponermin plus TD group compared to that of the TD group (3.1 months). A higher ORR and clinical benefit rate were also observed. To confirm the superiority of aponermin plus TD over placebo plus TD in patients with RRMM, a phase 3 trial (CPT-MM301) was performed.
Methods
Trial design
CPT-MM301 was a multicentre, randomized, double-blinded, placebo-controlled phase 3 trial conducted in China. The trial protocol was designed by sponsors and investigators, and was approved by the independent ethics committees of Beijing Chao-Yang Hospital Capital Medical University. The trial was conducted in accordance with the principles of Good Clinical Practice and the Declaration of Helsinki. All patients provided written informed consent before enrolment. The data were collected, analysed, and interpreted by investigators and sponsor. Investigators had full accessibility to all data. The trial was registered at http://www.chictr.org.cn as ChiCTR-IPR-15006024, 17/11/2014.
Patients
Eligible participants were enrolled in this study. Key inclusion criteria were as follows: participants' age: 18–75 years; previous treatment with two or more regimens for MM and not considered for bone marrow transplantation; M-protein levels needed to meet at least one of the following criteria: serum M-protein ≥ 10 g/L (IgG, IgM, IgD subtype), or ≥ 7.5 g/L (IgA subtype), or urinary M-protein ≥ 200 mg/24 h; absolute neutrophil count ≥ 1.0 × 109/L; platelet ≥ 50 × 109/L; aspartate transaminase (AST) ≤ 2.5 × upper limit of normal (ULN); alanine transaminase (ALT) ≤ 2.5 × ULN; alkaline phosphatase ≤ 2.5 × ULN; total bilirubin (TBIL) ≤ 1.5 × ULN; creatinine clearance rate ≥ 30 ml/min. Key exclusion criteria included: refractoriness to TD or lenalidomide plus dexamethasone (RD) regimens of the last treatment; received any anti-MM drug treatment within 4 weeks before the trial; participated in aponermin clinical trials previously; had serious organic or mental diseases. (see Supplementary file).
Randomization and masking
An allocation ratio preserving biases coin randomization was used in this trial (block size of 6) [23]. The random allocation sequence was generated using SAS 9.2 and uploaded to an interactive web response system (IWRS) by an unblinded system administrator. Eligible patients were enrolled and randomly assigned (2:1) by investigators via the IWRS to receive either aponermin plus TD (aponermin group), or placebo plus TD (placebo group). Randomization was stratified according to the number of prior therapeutic regimens (≤ 3 or > 3), the status of TD/RD therapy (yes vs. no), and International Staging System (ISS) stage (stage I vs. stage II or III). Aponermin and placebo were packaged in a blinded manner under the supervision of a statistician according to the drug list. The packaging and labels of aponermin and placebo were identical to ensure that they remained masked to the treatment assignment. The investigators, participants, research staff, members of the independent assessment committee (IAC), and sponsor study team were masked to the treatment location.
Procedures
In each cycle, patients were administered 10 mg/kg of aponermin or placebo via intravenous infusion on days 1–5, oral thalidomide 150 mg on days 1–28, and oral dexamethasone 40 mg on days 1–4 for 18 cycles (28 days for each cycle). In the first cycle, thalidomide was administered on days 2–28, and dexamethasone on days 2–5 to observe the changes in AST, ALT, and lactate dehydrogenase (LDH) after the first dose of aponermin alone. Treatment was continued for up to 18 cycles or until progressive disease (PD), unacceptable toxicities, or withdrawal from the study. After completing 18 cycles of treatment, patients might receive further treatment based on the investigators' opinions.
Outcomes and assessments
The primary endpoint was PFS, defined as the time from the date of the randomization to the date of the first documented PD or death from any cause during the study, whichever occurred first. Secondary endpoints included overall survival (OS), ORR, duration of response (DOR), time to response (TTR), time to progression (TTP), safety, and health-related quality of life (HRQoL). The exploratory endpoint was to evaluate the efficacy among high-risk patients defined as [t(14;16)], [t(4;14)], or [del(17p)] by fluorescence in situ hybridization, or chromosome 13 deletion with hypodiploidy by G-band staining [24].
Serum and urine monoclonal proteins and serum free light chain levels were measured at a central laboratory. Disease status were assessed by the investigators at the baseline and the end of each cycle. The International Myeloma Working Group consensus criteria (IMWG criteria) were used to assess responses and PD. For patients in remission, if treatment was discontinued due to intolerable adverse event (AE) or the completion of 18 cycles of treatment, the disease status was assessed every six weeks until PD, death or next anti-myeloma therapy started. We required all responses and PD be assessed by IAC. PFS, ORR, DOR, TTR, and TTP were calculated based on the responses and PD assessed by IAC.
The HRQoL questionnaires were completed at baseline and the end of every cycle using the European Organization for Research and Treatment of Cancer (EORTC) questionnaires: the generic EORTC QLQ-C30 and the myeloma-specific QLQ-MY20.
The AEs and serious adverse events (SAEs) were collected up to 28 days following the last treatment dose and graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events Version 4.03.
Statistical analysis
The sample size was determined based on a conservative estimation of the median PFS of 5.0 months in the aponermin group and 3.5 months in the placebo group. According to ethical opinion, the proportion of patients in the aponermin and placebo groups was 2:1. It was estimated that a total of 286 events of PD or death over a 30-months enrollment period and a 6-months follow-up would be required to have a statistical power of 80%. This is to show superiority at a hazard ratio (HR) of 0.70 using a log-rank test (one-sided alpha is 0.025). A total of 313 patients were needed based on the calculation before factoring in the dropout rate. However, considering a 25% dropout rate, at least 417 patients were required (278 in the aponermin group and 139 in the placebo group).
No interim analyse was done in this trial. A final analysis was performed when the last participant had been enrolled for 6 months.
In this study, all efficacy analyses were based on the modified intention-to-treat population (mITT), including all randomized patients who received at least one dose of the trial treatment. The primary endpoint, PFS, was compared using a prespecified stratified log-rank test. The Kaplan–Meier method was used to estimate the median PFS and depict the curve. The 95% confidence interval (CI) was estimated using the Brookmeyer-Crowley formula. HR and 95% CI were estimated using a stratified Cox proportional hazards model. The proportional hazards assumption was assessed and met by the Cox regression model, using time-dependent explanatory variables. The aforementioned strata variables were the same as those used in the randomization. Other time-to-event data, including OS, DOR, TTR, TTP, and subgroup analyses of PFS and OS, were analysed using a method similar to PFS. The ORR, clinical benefit rate and the rate of each response were compared between the groups using chi-squared tests. The Clopper–Pearson method was used to calculate the 95% CI. Subgroup analyses of ORR were performed using similar method. Scores for the EORTC QLQ-30 and MY20 were calculated according to the developer's scoring manual. The raw scores from the scales in both questionnaires were standardized by linear transformation to range 0–100. Descriptive statistics for the baseline scores for each domain were summarized, and the differences between groups were assessed using a group t-test. A mixed-model measure analysis was used to estimate the treatment effects over time for each domain (longitudinal analysis) and assess the differences between groups. The safety analysis included all patients who received at least one dose of the trial treatment. AEs were coded using MedDRA version 22.1. The frequency of AEs was reported.
All statistical analyses were conducted using SAS software (version 9.4). Two-sided P-values < 0.05 were considered statistically significant.
Results
Patients
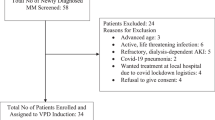
From February 25, 2015, to July 3, 2019, 508 patients were screened in 36 hospitals in China. A total of 417 patients were eligible and randomly assigned (2:1) to the aponermin group (278 patients) or the placebo group (139 patients). Of these, 276 patients in the aponermin group and 139 in the placebo group received the study treatment and were included in the analysis of efficacy and safety. By the final analysis date (January 3, 2020), 257 (93.1%) patients in the aponermin group and 135 (97.0%) patients in the placebo group had discontinued treatment. The reasons for the discontinuation of the intervention are shown in Fig. 1. The median treatment duration for the aponermin group was significantly longer than that of the placebo group (5 vs. 3 cycles).

CONSORT flow diagram
The baseline characteristics were well balanced between the two groups (Table 1). The median age was 59 years (range, 26–75), 42.2% were women. The median time since the initial diagnosis of MM was 2.6 years (range, 0.2–14.7). The median number of prior treatment regimens was three (range, 2–25), of which 46.0% had received at least four regimens. All patients had received previous glucocorticoids; 73.6%, proteasome inhibitor (PI); and 86.6%, immunomodulator (IMiD).
Efficacy
At a median follow-up of 17.2 months (95% CI, 15.1–28.2), 203 (73.6%) events of PD or death occurred in the aponermin group and 111 (79.9%) in the placebo group as assessed by the IAC. The median PFS was 5.5 months (95% CI, 4.7–6.5) in the aponermin group vs. 3.1 months (95% CI, 2.0–3.9) in the placebo group (HR, 0.62; 95% CI, 0.40–0.78; P < 0.001) (Fig. 2A). A significantly prolonged PFS was also observed based on the investigator's evaluations (Supplementary file: Table S1).

Progression-free Survival. A Kaplan–Meier analysis of progression-free survival (response assessed by Independent Assessment Committee) in the modified Intention-to-Treat Population, which included all patients who received at least one dose of trial treatment. B Subgroup analysis of progression-free survival
The prespecified subgroup analysis showed that the effect of aponermin on prolonging PFS compared to placebo was consistent for most subgroups (Fig. 2B). The PFS benefits of the aponermin group vs. placebo group were particularly evident in the subgroups of the patients with refractory MM (median 6.4 vs. 3.7 months; HR, 0.59; 95% CI, 0.38–0.89), the patients with prior therapy of IMiD and PI (median 4.7 vs. 2.1 months; HR, 0.55; 95% CI, 0.41–0.74), and the patients who were refractory to both IMiD and PI (median 3.7 vs. 1.8 months; HR, 0.36; 95% CI, 0.17–0.74).
At a median follow-up of 30.1 months (95% CI, 25.9–34.0), 151 (54.7%) deaths occurred in the aponermin group, and 88 (63.3%) in the placebo group. The median OS was 21.8 months (95% CI, 17.3–27.3) in the aponermin group and 17.0 months (95% CI, 12.2–23.2) in the placebo group (HR, 0.72; 95% CI, 0.56–0.94; P = 0.02) (Supplementary file: Figure S1). In an updated analysis of OS with a median follow-up of 48.0 months (95% CI, 40.0–55.7), 6.0 months extension was observed in the aponermin group compared to that in the placebo group (median, 22.4 vs. 16.4 months; HR, 0.70; 95% CI, 0.55–0.89; P = 0.003) (Fig. 3A). The prespecified subgroup analysis showed a significant effect of the aponermin group compared with the placebo group on OS for most of the subgroups (Fig. 3B).

Overall Survival. A. Kaplan–Meier analysis of overall survival in the modified Intention-to-Treat Population, which included all patients who received at least one dose of trial treatment. B Subgroup analysis of overall survival
The median DOR was 15.2 months (95% CI, 10.0–18.2) in the aponermin group vs. 9.8 months (95% CI, 4.4–14.7) in the placebo group (HR, 0.55; 95% CI, 0.29–1.05; P = 0.07). The median TTR were both 1.9 months in the two groups (HR, 0.89; 95% CI, 0.53–1.51; P = 0.67). TTP was longer in the aponermin group than in the placebo group (median, 5.8 vs. 3.5 months; HR, 0.61; 95% CI, 0.48–0.78; P < 0.001) (Table 2).
According to the assessment of the IAC, the ORR was 30.4% (95% CI, 25.1%–36.2%) in the aponermin group vs. 13.7% (95% CI, 8.4%–20.5%) in the placebo group (P < 0.001). The rates of very good partial response (VGPR) or better (14.1% vs. 2.2%), VGPR (12.0% vs. 1.4%), and clinical benefit (MR or better) (45.3% vs. 29.5%) were all superior in the aponermin group than those in the placebo group (Table 2). In a prespecified subgroup for patients who had achieved PR or better responses, greater benefits in PFS (median, 17.6 vs. 10.7 months; HR, 0.584; 95% CI, 0.31–1.11; P = 0.10) and OS (median, 42.9 vs. 31.6 months; HR, 0.41; 95% CI, 0.18–0.92; P = 0.03) were observed. The ORR and other response rates assessed by the investigators were similar to those assessed by the IAC (Supplementary file: Table S1).
Safety
AEs that occurred in 15% or more of the patients in either group are shown in Table 3. The main grade 3 or 4 AEs included neutropenia (26.8% vs. 26.6%), pneumonia (25.0% vs. 23.7%), and hyperglycemia (21.0% vs. 12.2%) in the aponermin and placebo groups. Hepatotoxicity occurred at a significantly higher frequency in the aponermin group than in the placebo group (ALT, 52.2% vs. 24.5%; AST, 51.1% vs. 19.4%; LDH, 44.9% vs. 21.6%). However, most of the ALT and AST elevations were grade 1 to 2, and approximately 10% of patients exhibited grade 3 or 4 elevations in the aponermin group. All hepatotoxicity events were transient and returned to normal or baseline levels before the next treatment. The incidence of dose adjustment or discontinuation due to hepatotoxicity was < 3%. No case of liver failure or death due to aponermin-related hepatotoxicity was reported.
The incidence of AEs leading to treatment termination were similar between the two groups (8.7% vs. 7.2%), and the most common AE was infectious pneumonia (1.8% vs. 3.6%). All of the AEs were transient and reversible.
SAEs were reported in 112 (40.6%) of 276 patients in the aponermin group and 52 (37.4%) of 139 patients in the placebo group. Pneumonia was the most common SAE (20.3% vs. 20.9%). SAEs occurred in 1% or more of the patients in either group are shown in Table S2.
HRQoL assessment
The mean scores for each domain of the EORTC QLQ-C30 and MY20 at baseline have no difference between the two groups (Supplementary file: Table S3). The Least-Squares (LS) mean changes in scores from baseline over the treatment cycles showed that the difference between groups favored the aponermin group over the placebo group for global health status, emotional functioning, social functioning, fatigue, constipation, and financial difficulties in the QLQ-C30 (all P-values < 0.05). For other domains, no significant differences between groups were observed. Future perspective, body image, and disease symptoms of QLQ-MY20 in the aponermin group were significantly better than those in the placebo group (all P-values < 0.05). The LS mean changes for disease symptoms and side effects of treatment were stable across treatments in the aponermin group and was not worse than that in the placebo group. (Supplementary file: Table S4).
Discussion
In this study, aponermin was combined with TD regimens. In China, thalidomide was more widely used than lenalidomide and bortezomib because of its affordability. Although a few new drugs for RRMM have been approved in China in the last five years, they are expensive, and some patients are deterred. Therefore, thalidomide remains an indispensable anti-myeloma drug. The superiority of aponermin plus TD over placebo plus TD in RRMM was confirmed in this study. The PFS, OS, and ORR were significantly improved in the aponermin group compared to the placebo group. The OS benefit of aponermin group vs. placebo group was further improved in the updated analysis than that in the first analysis, from 4.8 months to 6.0 months. In the aponermin group, more patients achieved deep remission, with the much higher rate of VGPR or better response compared to the placebo group. For patients who had achieved PR or better responses, greater benefits in PFS and OS were observed in the aponermin group vs. the placebo group, suggesting that patients who received aponermin treatment were able to maintain longer periods of remission and longer overall survival.
The benefits of the aponermin group regarding PFS and OS were observed in most prespecified subgroups, including those with poor prognosis, such as patients aged ≥ 65 years, previous exposure to TD/RD, refractory to PI and IMiD, or previous treated with more than three regimens. It is noteworthy that in patients with previous exposure to PI and IMiD, the risk of progression or death reduced by 45% and the risk of death reduced by 39% in the aponermin group compared to the placebo group. Of these, more than 50% patints had been treated with at least four regimens, and 38.3% had received lenalidomide. In the aponermin group, four patients had exposed to carfilzomib, bortezomib, and IMiD, one VGPR, one MR and two stable disease (SD) were observed; two patients had previously used monoclonal anti-CD38 antibody, bortezomib, and lenalidomide, one PR and one SD were obtained. The result suggests that aponermin combined with TD may be still an option even for patients with previous heavy treatment.
Overall, the efficacy outcomes in this study were consistent with the results of the phase 2 trial, in which improvements in PFS and ORR were observed in patients of the aponermin plus TD group compared with those in the TD group [22]. The safety profile in the study was also consistent with previous studies, with hepatotoxicity as the major adverse reaction of aponermin [18, 19, 22].
Treatment with aponermin may cause hepatotoxicity in some patients, as indicated by the elevated ALT and AST levels. The elevations of ALT and AST generally occurred after two days of treatment, reached a peak value after five days treatment of aponermin, and returned to normal or baseline levels before the next treatment cycle (the representative shown in Supplementary file: Figure S2A). No TBIL abnormalities accompanied by elevated ALT levels were observed. It is worth noting that approximately 28% of the patients in the aponermin group had an early transient elevation of the AST level on the second day of the first cycle, even reaching grade 3 or above. However, concurrently, the ALT level was not elevated or only slightly elevated (7% of patients) (the representative shown in Supplementary file: Figure S2B). The vast majority of these elevations were only detected in the first cycle and recovered quickly and spontaneously even though aponermin was not stopped. It is speculated that this transient elevation of AST may be associated with tumor lysis but not hepatotoxicity [18, 19, 22, 25].
The incidences of anemia and decreased lymphocyte count in the aponermin group were higher than that in the placebo group, but there was no statistical significance. After adjustment for drug exposure, rate of anemia was slightly lower in the aponermin group (132 vs. 140 events per 100 patient-years), and rates of decreased lymphocyte count were similar in the two groups (173 vs. 169 events per 100 patient-years). There was no decrease in leukocyte, platelet and neutrophil. The result suggests that aponermin has no hematological toxicity.
Pyrexia (grade 1/2) is a confirmed adverse reaction of aponermin, which was reported by 20.7% and 12.2% patients in the aponermin group and the placebo group, respectively. More patients reported positive urine leukocyte (15.9% vs. 8.6%) and increased monocyte cell count (15.2% vs. 5.0%) in aponermin group compared to the placebo group, but there was no difference in the laboratory test results between the two groups. Hypocalcemia, upper respiratory tract infections, hypertriglyceridemia and hyperglycemia are the known adverse reactions of dexamethasone. Increased incidences of these adverse events were observed in the aponermin group compare to the placebo group, which mainly related to the longer drug exposure (5 vs. 3 cycles). More patients with a history of diabetes (14.9% vs. 10.8%) may be another reason for the higher incidence of hyperglycemia. The higher incidence of hypocalcemia in aponermin group may also be related to tumor lysis caused by aponermin. Hypocalcemia, upper respiratory tract infections, hypertriglyceridemia and hyperglycemia have not been observed eigher in the preclinical study, or in clinical studies of aponermin monotherapy. Further researches are needed to determine whether the added of aponermin to TD will increase the risks.
There was no evidence that aponermin leads to nephrotoxicity, cardiotoxicity, or secondary tumors. The incidence of SAEs was similar between the two groups.
The clinical benefit of aponermin was further supported by the results of HRQoL. In half of the domains of the QLQ-C30 and MY20, the LS mean changes of the aponermin group were significantly better compared to the placebo group. Disease symptoms and side effects of treatment of the QLQ-MY20 were stable across treatments in the aponermin group and were not worse than those in the placebo group. The result suggests that aponermin owned a good safety profile in clinic, and there is a broad space for its combined application with other antitumor drugs.
In this study, efficacy analyses were based on a mITT population, with two patients excluded from the analysis for not receiving any study treatment. Sensitivity analysis of PFS for the mITT population was performed. The results of intention-to-treat population (417 patients) (median PFS 5.5 months for the aponermin group and 3.1 months for the placebo group, HR, 0.62; 95% CI, 0.49–0.78; P < 0.0001) were completely consistent with those of the mITT population.
The main limitation of this study is that the efficacy outcomes for both the aponermin group and the placebo groups were slightly weaker compared with the triplet regimens of novel drugs approved in recent years. In particular, the ORR of the placebo group was only 13.7%, which was lower than previously reported [26,27,28]. However, cross-trial comparisons are confounded by differences in patients populations and study designs. In this study, 46.0% patients had received at least four regimens, 73.6% had received PI, 86.6%, IMiD. Importantly, 74.6% of the patients had previously exposed to TD/RD treatment (no documented refractoriness to TD/RD regimens). Due to the above reasons, TD regimen of the placebo group showed weak efficacy in this trial. Although there was a significant improvement when adding aponermin to the TD regimen, the improvement was limited. In order to get better clinical benefit, it will be important to improve the overall response rate based on the application of biomarkers and combination with more potent anti-myeloma drugs. As a next step, we will design clinical trials using aponermin plus bortezomib/carfilzomib, lenalidomide/pomalidomide, or CD38-targeting antibody for the treatment of RRMM.
Overall, the results of this study indicate that aponermin plus TD had a favourable benefit-risk profile compared with placebo plus TD in RRMM. The role of the TRAIL signalling pathway in inducing apoptosis has been explored for a long time. But, at present, no drug have been approved for anti-tumor therapy targeting death receptors 4/5. To our knowledge, this is the first phase 3 trial shows that activation of the TRAIL pathway is a feasible approach for the treatment of RRMM. This represents a genuine breakthrough in cancer treatment and brings a novel weapon to the arsenal for fighting cancers. Additionally, this opens the door to exploring the applications of TRAIL family members in other cancers.
In conclusion, this phase 3 study demonstrated that aponermin plus TD has a favorable benefit-risk profile compared with placebo plus TD. Aponermin plus TD significantly improved PFS, OS, and ORR with manageable and reversible toxicity in RRMM patients with at least two prior therapies, and should be considered an effective treatment option for RRMM patients by targeting death receptors 4/5.
Availability of data and materials
The data that support the findings of this study are available upon reasonable request, by contact 13,381,075,598@163.com.
Abbreviations
- AE:
-
Adverse event
- ALT:
-
Alanine transaminase
- AST:
-
Aspartate transaminase
- CI:
-
Confidence interval
- CPT:
-
Recombinant circularly permuted human TRAIL
- CR:
-
Complete response
- DOR:
-
Duration of response
- DR4/5:
-
Death receptor 4 or 5
- EORTC:
-
European Organization for Research and Treatment of Cancer
- HR:
-
Hazard ratio
- HRQoL:
-
Health-related quality of life
- IAC:
-
Independent assessment committee
- IMiD:
-
Immunomodulator
- IMWG criteria:
-
International Myeloma Working Group consensus criteria
- ISS:
-
International Staging System
- IWRS:
-
Interactive web response system
- LDH:
-
Lactate dehydrogenase
- LS:
-
Least-Squares
- mITT:
-
Modified intention-to-treat population
- MM:
-
Multiple myeloma
- ORR:
-
Overall response rate
- OS:
-
Overall survival
- PD:
-
Progressive disease
- PFS:
-
Progression-free survival
- PI:
-
Proteasome inhibitor
- RD:
-
Lenalidomide plus dexamethasone
- RRMM:
-
Relapsed or refractory multiple myeloma
- SAEs:
-
Serious adverse events
- SD:
-
Stable disease
- TBIL:
-
Total bilirubin
- TD:
-
Thalidomide plus dexamethasone
- TRAIL:
-
Tumor necrosis factor-related apoptosis-inducing ligand
- TTP:
-
Time to progression
- TTR:
-
Time to response
- ULN:
-
Upper limit of normal
- VGPR:
-
Very good partial response
References
Rajkumar SV, Kumar S. Multiple myeloma current treatment algorithms. Blood Cancer J. 2020;10(9):94. https://doi.org/10.1038/s41408-020-00359-2.
Jagannath S, Miguel JS, Orlowski R, Palumbo A, Sezer O, Rajkumar SV, Durie BG, International Myeloma Working Group. Risk of progression and survival in multiple myeloma relapsing after therapy with IMiDs and bortezomib: a multicenter international myeloma working group study. Leukemia. 2012;26(1):149–57. https://doi.org/10.1038/leu.2011.196.
Genadieva-Stavric S, Cavallo F, Palumbo A. New approaches to management of multiple myeloma. Curr Treat Options Oncol. 2014;15(2):157–70. https://doi.org/10.1007/s11864-014-0276-6.
Liu J, Liu W, Mi L, Zeng X, Cai C, Ma J, Wang L. Union for China Lymphoma Investigators of the Chinese Society of Clinical Oncology; Union for China Leukemia Investigators of the Chinese Society of Clinical Oncology. Incidence and mortality of multiple myeloma in China, 2006-2016: an analysis of the Global Burden of Disease Study 2016. J Hematol Oncol. 2019;12(1):136. https://doi.org/10.1186/s13045-019-0807-5.
Vetma V, Guttà C, Peters N, Praetorius C, Hutt M, Seifert O, Meier F, Kontermann R, Kulms D, Rehm M. Convergence of pathway analysis and pattern recognition predicts sensitization to latest generation TRAIL therapeutics by IAP antagonism. Cell Death Differ. 2020;27(8):2417–32. https://doi.org/10.1038/s41418-020-0512-5.
Lemke J, von Karstedt S, Zinngrebe J, Walczak H. Getting TRAIL back on track for cancer therapy. Cell Death Differ. 2014;21(9):1350–64. https://doi.org/10.1038/cdd.2014.81.
Amarante-Mendes GP, Griffith TS. Therapeutic applications of TRAIL receptor agonists in cancer and beyond. Pharmacol Ther. 2015;155:117–31. https://doi.org/10.1016/j.pharmthera.2015.09.001.
Snajdauf M, Havlova K, Vachtenheim J Jr, Ozaniak A, Lischke R, Bartunkova J, Smrz D, Strizova Z. The TRAIL in the treatment of human cancer: an update on clinical trials. Front Mol Biosci. 2021;8:628332. https://doi.org/10.3389/fmolb.2021.628332.
Liu H, Su D, Zhang J, Ge S, Li Y, Wang F, Gravel M, Roulston A, Song Q, Xu W, Liang JG, Shore G, Wang X, Liang P. Improvement of pharmacokinetic profile of TRAIL via trimer-tag enhances its antitumor activity in vivo. Sci Rep. 2017;7(1):8953–63. https://doi.org/10.1038/s41598-017-09518-1.
Overdijk MB, Strumane K, Beurskens FJ, Ortiz Buijsse A, Vermot-Desroches C, Vuillermoz BS, Kroes T, de Jong B, Hoevenaars N, Hibbert RG, Lingnau A, Forssmann U, Schuurman J, Parren PWHI, de Jong RN, Breij ECW. Dual epitope targeting and enhanced hexamerization by DR5 antibodies as a novel approach to induce potent antitumor activity through DR5 agonism. Mol Cancer Ther. 2020;19(10):2126–38. https://doi.org/10.1158/1535-7163.MCT-20-0044.
Ratain MJ, Doi T, De Jonge MJ, LoRusso P, Calvo E. Phase 1, first-in-human study of TRAIL receptor agonist fusion protein ABBV-621. J Clin Oncol. 2019;37(15 suppl):3013.
Phillips DC, Buchanan FG, Cheng D, Solomon LR, Xiao Y, Xue J, Tahir SK, Smith ML, Zhang H, Widomski D, Abraham VC, Xu N, Liu Z, Zhou L, DiGiammarino E, Lu X, Rudra-Ganguly N, Trela B, Morgan-Lappe SE. Hexavalent TRAIL fusion protein eftozanermin alfa optimally clusters apoptosis-inducing TRAIL receptors to induce on-target antitumor activity in solid tumors. Cancer Res. 2021;81(12):3402–14. https://doi.org/10.1158/0008-5472.CAN-20-2178.
Wang BT, Kothambawala T, Wang L, Matthew TJ, Calhoun SE, Saini AK, Kotturi MF, Hernandez G, Humke EW, Peterson MS, Sinclair AM, Keyt BA. Multimeric Anti-DR5 IgM agonist antibody IGM-8444 is a potent inducer of cancer cell apoptosis and synergizes with chemotherapy and BCL-2 inhibitor ABT-199. Mol Cancer Ther. 2021;20(12):2483–94. https://doi.org/10.1158/1535-7163.MCT-20-1132.
Fang F, Wang AP, Yang SF. Antitumor activity of a novel recombinant mutant human tumor necrosis factor-related apoptosis-inducing ligand. Acta Pharmacol Sin. 2005;26(11):1373–81. https://doi.org/10.1111/j.1745-7254.2005.00206.x.
Ge Y, Yan D, Deng H, Chen W, An G. Novel molecular regulators of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in NSCLC cells. Clin Lab. 2015;61(12):1855–63. https://doi.org/10.7754/clin.lab.2015.150328.
Jian Y, Chen Y, Geng C, Liu N, Yang G, Liu J, Li X, Deng H, Chen W. Target and resistance-related proteins of recombinant mutant human tumor necrosis factor-related apoptosis-inducing ligand on myeloma cell lines. Biomed Rep. 2016;4(6):723–7. https://doi.org/10.3892/br.2016.650.
Wu Y, Giaisi M, Kohler R, Chen WM, Krammer PH, Li-Weber M. Rocaglamide breaks TRAIL-resistance in human multiple myeloma and acute T-cell leukemia in vivo in a mouse xenogtraft model. Cancer Lett. 2017;389:70–7. https://doi.org/10.1016/j.canlet.2016.12.010.
Hou J, Qiu L, Zhao Y, Zhang X, Liu Y, Wang Z, Zhou F, Leng Y, Yang S, Xi H, Wang F, Zhu B, Chen W, Wei P, Zheng X. A Phase1b dose escalation study of recombinant circularly permuted TRAIL in patients with relapsed or refractory multiple myeloma. Am J Clin Oncol. 2018;41(10):1008–14. https://doi.org/10.1097/COC.0000000000000404.
Geng C, Hou J, Zhao Y, Ke X, Wang Z, Qiu L, Xi H, Wang F, Wei N, Liu Y, Yang S, Wei P, Zheng X, Huang Z, Zhu B, Chen WM. A multicenter, open-label phase II study of recombinant CPT (Circularly Permuted TRAIL) plus thalidomide in patients with relapsed and refractory multiple myeloma. Am J Hematol. 2014;89(11):1037–42. https://doi.org/10.1002/ajh.23822.
Tan D, Lee JH, Chen W, Shimizu K, Hou J, Suzuki K, Nawarawong W, Huang SY, Sang Chim C, Kim K, Kumar L, Malhotra P, Chng WJ, Durie B, Asian Myeloma Network. Recent advances in the management of multiple myeloma: clinical impact based on resource-stratification. Consensus statement of the Asian Myeloma Network at the 16th international myeloma workshop. Leuk Lymphoma. 2018;59(10):2305–17. https://doi.org/10.1080/10428194.2018.1427858.
Cowan AJ, Allen C, Barac A, Basaleem H, Bensenor I, Curado MP, Foreman K, Gupta R, Harvey J, Hosgood HD, Jakovljevic M, Khader Y, Linn S, Lad D, Mantovani L, Nong VM, Mokdad A, Naghavi M, Postma M, Roshandel G, Shackelford K, Sisay M, Nguyen CT, Tran TT, Xuan BT, Ukwaja KN, Vollset SE, Weiderpass E, Libby EN, Fitzmaurice C. Global burden of multiple myeloma: a systematic analysis for the Global Burden of Disease Study 2016. JAMA Oncol. 2018;4(9):1221–7. https://doi.org/10.1001/jamaoncol.2018.2128.
Leng Y, Hou J, Jin J, Zhang M, Ke X, Jiang B, Pan L, Yang L, Zhou F, Wang J, Wang Z, Liu L, Li W, Shen Z, Qiu L, Chang N, Li J, Liu J, Pang H, Meng H, Wei P, Jiang H, Liu Y, Zheng X, Yang S, Chen W. Circularly permuted TRAIL plus thalidomide and dexamethasone versus thalidomide and dexamethasone for relapsed/refractory multiple myeloma: a phase 2 study. Cancer Chemother Pharmacol. 2017;79(6):1141–9. https://doi.org/10.1007/s00280-017-3310-0.
Kuznetsova OM, Tymofyeyev Y. Preserving the allocation ratio at every allocation with biased coin randomization and minimization in studies with unequal allocation. Stat Med. 2012;31(8):701–23. https://doi.org/10.1002/sim.4447.
Sonneveld P, Avet-Loiseau H, Lonial S, Usmani S, Siegel D, Anderson KC, Chng WJ, Moreau P, Attal M, Kyle RA, Caers J, Hillengass J, San Miguel J, van de Donk NW, Einsele H, Bladé J, Durie BG, Goldschmidt H, Mateos MV, Palumbo A, Orlowski R. Treatment of multiple myeloma with high-risk cytogenetics: a consensus of the International Myeloma Working Group. Blood. 2016;127(24):2955–62. https://doi.org/10.1182/blood-2016-01-631200.
Chen WM, Wei P, Yang SF, Zheng XJ, Qiu LG, Hou J, Zhang XJ, Wang Z, Ke XY, Pan L, Pang HY. Transiently elevated AST/LDH are associated with clinical response to recombinant circularly permuted TRAIL (CPT) plus thalidomide in patients with relapsed and/or refractory multiple myeloma [Conference session]. San Francisco: ASH annual meeting; 2014.
Cavo M, Tacchetti P, Patriarca F, Petrucci MT, Pantani L, Galli M, Di Raimondo F, Crippa C, Zamagni E, Palumbo A, Offidani M, Corradini P, Narni F, Spadano A, Pescosta N, Deliliers GL, Ledda A, Cellini C, Caravita T, Tosi P, Baccarani M, GIMEMA Italian Myeloma Network. Bortezomib with thalidomide plus dexamethasone compared with thalidomide plus dexamethasone as induction therapy before, and consolidation therapy after, double autologous stem-cell transplantation in newly diagnosed multiple myeloma: a randomised phase 3 study. Lancet. 2010;376(9758):2075–85. https://doi.org/10.1016/S0140-6736(10)61424-9.
Rosiñol L, Oriol A, Teruel AI, Hernández D, López-Jiménez J, de la Rubia J, Granell M, Besalduch J, Palomera L, González Y, Etxebeste MA, Díaz-Mediavilla J, Hernández MT, de Arriba F, Gutiérrez NC, Martín-Ramos ML, Cibeira MT, Mateos MV, Martínez J, Alegre A, Lahuerta JJ, San Miguel J, Bladé J, Programa para el Estudio y la Terapéutica de las Hemopatías Malignas/Grupo Español de Mieloma (PETHEMA/GEM) group. Superiority of bortezomib, thalidomide, and dexamethasone (VTD) as induction pre-transplantation therapy in multiple myeloma: a randomized phase 3 PETHEMA/GEM study. Blood. 2012;120(8):1589–96. https://doi.org/10.1182/blood-2012-02-408922.
Iida S, Wakabayashi M, Tsukasaki K, Miyamoto K, Maruyama D, Yamamoto K, Takatsuka Y, Kusumoto S, Kuroda J, Ando K, Kikukawa Y, Masaki Y, Kobayashi M, Hanamura I, Asai H, Nagai H, Shimada K, Tsukamoto N, Inoue Y, Tobinai K. Bortezomib plus dexamethasone vs thalidomide plus dexamethasone for relapsed or refractory multiple myeloma. Cancer Sci. 2018;109(5):1552–61. https://doi.org/10.1111/cas.13550.
Acknowledgements
We thank all the patients who participated in this trial and their families. We also thank the investigators and staff members involved in this clinical trial at each trial site. Finally, we thank Janus for their support with the data analysis.
Funding
This study was funded by the National Major Science and Technology Projects of China (grant number: 2018ZX09733003) and Beijing Sunbio Biotech. Co., Ltd., a wholly-owned subsidiary of Wuhan Hiteck Biological Pharma Co., Ltd. The funder, National Major Science and Technology Projects of China, had no role in the design, analysis, or interpretation. The funder, Beijing Sunbio, in collaboration with the investigators, designed the trial and collected and interpreted the data.
Author information
Authors and Affiliations
Contributions
WC contributed to trial design, patient recruitment, data collection, interpretation, and writing. ZX, YL, BF, YL, WL, CF, LY, XK, HJ, JW, LL, YZ, XZ, ZH, AL, QS, YG, XC, LP, ZC, ZW, YW, YF, MH, YM, JH, JL, JZ, XZ, HM, XL, FL, HR, BH, ZS, HZ, YH, YL, SG, LY, YM, HJ, JD, WL, JZ, WS, FW, and XL contributed to the patient recruitment and data collection. SY, XZ, PW, HP and WY contributed to trial design and interpretation. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The trial protocol was approved by the independent ethics committees of Beijing Chao-Yang Hospital Capital Medical University. The trial was conducted in accordance with the principles of Good Clinical Practice and the Declaration of Helsinki. All patients provided written informed consent before enrolment.
Consent for publication
Not applicable.
Competing interests
SY, XZ, PW, HP and WY are employees of Beijing Sunbio Biotech Co. Ltd. The other authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xia, Z., Leng, Y., Fang, B. et al. Aponermin or placebo in combination with thalidomide and dexamethasone in the treatment of relapsed or refractory multiple myeloma (CPT-MM301): a randomised, double-blinded, placebo-controlled, phase 3 trial. BMC Cancer 23, 980 (2023). https://doi.org/10.1186/s12885-023-11489-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-023-11489-8