
Abstract
Background
Exerkines are all peptides, metabolites, and nucleic acids released into the bloodstream during and after physical exercise. Exerkines liberated from skeletal muscle (myokines), the heart (cardiokines), liver (hepatokines), white adipose tissue (adipokines), brown adipose tissue (batokines), and neurons (neurokines) may benefit health and wellbeing. Cancer-related cachexia is a highly prevalent disorder characterized by weight loss with specific skeletal muscle and adipose tissue loss. Many studies have sought to provide exercise strategies for managing cachexia, focusing on musculoskeletal tissue changes. Therefore, understanding the responses of musculoskeletal and other tissue exerkines to acute and chronic exercise may provide novel insight and recommendations for physical training to counteract cancer-related cachexia.
Methods
For the purpose of conducting this study review, we made efforts to gather relevant studies and thoroughly discuss them to create a comprehensive overview. To achieve this, we conducted searches using appropriate keywords in various databases. Studies that were deemed irrelevant to the current research, not available in English, or lacking full-text access were excluded. Nevertheless, it is important to acknowledge the limited amount of research conducted in this specific field.
Results
In order to obtain a comprehensive understanding of the findings, we prioritized human studies in order to obtain results that closely align with the scope of the present study. However, in instances where human studies were limited or additional analysis was required to draw more robust conclusions, we also incorporated animal studies. Finally, 295 studies, discussed in this review.
Conclusion
Our understanding of the underlying physiological mechanisms related to the significance of investigating exerkines in cancer cachexia is currently quite basic. Nonetheless, this demonstrated that resistance and aerobic exercise can contribute to the reduction and control of the disease in individuals with cancer cachexia, as well as in survivors, by inducing changes in exerkines.
Similar content being viewed by others
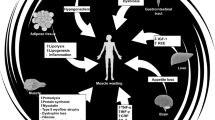


Introduction
Cancer-related cachexia (CC) is a devastating, multifactorial, and often irreversible syndrome that affects approximately 50–80% of cancer patients, depending on the type of tumor [1]. CC is a disorder characterized by weight loss with specific loss of skeletal muscle and sometimes also loss of adipose tissue [2]. Symptoms of CC include severe body mass loss, anorexia, generalized inflammation, and marked muscle wasting, leading to a severe reduction in quality of life [3]. These can also limit treatment options, as CC patients are usually less tolerant of radiotherapy and chemotherapy due to general weakness and discomfort [4]. Unlike starvation, which mainly affects adipose tissue, skeletal muscle is the main target of wasting in CC patients, suggesting a different mechanism that targets muscle loss [5]. Although the primary tissue affected by cachexia is skeletal muscle, CC cannot be reduced and eventually affects other tissues such as the liver, heart, adipose tissue, and brain [6]. Epidemiological data show that the prevalence of cachexia varies among different cancers, and overall, CC is believed to be directly responsible for up to 20% of cancer-related deaths [7]. When CC occurs is still not completely clear; however, it is known that CC can be considered one of the symptoms of metastatic cancer [8].
The effects of exercise and physical activity on health and fitness are well known [9]. Exercise is the number one priority for good health and physical fitness, but when it comes to many diseases, exercise is seen as a secondary therapeutic strategy [10]. Clinical benefits of exercise during cancer can include reducing the severity of the disease, reducing the side effects of chemotherapy, and managing negative physiological changes, and after cancer or discharge from the hospital, the return of quality of life [11, 12]. However, the molecular mechanisms underlying the beneficial effects of exercise in cancer are still poorly understood [13]. Nonetheless, it has recently been proposed that exercise-induced secretory factors, termed exerkines [14], have the potential to improve numerous aspects of health [13] and may, therefore, be involved in mediating or moderating the beneficial effects of exercise in cancer. Exerkines encompass a wide variety of signaling moieties released in response to acute and/or chronic exercise that exert their effects through endocrine, paracrine, and/or autocrine pathways [13]. These include factors released during exercise from skeletal muscle (myokines), the heart (cardiokines), liver (hepatokines), white adipose tissue (adipokines), brown adipose tissue (batokines) and neurons (neurokines) [13]. It has been suggested that the exerkine response can provide an insight into multi-organ changes following exercise [14]. In recent years, several studies related to exerkines have been published, the results of which focus on health and disease management [13, 15, 16]. Researchers responsible for these studies believe that the specificity of the exerkine responses could provide a new approach to developing therapeutic exercise recommendations targeted toward particular diseases [13, 15, 16].
In this review we intend to address the issue of whether exerkines can be effective in managing CC, with a unique look at the influence of exercise programming variables on exerkine responses; and how exerkines can lead to CC management. This novel insight could create direction for new research addressing exercise regimens targeted at beneficial exerkine responses for various clinical situations (See Fig. 1). For the purpose of conducting this study review, we made efforts to gather relevant studies and thoroughly discuss them to create a comprehensive overview. To achieve this, we conducted searches using appropriate keywords in various databases. Studies that were deemed irrelevant to the current research, not available in English, or lacking full-text access were excluded. Nevertheless, it is important to acknowledge the limited amount of research conducted in this specific field. In order to obtain a comprehensive understanding of the findings, we prioritized human studies in order to obtain results that closely align with the scope of the present study. However, in instances where human studies were limited or additional analysis was required to draw more robust conclusions, we also incorporated animal studies.

Association between CC and exerkines
Highlighting exerkines in cancer cachexia
Myokines
Skeletal muscle corresponds to approximately 40% of the total body weight. This tissue secretes several factors that act in an autocrine, paracrine, and/or endocrine manner to regulate the physiology of muscles and distant organs [17]. These secretory agents were named myokines, which are synthesized and released by myocytes during muscle contractions. Secretory analysis of human myocyte culture media has identified more than 600 myokines [18]. However, most of these myokines are still not well characterized. Only a few have been studied for their biological activity and function, providing clear evidence that they are released directly from muscle contraction [18].
Epidemiological data show that skeletal muscle wasting is seen in most cancers [19, 20]. Skeletal muscles are more affected than other organs during CC [21, 22], however our understanding of why skeletal muscle is more affected by CC is incomplete. For this reason, researchers have shown an increasing interest in investigating the underlying causes of skeletal muscle wasting in cancer patients [23]. Most studies have focused their attention on muscle atrophy pathways and key proteins involved in muscle protein degradation, such as atrogin-1 and MuRF-1 [24]. It has been found that targeting myokines can be a therapeutic strategy for CC management [25]. This could indicate that, in cancer patients, skeletal muscle myokines and/or their biological effectiveness are affected by the adverse effects of CC. Thus far, only a few myokines are known to be affected by CC e.g., ↑myostatin, ↓follistatin, ↑irisin, ↑IL-6, ↑FGF-21, and possibly ↓myonectin [25,26,27,28,29,30,31] (Table 1).
Myostatin is a negative regulator of skeletal muscle mass, with high levels causing muscle atrophy, while low or no expression causes muscle hypertrophy [32]. Skeletal muscle atrophy and adipose tissue loss due to myostatin overexpression suggests potential for a role in the pathogenesis of CC [21]. In support, some studies report myostatin overexpression in CC [1, 27, 33]. High levels of myostatin inhibit proliferation and differentiation of satellite cells and block muscle protein synthesis [34]. It has been determined that reduction of myostatin reduces circulating inflammatory cytokine concentrations, including TNF-α and IL-6 [35] which could be useful in CC given the chronic inflammation present. On the other hand, follistatin has an opposing biological role to myostatin. Follistatin binds to and inhibits activin A and myostatin, increasing skeletal muscle hypertrophy [36, 37]. In CC, the level of follistatin is significantly reduced, and so therapeutic strategies to increase follistatin could reduce or prevent atrophy [38]. Follistatin also appears to act as an anti-inflammatory agent, reducing IL-6 [39] (Table 1).
Irisin is one of the newest myokines that many researchers are interested in investigating. Irisin may be a key molecule involved in crosstalk between muscle and fat tissue [40]. The expression of irisin in muscle is 200 times that of adipose tissue, and it has an important role in lipid metabolism by turning white adipose tissue into brown adipose tissue to increase metabolism and thermogenesis and reduce body fat percentage [41]. Given that muscle loss is often accompanied by fat loss in CC, it is of interest to investigate alterations in irisin [40]. It has been found that, in cancer patients, the increase of irisin can lead to the proliferation of cancer cells [42, 43]. However, until now, no study has been conducted that has investigated this myokine with the aim of determining an effect on cachexia, with only Altay et al. showing that irisin is increased in CC [40] (Table 1).
IL-6 is another myokine that is affected in CC. IL-6 is a cytokine with pleiotropic functions in various tissues and organs. After prolonged exercise, skeletal muscle produces and releases significant levels of IL-6, which is consequently considered a myokine [44]. In this regard, it has been found that IL-6 plays an important role in the development of CC [45]. Interleukin 6 can stimulate muscle protein synthesis, is an anti-inflammatory factor, but can also be a risk factor in CC [46,47,48]. These conflicting effects of IL-6 have been attributed to a temporal function based on acute versus chronic IL-6 exposure [45]. It appears that when the body experiences cachexia due to chronic inflammation, the level of IL-6 is elevated, and IL-6 can suppress protein synthesis and activate several protein degradation pathways [49, 50]. However, studies have cautiously reported that IL-6 may directly contribute to decreased protein synthesis [51, 52]. Other studies have suggested that increased IL-6 in CC may lead to suppression of IGF-1 and inhibition of AMPK activation [50, 53] (Table 1).
In CC, along with increased IL-6, FGF-21 is also increased, which appears to be a new player in the regulation of muscle mass [54]. FGF-21 is a secretory myokine that can also be released into the bloodstream by other organs, such as the liver, heart, WAT, and BAT. In skeletal muscle, FGF-21 expression is almost undetectable under healthy conditions and circulating FGF-21 is mainly produced and released by the liver [54]. In a study by Franz et al. 2019, it was found that patients with CC had the highest levels of FGF-21 compared to healthy individuals [55]. Elevated FGF-21 in CC appears to lead to the activation of muscle atrophy pathways [54]. Deletion of FGF-21 can lead to the prevention of muscle atrophy, and the overexpression of FGF-21 can lead to the induction of autophagy, and muscle loss of up to 15% [54] (Table 1).
Recently, researchers have identified myonectin as a myokine. Myonectin (CTRP15) is mainly expressed and secreted by skeletal muscle [56]. Myonectin appears to mediate cross-talk between skeletal muscle and other metabolic compartments, such as adipose tissue and the liver, to coordinate the integration of whole-body metabolism [56]. Consistent with this concept, myonectin expression and secretion by the skeletal muscle are highly responsive to acute nutritional and metabolic changes (e.g., fasting/re-feeding cycles and exercise) and chronic changes in the animal’s energy status (e.g., diet) [56]. It has been determined that myonectin is released into the bloodstream through muscle contraction. Myonectin increases the expression of fatty acid transfer genes such as CD36, FATP1, Fabp1, and Fabp4, which in turn enhances the absorption of fatty acids into cells [56,57,58]. It is known that autophagy levels are high in skeletal muscles of patients with CC. Autophagy is a mechanism that causes muscle atrophy [59]. Myonectin has been shown to suppress autophagy via the PI3K/Akt/mTOR signaling pathway [56,57,58]. ollowing mtDNA depletion, myonectin levels are significantly increased, and it enhances glucose uptake and fatty acid oxidation through activation of the AMPK signaling pathway in mouse skeletal myocytes [60, 61]. Consequently, research conducted on this emerging myokine has demonstrated that it can reduce autophagy and inflammation via the PI3K/Akt/mTOR signaling pathway [21]. This mechanism has the potential to prevent muscle atrophy in CC patients and promote increased synthesis of muscle proteins. Furthermore, it also has an impact on mitochondrial biogenesis [21] (Table 1).
Adipokines
Adipose tissue produces pro-inflammatory and anti-inflammatory mediators that affect local and systemic inflammation. Among these mediators are adipokines, proteins produced by white adipose tissue cells that act as hormones [62]. Adipokines include leptin, adiponectin, resistin, chemerin, visfatin, omentin, vaspin, progranulin and CTRP-4 [62]. Although skeletal muscle wasting due to increased protein breakdown is recognized as the main characteristic of CC, adipose tissue depletion and remodeling is also an important clinical feature in cancer patients. Recently, adipose tissue breakdown has been shown to occur before the appearance of other classic markers of cachexia (i.e., fat loss is a more rapid event compared to muscle loss) [63, 64]. CC causes inflammation and lipid dysfunction, which leads to impaired synthesis and secretion of several pro-inflammatory and anti-inflammatory adipokines [65]. However, the role of adipokine dysregulation in cancer-induced cachexia has not yet been fully elucidated. Most of the studies on this topic are limited to adipokine changes and only relate their plasma concentrations to tumor size, while changes in synthesis, secretion and signaling in adipose tissue remodeling during CC have not yet been studied in depth [65]. Available data appears to show that only a few adipokines play a significant role in the development of cachexia in cancer (↑leptin, ↓adiponectin, and ↑resistin) [66, 67]. It has been found that leptin has the greatest effect on causing inflammation and fat loss in CC [67]. Leptin is a 16 kDa non-glycosylated protein produced by subcutaneous adipose tissue [68]. Lep-R is expressed on immune cells, and leptin mainly binds to them through the JAK-STAT and NF-kB dependent pathway in order to affect the immune response [69]. In CC, leptin increases several inflammatory factors, such as, TNF-α, IL-6 and IL-1β [70]. In confirmation of these findings, Santos-Alvarez et al. reported that leptin increases the proliferation of monocytes and causes the expression of inflammatory cytokines (TNF-α and IL-6) in the body [71]. Adiponectin has an anti-inflammatory function [72], and in cancer patients there is an observed decline in adiponectin and an increase in inflammation [73]. It has been found that mice deficient in adiponectin have increased numbers of classically activated M1 macrophages in their adipose tissue. M1 macrophages increase the production of cytokines TNF-α and IL-6 [74]. In the meantime, it has been found that the increase of adiponectin can suppress the function of eosinophils. Eosinophils are responsible for activating M1 and IL-4, which can increase inflammation [75]. It has also been found that the increase of adiponectin can be one of the main factors preventing the suppression of neutrophils [76]. Consistently, adiponectin suppresses neutrophil membrane ceramide accumulation and inhibits neutrophil apoptosis via AMPK [77]. In monocytes/macrophages, adiponectin suppresses the production of TNF-α and IL-6 and induces the production of IL-10 and IL-1 receptor antagonist anti-inflammatory mediators [78, 79]. Cancer-induced chronic inflammation has emerged as a key driver of CC, due to the multiorgan pathology and associated wide range of metabolic and endocrine disorders that lead to tissue dysfunction [65]. High levels of circulating proinflammatory cytokines, such as TNF-α and IL-6, have been observed in cachectic patients [80]. This could be due to changes in the concentration of leptin, adiponectin, and other adipokines [80]. Interestingly, increased expression of IL-6 in adipose tissue is positively correlated with increased circulating levels of IL-6. Thus, it suggests that adipose tissue, especially subcutaneous adipose tissue, may act as a key source of inflammatory mediators during the progression of CC [65]. In mouse models of CC, the secretion of pro-inflammatory cytokines by white adipose tissue, such as TNFα and IL-6, has been observed. These molecules can cause a decrease in fat storage and as a result atrophy of adipose tissue [65]. On the other hand, the deformation of fat cells and the gradual transformation of white fat cells into thermogenic fat cells (called browning) is one of the main developments of CC, which indicates high energy expenditure [81] (Table 1).
In cancer patients with CC, the amount of omentin in the blood is low [82]. Importantly, omentin suppresses monocyte adhesion to TNF-α-activated endothelial cells by inhibiting the expression of ICAM-1 and VCAM-1 via PI3K-AKT signaling by blocking the ERK/NF-κB pathway [83]. Other studies have indicated that omentin may play an anti-inflammatory and vasoprotective role in reducing obesity-related vascular complications [83, 84]. It has also been reported that omentin levels in subjects with respiratory infections are associated with the inflammatory response, and overexpression of omentin can decrease the expression of IL-6 and TNF-α and decrease the activation of the NF-κB Rel subunit [85] (Table 1).
Cardiokines
A body of evidence shows that peptides or proteins secreted from cardiac cells could be considered cardiokines [86]. Most cardiokines, as mediators, play an essential role in maintaining the homeostasis of a healthy heart or responding to myocardial injury [86]. It has been reported that cardiokines are physiologically involved in stress response, damage repair, and myocardial regeneration and can participate in protein synthesis in end-organ tissues and systemic metabolic processes [86]. In addition, cardiokines have differential expression in response to varying physiological conditions of the heart. Secreted cardiokines are thought to maintain healthy cardiac function through paracrine/autocrine pathways or influence the response of cardiomyocytes and cardiac fibroblasts (CFs) to pathological abnormalities caused by cardiac injury or damage [86]. Natriuretic peptides, and in particular ANP and BNP, secreted by the cardiovascular system, have a particularly large impact on the occurrence and development of CVD in a paracrine/autocrine manner [87]. In addition to skeletal muscle wasting, CC is also associated with cardiac muscle dysfunction. Cardiac abnormalities are commonly seen in cancer patients and are the leading cause of death in at least one-third of cancer patients [81]. Cardiac abnormalities manifest as cardiac muscle atrophy, fibrosis, and ultimately cardiac dysfunction. This condition significantly impacts the quality of life and reduces overall survival [88]. It has been established that cancer patients commonly experience symptoms of chronic heart failure, including fatigue, shortness of breath, and decreased exercise tolerance [81]. Additionally, cardiac cachexia involves metabolic alterations, primarily including increased energy expenditure, activation of the UPR system leading to proteolysis, ubiquitin-mediated proteasome degradation, and autophagy [66, 89]. Cardiokines include atrial natriuretic peptide, brain natriuretic peptide, IL-33, IL-6, IL-18, IL-1β, follistatin, TGF-β, Ang-II, and TNF-α [90]. Limited research has examined the significance of cardiokines in CC. However, it is evident that several cardiokines undergo significant alterations in the context of CC (↓ANP, ↓BNP, ↓IL-33, ↑IL-6, ↓IL-18, ↑IL-1β, ↓follistatin, ↑TGF-β, ↓Ang-II, and ↑TNF-α) [5, 21, 91]. Follistatin and TNF-α levels have been found to increase in cardiac cachexia and can effectively increase inflammation and atrophy of the heart muscle [92] (Table 1).
Hepatokines
Several hepatokines have been identified and investigated for their role in the development of obesity, insulin resistance, and non-alcoholic fatty liver disease [93]. However, the role of hepatokines in CC has not been investigated in detail yet. Hepatokines include Activin-E, ANGPTL3, ANGPTL4, ANGPTL6, ANGPTL8, Fetuin-A, FGF-21, Follistatin, GDF15, Hepassocin, IGF1, LECT2, Lipocalin 13, Selenoprotein-P, SMOC1 and Tsukushi [94] with several of them shown to be altered in CC (↓Activin-E, ↑ANGPTL-4, ↑FGF-21, ↓Follistatin and ↓IGF1) [95,96,97,98,99]. These changes can lead to increased inflammation, a negative effect on muscle myokines, a negative effect on adipokines and the development of cachexia [100,101,102] Table 1. Finally, it should be noted that while the importance of hepatokines in CC has not been extensively studied, the expression of these proteins can lead to the exacerbation and increase of cachexia during cancer.
Acute and chronic exercise effects on exerkines
As shown in the above section, numerous kines from multiple organs and tissues are affected by and may contribute to CC. There is growing evidence that the abundance of many of these kines are favourably altered by exercise and this may be one of the mechanisms by which exercise improves health. As such, an understanding of the effects of exercise on these kines may help in the development of effective exercise interventions for treating CC and will be the focus of the following section.
Exercise effects on muscle-derived exerkines (myokines)
There have been numerous studies examining the impact of resistance and aerobic exercise, both in acute and chronic forms, on myokines. However, there is limited research regarding the effects of exercise on myokines in cancer patients. Nonetheless, a review conducted by Kim et al. (2021) [103] revealed that alterations in myokine levels could directly inhibit cancer growth by impeding proliferation. Moreover, myokines induced by exercise play a crucial role in enhancing cytotoxicity and facilitating immune cell infiltration into tumors. In line with this, findings from other review studies validate the potential effectiveness of exercise-induced myokines in cancer [104,105,106]. Nevertheless, these studies did not primarily focus on investigating the impact of exercise-induced myokines on the management of CC and its survivors.
Responses to acute exercise
Most, but not all studies indicate that myostatin is reduced following a single bout of either aerobic, HIIT or resistance exercise. As an example, Gholamali et al. (2015) observed that cycling at 70% of VO2max in a single session resulted in an immediate 45% reduction in myostatin levels after exercise, followed by a further 54% decrease four hours post-exercise [107]. They proposed that one possible reason for the reduction in plasma myostatin levels following endurance activity is the concurrent increase in insulin-like growth factor-1. This increase in IGF-1 levels within skeletal muscle leads to a decrease in FoxO pathway activity, which is a crucial cellular pathway involved in promoting apoptosis [107, 108]. In line with this study, Pugh et al. (2015) showed that high-intensity interval training at 90% HRmax could decrease myostatin immediately, 2 h, and 6 h after exercise [109]. These results are consistent with other studies that reported that acute high-intensity exercise between 80 and 90% of VO2max or at an intensity of 90% HRmax could decrease myostatin [110,111,112,113]. In contrast to these results, Kabak et al. (2018) showed that high-intensity interval exercise (Wingate test) could increase myostatin immediately after exercise [114]. These researchers reported that the increase in myostatin was due to a decrease or lack of IGF-1 secretion and the increase of IL-6 [114]. Dalbo et al. (2011) showed in their study that three sets of 10 repetitions (80% 1RM) could decrease myostatin [115]. In line with this study, Gonzalo et al. (2013) showed that resistance exercise of three sets of 12 repetitions (80–85% 1RM) could decrease myostatin [116]. In their study, Matthew et al. 2013 investigated the effect of acute resistance exercise of 4 sets of 10 repetitions (with an intensity of 90% 1RM) on the transcriptional activity of myostatin [117]. These researchers stated in their results that acute resistance exercise reduces myostatin signaling through the activation of the TGFβ Notch inhibitor and leads to a decrease in the transcriptional activity of myostatin. The results are consistent with other studies that stated that an acute resistance training session (between three to four sets and 10 to 15 repetitions at 80 to 95% of 1RM) could reduce myostatin [118, 119]. For instance, in the study conducted by Shabkhiz et al. (2021), it was discovered that acute resistance training leads to an increase in myostatin levels when performed at an intensity of 80% of 1RM, with 10–12 repetitions and 3–4 sets [118]. It also seems that low-intensity resistance training (60% of 1RM) with blood-flow restriction can also be effective in decreasing myostatin [120]. However, in another study, it was reported that low-intensity training (40–50% of 1RM) combined with blood flow restriction could not affect the reduction of myostatin [121].
There are various reports regarding follistatin. Most studies have reported stimulation of follistatin in response to acute resistance exercise with intensity of 70–90% 1RM, 8–15 repetitions and between 3 and 4 sets [117, 122,123,124]. Endurance exercise also stimulates follistatin release. For example, running on a treadmill at a speed of 15–30 m/min for 35 min lead to follistatin stimulation [125]. Another study found that an acute high-intensity SIT cycle session consisting of four 30-second maximal efforts with a 4-minute recovery between sets could induce follistatin [126].
Irisin appears to be stimulated by resistance exercises with an intensity between 70 and 90% 1RM, but high-intensity exercises with an intensity of 70 to 95% VO2max, such as running or cycling, have a greater effect on irisin stimulation and duration [127,128,129]. It seems that this intensity can keep irisin levels high for 2–3 h after exercise [130,131,132]. Another study demonstrated that circuit training, consisting of 12–15 repetitions at 65–70% of 1RM, and performed for 3 sets, did not have any effect on irisin levels [133]. However, other studies stated that resistance training with an intensity of 70–85% 1RM and 6–12 rep can be effective in increasing irisin [134, 135].
In relation to IL-6 it seems that acute exercise, such as running or cycling at 70 to 95% of VO2max, can lead to an increase of IL-6 [136,137,138,139,140,141], Regarding resistance training, research has indicated that acute resistance training sessions with intensities ranging from 70 to 85% of 1RM and 6–12 repetitions can potentially lead to an increase in muscle IL-6 levels [141, 142]. However, it is important to note that the existing studies are limited and yield contradictory findings, emphasizing the need for further investigation in the future. Nonetheless, it can be cautiously suggested that acute and short-term resistance training may result in the release of IL-6. This assertion finds support in the fact that exercise intensity and mechanical stress play a role in mediating the release of IL-6 from skeletal muscle fibers. Additionally, lactate production in skeletal muscle has been proposed as a mediator of the IL-6 response to exercise [142]. On the other hand, there is evidence that an exercise-induced increase in AMPK activity in skeletal muscle correlates with IL-6 production [143]. It is worth noting that IL-6 released from skeletal muscle fibers can act in an autocrine manner, stimulating further release of IL-6 in a positive feedback loop. This could explain the observation that IL-6 activates AMPK, which, in turn, induces exocytosis of IL-6 vesicles and potentially accounts for the exponential rise in plasma IL-6 levels observed during exercise. It is important to consider that acute high-intensity resistance training, which effectively activates AMPK and increases lactate production, seems to be associated with an increase in IL-6 release from muscle [144, 145].
Resistance training with intensity between 70 and 90% 1RM can have a greater effect on FGF-21 between 2 and 3 h after training than cycling or running [146, 147] (Table 2; Fig. 2).
Responses to chronic exercise
Studies have shown that chronic exercise can significantly reduce myostatin [103, 148,149,150,151], leading to the inactivation of muscle atrophy pathways and activation of muscle hypertrophy pathways [152]. Hittel et al. (2010) reported that aerobic exercise with an intensity of 40-55% of VO2peak for 8 weeks could decrease myostatin levels [110]. In another study, the researchers reported that aerobic exercise with an intensity of 50–65% VO2max for 30–50 min for 12 weeks decreased myostatin [149]. Ko et al. (2014) showed that 30 min of running on a treadmill for 6 weeks at 45–55% VO2max reduced myostatin and improved muscle function [153]. In confirmation of these results, another study showed that running on a treadmill 3 times a week for 6 months was effective in reducing myostatin [154]. Aerobic training with an intensity of 50–60% of the reserve heart rate for 20–30 min continuously in the first 4 weeks and increasing the intensity to > 60% VO2max and up to 80% VO2max with a duration of 50 min can also decrease myostatin [154]. In relation to aerobic exercises, it has been discovered that engaging in these exercises at intensities ranging from a maximum of 50–65% of VO2max or 40–55% of VO2peak, with a duration of 30 to 60 min over a period of 6 to 12 weeks, can result in a reduction in myostatin levels [103, 110, 148,149,150,151,152,153,154].
In the study of Shahrokhian et al. (2022), it was found that resistance training can lead to a significant decrease of 15–23% in myostatin in women with breast cancer. In this study, participants performed resistance training three times per week for 12 weeks at 40-90% 1RM [155]. Furthermore, in elderly men with and without type 2 diabetes, resistance training at 70% 1RM for three sets of 10 repetitions, three times per week for 12 weeks, decreased myostatin by 5.5% (diabetic group) to 17% (non-diabetic group) relative to the non-exercising controls [118]. These findings support other studies that have also reported a decrease in myostatin levels following resistance training [112, 118, 156,157,158,159,160]. Chronic resistance training for 8–12 weeks and 3–4 training sessions per week, at 60-90% 1RM for 10–12 repetitions in 3 sets can be a suitable approach to reduce myostatin [112, 118, 155,156,157,158,159,160].
Six to twelve weeks of resistance training (3–5 sessions per week) at an intensity of 70–90% of 1RM between 3 and 4 sets of 10–12 repetitions can change other myokines, such as↑follistatin, ↑irisin, ↓IL-6, ↓FGF-21, and ↑myonectin [132, 156, 161,162,163,164,165,166,167,168,169]. Along with the reported reduction of myostatin in resistance exercise, an increase in follistatin has been seen, which can lead to activation of muscle hypertrophy [156, 158, 170, 171]. Based on the designs and outcomes of these interventions, it is evident that the intensity and duration of training play crucial roles in the observed increase in follistatin levels. Low intensity (40–50% 1RM) seems to have no effect whereas moderate (55–65% 1RM) to high intensity (70–100%1RM) can be chronically effective [156, 158, 170, 171]. Regarding irisin and FGF-21, research has indicated that resistance training can result in an increase in irisin levels and a decrease in FGF-21 levels [128, 172]. For instance, it has been found that resistance training between 6 and 26 weeks, with intensities between 60 and 95% of 1RM or an activity score of 12 to 13 on the BORG scale, can significantly increase irisin levels [173,174,175].
While physical activities such as Total Body Resistance Exercise (TRX) training [176,177,178,179,180], yoga [181,182,183,184,185], swimming [186,187,188,189,190,191,192,193,194,195], walking [196,197,198,199,200,201,202,203,204,205,206,207,208] and cycling [199, 209,210,211,212] are recommended for cachexia patients, it seems that only some of these modalities might be effective in positively changing myokines given the relative intensity involved in each.
Because of the limited results concerning myonectin in human studies, we also looked at animal studies. In animal studies it was found that exposure to three weeks of free wheel running enhanced the expression of the myonectin gene [56]. Supporting these findings, Vosadi et al. (2016) reported that moderate-intensity aerobic exercise in rats can increase myonectin levels [213]. However, there are only a limited number of studies that have reported circulating myonectin. On the other hand, it was found in human studies that eight weeks of aerobic training (50–70% of maximal heart rate for 45 min) can boost myonectin levels in women [214]. In contrast, Lim et al. (2012) found that cycling exercise at 60–80% of maximum oxygen consumption (3 sessions of 1 h per week) decreased myonectin levels [61] (Table 2; Fig. 2).
Exercise effects on adipose tissue-derived exerkines (Adipokines)
Responses to acute exercise
Similar to the different responses seen in myokines due to exercise, these differences are also observed in relation to adipokines. In this regard, exercise appears to be an effective intervention for decreasing leptin, with many studies reporting a decrease in leptin after acute exercise [215,216,217,218,219,220,221, 205]. In general, these studies showed that high-intensity exercises of 70–95% VO2max and resistance exercises with an intensity of 60–80% 1RM (3 sets of 12 repetitions) can be effective in decreasing leptin by 12–26% [215,216,217,218,219,220,221]. Leptin levels seem to decrease after acute exercise for up to 12 h and in some studies for up to 24 h [215,216,217,218,219,220,221]. Also, Leal-Cerro et al., who reported that significant changes in energy expenditure may change leptin levels, have concluded that after marathon running, which caused expenditure of 2800 calories, leptin level starts to decrease [222]. On the other hand, other studies reported no leptin reduction. Dündaret al. (2019), in their study, stated that a single short-term high-intensity exercise session at 80 to 95% VO2max did not lead to a decrease in leptin levels [223].
The results of studies examining the effects of acute exercise on adiponectin levels are contradictory, with studies showing an increase in adiponectin or no effect on its levels [224,225,226,227,228,229]. It has been observed that both resistance training (at intensities of 70–90% 1RM, 8–15 repetitions) and aerobic training (at intensities of 70 to 95% VO2max) can lead to an increase in plasma adiponectin [224,225,226,227,228,229,230,231,232]. For instance, one study demonstrated that engaging in moderate and high-intensity exercises for 60 min, with intensities set at 50% and 70% of peak oxygen uptake, respectively, can lead to an elevation in adiponectin levels [230]. However, the majority of studies support the notion that acute exercise promotes an increase in adiponectin, with the intensity of the exercises being a contributing factor.
Regarding chemerin, visfatin, omentin and vaspin, the results of acute studies are limited. The currently available studies indicate that resistance exercises with an intensity of 65 to 90% (3 to 5 sets of 10 to 15 repetitions) and aerobic exercises with an intensity of 75 to 95% VO2max, 70 to 90% HRmax or 60 to 65% VO2peak (20 to 30 min) can affect these adipokines (↓ 20–38% chemerin, ↑ 16–28% visfatin, ↓ 18.3–32.6% omentin and ↓ 6–16% vaspin) [233,234,235,236,237,238,239,240,241,242,243] (Table 2; Fig. 2).
Responses to chronic exercise
With respect to exercise type, many researchers have studied the response of adiponectin in overweight and obese humans and animals after 8–20 weeks of aerobic or resistance exercise. It seems that either aerobic exercise or resistance exercise can lead to an increase in adiponectin levels [244,245,246,247]. The results of most studies related to aerobic training indicate that 6 to 12 weeks of aerobic training with an intensity of 40 to 65% VO2max or 64 to 75% HRmax, for 30 to 50 min, 3 to 4 sessions per week can alter the abundance of numerous adipokines (↓leptin, ↑adiponectin, ↓chemerin, ↑visfatin, ↓omentin, ↓vaspin, ↓progranulin) [206, 207, 248,249,250,251,252,253]. In connection with resistance training, it seems that training with an intensity of 60 to 80% 1RM with 10 to 15 repetitions between 3 and 5 sets for 3 to 5 training sessions per week can also be effective in stimulating changes of these adipokines [207, 254,255,256,257,258,259] (Table and Fig. 2).
A meta-analysis of the magnitude of change in leptin levels following participation in exercise interventions lasting ≥ 2 weeks indicated that engaging in chronic exercise training is associated with a 26% decrease in leptin levels for individuals regardless of age and sex and a greater reduction in leptin occurred with a decreased percentage of body fat [260]. Also, resistance training with an intensity of 30 to 50% 1RM, 50 to 70% 1RM and 75 to 95% 1RM during a 12-month intervention could lead to a 20% decrease in leptin [199]. The authors also reported that leptin changes were strongly associated with changes in resting metabolic rate (RMR) and body mass index [261]. In contrast, 12 weeks of aerobic exercise [262], 12 weeks of resistance training [263], or 3-week combined aerobic and resistance training yielded no change in leptin levels [264]. These studies stated that the reason for the lack of effect could be due to the intensity of exercise, gender, type of measuring devices or measurement time. Finally, chronic exercise training programs have shown little effect on plasma leptin concentrations in the absence of weight loss or decreased adiposity [245, 265, 266].
Zarei et al. (2018) conducted a study to investigate the effect of eight weeks of high-intensity interval training on the serum levels of chemerin and omentin-1. In this study, rats performed HIIT five days a week for eight weeks. The results showed no significant difference between chemerin serum levels in rats undergoing an intensive exercise program compared to the control group [267]. Twelve weeks of combined aerobic and resistance exercise training, which consisted of 3 weekly sessions at an intensity of 60–70% maximum heart rate and 60–70% 1RM decreased the levels of chemerin, but did not affect the level of omentin [268]. Alternatively, Neuparth et al. (2014) evaluated, in patients with T2DM, the effect of regular moderate walking exercise (practiced for at least 30–60 min, 3–5 times a week, for a year) on chemerin. The active T2DM patients showed significantly lower levels of chemerin than those of the inactive T2DM patients [269]. Saremi et al. (2010) found that chemerin levels decreased in men performing 50 to 60 min, five days a week, of aerobic exercise for 12 weeks,, which included 15 to 50 min of walking-running, increasing weekly the exercise intensity [270]. Twelve weeks of treadmill walking and cycle ergometer exercise (5 days/week for 60 min at 85% HRmax) lead to a decrease of 18.3% of chemerin [271]. Aghapour and Farzangi (2012) reported that six weeks of running at 50% HRmax, 3 times a week, 60 min per day in obese women lead to a 26.6% reduction of chemerin [272]. Supriya et al. (2018) reported that yoga (once a week, 60 min per session for one year) in people with metabolic syndrome and high-normal blood pressure decreased chemerin [184]. Engaging in three 60-minute sessions per week for a duration of 12 weeks, consisting of rhythmic aerobic exercise performed at intensities ranging from 55 to 85% of maximum heart rate, along with core stability training, resulted in a significant reduction of 12% in chemerin levels and 18% in vaspin levels. However, there was no observed impact on plasma omentin levels [273]. A study conducted by Ribeiro Costa et al. demonstrated that an eight-week period of high-intensity interval training (HIIT) did not have any significant impact on omentin and vaspin levels in obese rats [274]. Asadi et al. (2019) compared the effect of 12 weeks aerobic exercise (70% VO2max), resistance exercise (11 exercises at 20% 1RM), and HIIT (six three-minute sets of running at 90% of VO2max) on omentin and vaspin in obese young men, finding no significant difference between the different training programmes [275] (Table 2; Fig. 2).
Exercise effects on cardiac-derived exerkines (cardiokines)
Some studies have pointed to the importance of exercise in cardiac cachexia, and reports suggest that exercise can reduce cardiac cachexia [276,277,278]. Antunes et al. reported that both aerobic and resistance exercise could lead to a reduction in heart inflammation and a reduction in cardiac cachexia; however, it is unclear what exercise type (aerobic or resistance), intensity or duration are needed to affect cardiokines in cardiac cachexia [278]. Therefore, it is not possible to make a precise recommendation about what mode of exercise training and associated exercise programming variables are most suitable for stimulating cardiokines in CC and cardiac cachexia (Table 2; Fig. 2).
Responses to acute exercise
In patients with atrial fibrillation and healthy controls both ANP and BNP were increased at peak exercise during a graded exercise test on a cycle ergometer, although the increase was greater in atrial fibrillation patients [279]. In another study, Tanaka et al. found that plasma ANP and BNP increased in patients with hypertension and healthy controls after exercise consisting of 4 min cycling at each of 25, 50 and 75 W [87] (Table 2; Fig. 2).
Responses to chronic exercise
Xi et al. (2016) demonstrated that exercise increased the cross-sectional area of myocytes and the expression of follistatin. In their study, rats underwent high-intensity exercise, which involved alternating between 7 min and 25 m per minute (85–90% VO2max), and moderate-intensity exercise, consisting of 3 min at 15 m per minute (50–60% VO2max), for a total duration of 1 h. The protocol for this study involved exercising once a day, 5 days a week, for a period of 4 weeks [280].
Kamiński reported in their study that swimming at 55–75% VO2max for 30 min, 3 sessions per week for 6 weeks can lead to an increase in IL-6 [281]. The results of this study are in line with the results of McGinnis et al., who reported that aerobic exercise with an intensity of 60–75% VO2max for 30 min performed three times per week for 12 weeks can lead to an increase in IL-6 [282].
Pedersen et al. referred to IL-1β as a negative factor for the heart and reported that physical activity can limit IL-1β signaling. These researchers stated in this review that moderate intensity aerobic physical activities between 6 and 12 weeks can be a good strategy to reduce heart inflammation and IL-1β [283] (Table 2; Fig. 2).
Exercise effects on liver-derived exerkines (hepatokines)
In this section, we will delve into the significance of exercise in relation to hepatokines. However, it is worth noting that there is a scarcity of research in this particular field. We have included animal studies alongside the human studies in order to gain a comprehensive understanding of the findings.
Responses to acute exercise
A study by Kersten et al. (2009) showed that an aerobic exercise intervention (2 h cycling at 50% VO2max) increased circulating levels of ANGPTL4 in healthy adults in the fasted but not fed state [284]. Another study found that 2 h of single-leg knee extensions at 50% of maximal workload stimulated ANGPTL4 secretion from the liver in humans [285].
In another study, the researchers stated that 2 h of aerobic training on a bicycle (intensity 60% of VO2 max) could lead to an increase in liver follistatin [286]. Hansen et al. (2011) reported that exercise on a bicycle ergometer with an intensity of 50% VO2 max lead to an increase in plasma follistatin 3 h after recovery [192].
Regarding FGF-21 changes with exercise, aerobic exercise (50% of VO2max for 45 min) and high-intensity interval exercise (90% of VO2max, 4 bouts of 2 min) could lead to an increase in obese mice liver FGF-21 [287]. On the other hand, Willis et al. reported in their study that moderate intensity exercise (55% peak oxygen uptake) and high intensity exercise (75% peak oxygen uptake) on 10 healthy young men could lead to an increase in plasma FGF-21 up to 4 h after training [288] (Table 2; Fig. 2).
Responses to chronic exercise
Catoire et al., (2014) showed that 12 weeks of endurance training had no significant effect on systemic levels of ANGPTL4 in healthy adults [289]. However, another report showed that in obese participants, 6 months of endurance training reduced body mass and increased systemic ANGPTL4 levels [290]. Nevertheless, research on chronic effects and in line with the objective of the current review was limited (Table 2; Fig. 2).

proposed exercise guide in CC. RT: Resistance training, TRX: Total Resistance Exercises, AE: Aerobic Exercise, MFO: Maximum fat oxidation
Measuring exerkines in studies and research gaps
The studies conducted on exercise and exerkines revealed that, in accordance with ethical guidelines for human research, blood sampling was used to measure the desired factors. However, certain exerkines, such as follistatin or interleukin 6, are derived from more than one tissue, and measuring them in blood may not yield accurate conclusions. Therefore, animal studies played a significant role in this research to determine the impact of exercise or CC on exerkines.
As always, care needs to be taken when interpreting results from animal studies as the findings may not extrapolate to humans. In Table 3, the measurement of tissues to obtain exerkine results is specified for both humans and animals. Factors marked as “Unclear” indicate variables that should be assessed through tissue analysis to obtain more accurate results. This may serve as a research gap for future studies.
Discussion and limitations of exerkine-based exercise prescription during cancer cachexia
Although exercise has demonstrated beneficial effects on various diseases and organs, our understanding of the underlying mechanisms contributing to these benefits is still limited. Additionally, the significance of exercise effects on cachexia in cancer patients and survivors has received little attention. Therefore, the objective of this study is to explore the potential role of exerkines in managing CC during and after the disease.
Currently, researchers are expanding their investigations to explore the impact of physical exercise on organs beyond skeletal muscle in CC patients. This exploration aims to establish a better understanding of the relationship and effects of these organs, ultimately leading to the development of suitable exercise protocols or strategies. Exerkines have gained increasing recognition as crucial mediators of exercise-induced changes and health benefits, particularly in terms of inter-organismic and systemic communication and coordination. However, prescribing exercise protocols for CC patients or survivors requires careful consideration due to the sensitivity of their condition. The sensitivity of prescribing exercise protocols for CC patients or survivors stems from the presence of inflammation and immunological changes in these individuals. According to Webster et al.‘s 2020 review [291], CC patients experience elevated levels of chronic inflammation, characterized by an increase in inflammatory cytokines and a decrease in anti-inflammatory cytokines. This inflammatory process is influenced by negative alterations in myokines, cardiokines, hepatokines, and adipokines. Consequently, even the slightest stress may exacerbate inflammation and contribute to the progression of cachexia. For this reason, caution should be exercised when prescribing a systemic stressor such as exercise. High-intensity exercises have been found to temporarily suppress the immune system [292, 293]. Conversely, moderate-intensity exercise appears to be a favorable strategy for maintaining or enhancing immune function [293], while low-intensity walking may have anti-inflammatory effects [294]. In this regard, a systematic review by Lavín-Pérez et al. (2023) documented that both aerobic exercise and moderate-intensity resistance exercise did not result in impaired immune system function or tumor-specific immune cell activity. Consequently, moderate-intensity resistance and aerobic exercises can be cautiously employed to enhance physiological, immunological, and psychological adaptations in cancer patients [295].
Therefore, this review aimed to comprehensively investigate the significance of exerkines in CC and its survivors, divided into two parts. The first part extensively discussed the relationship and alterations of exerkines in CC. Findings revealed that muscle (myokines), adipose tissue (adipokines), heart (cardiokines), and liver (hepatokines) are affected during CC (Figure and Table 1). These organs were identified as the primary targets of CC, leading to muscle atrophy and muscle inflammation, adverse changes in adipokines in adipose tissue resulting in chronic inflammation, negative alterations in cardiokines impacting heart function and contributing to cardiac cachexia, and hepatokines exhibiting detrimental changes in the liver that can exacerbate the negative effects of myokines and adipokines, ultimately promoting inflammation and worsening cachexia (Figure and Table 1). These negative changes not only have an impact during CC but also significantly affect the quality of life in survivors.
The second part discussed the importance of exercise on these organs. The results presented in Table 2 demonstrate that most chronic resistance and aerobic exercises have a positive effect on these organs. These exercises, characterized by chronic responses, were primarily based on moderate intensities. Based on these findings, our recommended exercise protocols are outlined in Fig. 2. Figure 2 illustrates that low-intensity exercises during cancer and moderate-intensity exercises after CC can be suitable strategies with positive effects on muscle (myokines), adipose tissue (adipokines), heart (cardiokines), and liver (hepatokines), while emphasizing caution to prevent inflammation and immune system function decline.
The present review encountered certain limitations that warrant investigation in future studies, in order to enhance the significance of conclusions pertaining to exercise, exerkines, and cachexia cancer. One of the primary limitations of this study was the scarcity of research on hepatokines and cardiokines. Therefore, it is recommended that future studies focus on investigating hepatokines and cardiokines in relation to exercise, exerkines, and the signaling pathways associated with cachexia cancer. Additionally, the current study revealed measurement limitations for certain variables, as indicated by the “Unclear” marking in Table 3. This suggests that in human studies, some variables are explained based on data derived from multiple organs, which complicates the drawing of definitive conclusions. To overcome this challenge, it is suggested that future studies in animal models, specifically those investigating exercise and exerkines in CC, measure these variables directly from tissue samples. Lastly, another limitation of the study was the focus on variables with the greatest impact on CC. To further emphasize the role of exerkines, it is recommended that future studies explore additional variables, such as GDF15. By addressing these limitations in future research, a more comprehensive understanding of the relationship between exercise, exerkines, and cachexia cancer can be achieved, leading to more prominent findings and insights.
Considering the significant knowledge gap surrounding exerkines in cancer patients, it is highly recommended that future studies concentrate on investigating the role of exerkines in cachexia cancer (CC) and their impact on various organs. These studies should also delve into the underlying immunological and physiological mechanisms involved.
In summary, exerkines present a promising avenue for future research endeavors. They hold immense potential as biomarkers for predicting outcomes and facilitating the development of personalized exercise programs aimed at improving overall health, mitigating disease, and promoting resilience across all stages of life. Furthermore, exploring the connection between exerkines and cancer could lead to a substantial breakthrough in the field of exercise oncology.
Data Availability
All data generated or analysed during this study are included in this published article.
Abbreviations
- AMPK:
-
AMP-activated protein kinase
- Ang-II:
-
Angiotensin II
- ANGPTL4:
-
Angiopoietin-like 4
- AKT:
-
Protein kinase B
- ANP:
-
Atrial natriuretic peptide
- BAT:
-
Brown adipose tissue
- BDT:
-
Brown adipose tissue
- BNP:
-
Brain natriuretic peptide
- CC:
-
Cancer-related cachexia
- CTRP15:
-
C1q/TNF-related protein 15
- CTRP-4:
-
C1q/TNF-related protein 4
- CFs:
-
Cardiomyocytes and cardiac fibroblasts
- FGF-21:
-
Fibroblast growth factor 21
- HIIT:
-
High intensity interval training
- HRmax:
-
Maximum heart rate
- NF-Κb:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- IGF-1:
-
Insulin-like Growth Factor-1
- IL-33:
-
Interleukin 33
- IL-6:
-
Interleukin 6
- IL-18:
-
Interleukin 18
- IL-1β:
-
Interleukin 1β
- ICAM-1:
-
Intercellular Adhesion Molecule 1
- Lep-R:
-
Leptin receptor
- MuRF-1:
-
Muscle RING-finger protein-1
- MTOR:
-
Mammalian target of rapamycin
- NF-Kb:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- SIT:
-
Sprint interval training
- TGF-β:
-
Transforming growth factor beta
- TNF-α:
-
Tumor necrosis factor
- UPR:
-
Unfolded protein response
- VO2max:
-
Maximal oxygen consumption
- VCAM-1:
-
Vascular cell adhesion protein 1
- WAT:
-
White adipose tissue
- 1RM:
-
One-repetition maximum
References
Argilés JM, Busquets S, Stemmler B, López-Soriano FJ. Cancer cachexia: understanding the molecular basis. Nat Rev Cancer. 2014;14(11):754–62.
Baracos VE, Martin L, Korc M, Guttridge DC, Fearon KCH. Cancer-associated cachexia. Nat Reviews Disease Primers. 2018;4(1):17105.
Baazim H, Antonio-Herrera L, Bergthaler A. The interplay of immunology and cachexia in infection and cancer. Nat Rev Immunol. 2022;22(5):309–21.
Mitsunaga S. Development of Therapy for Cancer Cachexia in Pancreatic Cancer. Gan to Kagaku Ryoho Cancer & Chemotherapy. 2022;49(7):728–31.
Bordignon C, Dos Santos BS, Rosa DD. Impact of cancer cachexia on cardiac and skeletal muscle: role of exercise training. Cancers. 2022;14(2):342.
Porporato PE. Understanding cachexia as a cancer metabolism syndrome. Oncogenesis. 2016;5(2):e200–e.
Tomasin R, Martin A, Cominetti MR. Metastasis and cachexia: alongside in clinics, but not so in animal models. J Cachexia Sarcopenia Muscle. 2019;10(6):1183–94.
Vanhoutte G, van de Wiel M, Wouters K, Sels M, Bartolomeeussen L, De Keersmaecker S, et al. Cachexia in cancer: what is in the definition? BMJ open Gastroenterology. 2016;3(1):e000097.
Murphy RM, Watt MJ, Febbraio MA. Metabolic communication during exercise. Nat Metabolism. 2020;2(9):805–16.
Lakoski SG, Eves ND, Douglas PS, Jones LW. Exercise rehabilitation in patients with cancer. Nat Reviews Clin Oncol. 2012;9(5):288–96.
Hayes SC, Newton RU, Spence RR, Galvão DA. The Exercise and Sports Science Australia position statement: Exercise medicine in cancer management. J Sci Med Sport. 2019;22(11):1175–99.
Thomas R, Kenfield SA, Yanagisawa Y, Newton RU. Why exercise has a crucial role in cancer prevention, risk reduction and improved outcomes. Br Med Bull. 2021;139(1):100–19.
Chow LS, Gerszten RE, Taylor JM, Pedersen BK, van Praag H, Trappe S, et al. Exerkines in health, resilience and disease. Nat Reviews Endocrinol. 2022;18(5):273–89.
Safdar A, Saleem A, Tarnopolsky MA. The potential of endurance exercise-derived exosomes to treat metabolic diseases. Nat Reviews Endocrinol. 2016;12(9):504–17.
Magliulo L, Bondi D, Pini N, Marramiero L, Di Filippo ES. The wonder exerkines—novel insights: a critical state-of-the-art review. Mol Cell Biochem. 2022;477(1):105–13.
Lee TH-Y, Formolo DA, Kong T, Lau SW-Y, Ho CS-L, Leung RYH, et al. Chapter fourteen - potential exerkines for physical exercise-elicited pro-cognitive effects: insight from clinical and animal research. In: Yau S-Y, So K-F, editors. International Review of Neurobiology. Volume 147. Academic Press; 2019. pp. 361–95.
Hargreaves M, Spriet LL. Skeletal muscle energy metabolism during exercise. Nat Metabolism. 2020;2(9):817–28.
Lee JH, Jun H-S. Role of myokines in regulating skeletal muscle mass and function. Front Physiol. 2019;10:42.
Amitani M, Oba T, Kiyosawa N, Morikawa H, Chino T, Soma A, et al. Skeletal muscle loss during neoadjuvant chemotherapy predicts poor prognosis in patients with breast cancer. BMC Cancer. 2022;22(1):1–11.
Spetsieris N, Bobba G, Bhandari P, Patel S, Tharayil Z, Gupta R. Pancreatic cancer with a skeletal muscle metastasis-A case presentation and literature review. J Community Hosp Intern Med Perspect. 2022;12(5):60–4.
Rausch V, Sala V, Penna F, Porporato PE, Ghigo A. Understanding the common mechanisms of heart and skeletal muscle wasting in cancer cachexia. Oncogenesis. 2021;10(1):1–13.
Riccardi DMdR, das, Neves RX, de Matos-Neto EM, Camargo RG, Lima JDCC, Radloff K et al. Plasma lipid profile and systemic inflammation in patients with cancer cachexia. Frontiers in nutrition. 2020;7:4.
Wang G, Biswas AK, Ma W, Kandpal M, Coker C, Grandgenett PM, et al. Metastatic cancers promote cachexia through ZIP14 upregulation in skeletal muscle. Nat Med. 2018;24(6):770–81.
Rom O, Reznick AZ. The role of E3 ubiquitin-ligases MuRF-1 and MAFbx in loss of skeletal muscle mass. Free Radic Biol Med. 2016;98:218–30.
Pin F, Bonewald LF, Bonetto A. Role of myokines and osteokines in cancer cachexia. Experimental Biology and Medicine. 2021;246(19):2118–27.
McFarlane C, Plummer E, Thomas M, Hennebry A, Ashby M, Ling N, et al. Myostatin induces cachexia by activating the ubiquitin proteolytic system through an NF-κB‐independent, FoxO1‐dependent mechanism. J Cell Physiol. 2006;209(2):501–14.
Graca FA, Rai M, Hunt LC, Stephan A, Wang Y-D, Gordon B, et al. The myokine Fibcd1 is an endogenous determinant of myofiber size and mitigates cancer-induced myofiber atrophy. Nat Commun. 2022;13(1):1–22.
Korzun T, Moses AS, Kim J, Patel S, Schumann C, Levasseur PR et al. Nanoparticle-based Follistatin Messenger RNA therapy for reprogramming metastatic ovarian Cancer and ameliorating Cancer‐Associated Cachexia. Small. 2022:2204436.
Barton BE. IL-6-like cytokines and cancer cachexia. Immunol Res. 2001;23(1):41–58.
Refsgaard Holm M, Christensen H, Rasmussen J, Johansen ML, Schou M, Faber J, et al. Fibroblast growth factor 21 in patients with cardiac cachexia: a possible role of chronic inflammation. ESC Heart Failure. 2019;6(5):983–91.
Konishi M, Ishida J, Saito M, Springer J. Irisin–a myokine potentially bridging muscle and fat tissue in cachexia. J Cachexia Sarcopenia Muscle. 2015;6(4):396.
Bogdanovich S, Krag TO, Barton ER, Morris LD, Whittemore L-A, Ahima RS, et al. Functional improvement of dystrophic muscle by myostatin blockade. Nature. 2002;420(6914):418–21.
Liu C, Yang Z, Liu C, Wang R, Tien P, Dale R, et al. Myostatin antisense RNA-mediated muscle growth in normal and cancer cachexia mice. Gene Ther. 2008;15(3):155–60.
Allen DL, Hittel DS, McPherron AC. Expression and function of myostatin in obesity, diabetes, and exercise adaptation. Med Sci Sports Exerc. 2011;43(10):1828.
Zhang L, Rajan V, Lin E, Hu Z, Han H, Zhou X, et al. Pharmacological inhibition of myostatin suppresses systemic inflammation and muscle atrophy in mice with chronic kidney disease. FASEB J. 2011;25(5):1653–63.
Winbanks CE, Weeks KL, Thomson RE, Sepulveda PV, Beyer C, Qian H, et al. Follistatin-mediated skeletal muscle hypertrophy is regulated by Smad3 and mTOR independently of myostatin. J Cell Biol. 2012;197(7):997–1008.
Nakatani M, Takehara Y, Sugino H, Matsumoto M, Hashimoto O, Hasegawa Y, et al. Transgenic expression of a myostatin inhibitor derived from follistatin increases skeletal muscle mass and ameliorates dystrophic pathology in mdx mice. FASEB J. 2008;22(2):477–87.
Loumaye A, De Barsy M, Nachit M, Lause P, Frateur L, Van Maanen A, et al. Role of activin A and myostatin in human cancer cachexia. J Clin Endocrinol Metabolism. 2015;100(5):2030–8.
Diller M, Frommer K, Dankbar B, Tarner I, Hülser M-L, Tsiklauri L, et al. The activin-follistatin anti-inflammatory cycle is deregulated in synovial fibroblasts. Arthritis Res Therapy. 2019;21(1):1–11.
Us Altay D, Keha EE, Ozer Yaman S, Ince I, Alver A, Erdogan B, et al. Investigation of the expression of irisin and some cachectic factors in mice with experimentally induced gastric cancer. QJM: An International Journal of Medicine. 2016;109(12):785–90.
Ma C, Ding H, Deng Y, Liu H, Xiong X, Yang Y. Irisin: a new code uncover the relationship of skeletal muscle and cardiovascular health during exercise. Front Physiol. 2021;12:620608.
Nowinska K, Jablonska K, Pawelczyk K, Piotrowska A, Partynska A, Gomulkiewicz A, et al. Expression of Irisin/FNDC5 in cancer cells and stromal fibroblasts of non-small cell lung cancer. Cancers. 2019;11(10):1538.
Pinkowska A, Podhorska-Okołów M, Dzięgiel P, Nowińska K. The role of irisin in cancer disease. Cells. 2021;10(6):1479.
Muñoz-Cánoves P, Scheele C, Pedersen BK, Serrano AL. Interleukin‐6 myokine signaling in skeletal muscle: a double‐edged sword? FEBS J. 2013;280(17):4131–48.
Narsale AA, Carson JA. Role of IL-6 in cachexia–therapeutic implications. Curr Opin Support Palliat Care. 2014;8(4):321.
Wang X, Li J, Liu W, Zhang X, Xue L. The diagnostic value of interleukin 6 as a biomarker for gastric cancer: a meta-analysis and systematic review. Medicine. 2021;100:47.
Rose-John S. Local and systemic effects of interleukin‐6 (IL‐6) in inflammation and cancer. FEBS Lett. 2022;596(5):557–66.
Hazgui M, Weslati M, Ounissi D, Boughriba R, Bacha D, Loueslati BY. Interleukin-1β, interleukin-6 and interleukin-10 polymorphisms in tnisian patients with colorectal cancer and liver metastasis. Archives of Biological Sciences. 2022(00):32-.
Puppa MJ, Murphy EA, Fayad R, Hand GA, Carson JA. Cachectic skeletal muscle response to a novel bout of low-frequency stimulation. J Appl Physiol. 2014;116(8):1078–87.
Suzuki H, Asakawa A, Amitani H, Nakamura N, Inui A. Cancer cachexia—pathophysiology and management. J Gastroenterol. 2013;48(5):574–94.
Castell JV, Gómez-Lechón MJ, David M, Andus T, Geiger T, Trullenque R, et al. Interleukin-6 is the major regulator of acute phase protein synthesis in adult human hepatocytes. FEBS Lett. 1989;242(2):237–9.
Song M, Kellum JA. Interleukin-6. Crit Care Med. 2005;33(12):463–S5.
White JP, Baynes JW, Welle SL, Kostek MC, Matesic LE, Sato S, et al. The regulation of skeletal muscle protein turnover during the progression of cancer cachexia in the ApcMin/+ mouse. PLoS ONE. 2011;6(9):e24650.
Oost LJ, Kustermann M, Armani A, Blaauw B, Romanello V. Fibroblast growth factor 21 controls mitophagy and muscle mass. J cachexia Sarcopenia Muscle. 2019;10(3):630–42.
Franz K, Ost M, Otten L, Herpich C, Coleman V, Endres A-S, et al. Higher serum levels of fibroblast growth factor 21 in old patients with cachexia. Nutrition. 2019;63:81–6.
Seldin MM, Peterson JM, Byerly MS, Wei Z, Wong GW. Myonectin (CTRP15), a novel myokine that links skeletal muscle to systemic lipid homeostasis. J Biol Chem. 2012;287(15):11968–80.
Seldin MM, Wong GW. Regulation of tissue crosstalk by skeletal muscle-derived myonectin and other myokines. Adipocyte. 2012;1(4):200–2.
Seldin MM, Lei X, Tan SY, Stanson KP, Wei Z, Wong GW. Skeletal muscle-derived myonectin activates the mammalian target of rapamycin (mTOR) pathway to suppress autophagy in liver. J Biol Chem. 2013;288(50):36073–82.
Penna F, Baccino FM, Costelli P. Coming back: autophagy in cachexia. Curr Opin Clin Nutr Metabolic Care. 2014;17(3):241–6.
Park S-Y, Choi JH, Ryu HS, Pak YK, Park KS, Lee HK, et al. C1q tumor necrosis factor α-related protein isoform 5 is increased in mitochondrial DNA-depleted myocytes and activates AMP-activated protein kinase. J Biol Chem. 2009;284(41):27780–9.
Lim S, Choi SH, Koo BK, Kang SM, Yoon JW, Jang HC, et al. Effects of aerobic exercise training on C1q tumor necrosis factor α-related protein isoform 5 (myonectin): association with insulin resistance and mitochondrial DNA density in women. J Clin Endocrinol Metabolism. 2012;97(1):E88–E93.
Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol. 2011;11(2):85–97.
Arner P. Lipases in cachexia. Science. 2011;333(6039):163–4.
Das SK, Hoefler G. The role of triglyceride lipases in cancer associated cachexia. Trends Mol Med. 2013;19(5):292–301.
Batista M Jr, Olivan M, Alcantara P, Sandoval R, Peres S, Neves R, et al. Adipose tissue-derived factors as potential biomarkers in cachectic cancer patients. Cytokine. 2013;61(2):532–9.
Argilés JM, Stemmler B, López-Soriano FJ, Busquets S. Inter-tissue communication in cancer cachexia. Nat Reviews Endocrinol. 2019;15(1):9–20.
Mak RH, Cheung W, Cone RD, Marks DL. Mechanisms of Disease: cytokine and adipokine signaling in uremic cachexia. Nat Clin Pract Nephrol. 2006;2(9):527–34.
Rebello CJ, Kirwan JP, Greenway FL. Obesity, the most common comorbidity in SARS-CoV-2: is leptin the link? Int J Obes. 2020;44(9):1810–7.
Maurya R, Sebastian P, Namdeo M, Devender M, Gertler A. COVID-19 severity in obesity: leptin and inflammatory cytokine interplay in the link between high morbidity and mortality. Front Immunol. 2021;12:2349.
Naylor C, Petri WA Jr. Leptin regulation of immune responses. Trends Mol Med. 2016;22(2):88–98.
Santos-Alvarez J, Goberna R, Sánchez-Margalet V. Human leptin stimulates proliferation and activation of human circulating monocytes. Cell Immunol. 1999;194(1):6–11.
Khoramipour K, Chamari K, Hekmatikar AA, Ziyaiyan A, Taherkhani S, Elguindy NM, et al. Adiponectin: structure, physiological functions, role in diseases, and effects of nutrition. Nutrients. 2021;13(4):1180.
Wolf I, Sadetzki S, Kanety H, Kundel Y, Pariente C, Epstein N, et al. Adiponectin, ghrelin, and leptin in cancer cachexia in breast and colon cancer patients. Cancer. 2006;106(4):966–73.
Ohashi K, Parker JL, Ouchi N, Higuchi A, Vita JA, Gokce N, et al. Adiponectin promotes macrophage polarization toward an anti-inflammatory phenotype. J Biol Chem. 2010;285(9):6153–60.
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Investig. 2003;112(12):1796–808.
Wright HL, Moots RJ, Bucknall RC, Edwards SW. Neutrophil function in inflammation and inflammatory diseases. Rheumatology. 2010;49(9):1618–31.
Rossi A, Lord JM. Adiponectin inhibits neutrophil apoptosis via activation of AMP kinase, PKB and ERK 1/2 MAP kinase. Apoptosis: An International Journal on Programmed cell Death. 2013;18(12):1469–80.
Shaty MH, Al-Ezzi MI, Arif IS, Basil D. Effect of Metformin on inflammatory markers involved in cardiotoxicity induced by Doxorubicin. Res J Pharm Technol. 2019;12(12):5815–21.
Wolf AM, Wolf D, Rumpold H, Enrich B, Tilg H. Adiponectin induces the anti-inflammatory cytokines IL-10 and IL-1RA in human leukocytes. Biochem Biophys Res Commun. 2004;323(2):630–5.
Homa-Mlak I, Pigoń-Zając D, Wawrejko P, Małecka-Massalska T, Mlak R. Three Pathways of Cancer Cachexia: inflammation, changes in adipose tissue and loss of muscle Mass—The role of miRNAs. J Personalized Med. 2022;12(9):1438.
Porporato P. Understanding cachexia as a cancer metabolism syndrome. Oncogenesis. 2016;5(2):e200–e.
Christodoulatos GS, Antonakos G, Karampela I, Psallida S, Stratigou T, Vallianou N et al. Circulating Omentin-1 as a biomarker at the intersection of postmenopausal breast Cancer occurrence and cardiometabolic risk: an observational cross-sectional study. Biomolecules. 2021;11(11).
Yamawaki H, Kuramoto J, Kameshima S, Usui T, Okada M, Hara Y. Omentin, a novel adipocytokine inhibits TNF-induced vascular inflammation in human endothelial cells. Biochem Biophys Res Commun. 2011;408(2):339–43.
Zhong X, Li X, Liu F, Tan H, Shang D. Omentin inhibits TNF-α-induced expression of adhesion molecules in endothelial cells via ERK/NF-κB pathway. Biochem Biophys Res Commun. 2012;425(2):401–6.
Qi D, Tang X, He J, Wang D, Zhao Y, Deng W, et al. Omentin protects against LPS-induced ARDS through suppressing pulmonary inflammation and promoting endothelial barrier via an Akt/eNOS-dependent mechanism. Cell Death Dis. 2016;7(9):e2360–e.
Senesi P, Luzi L, Terruzzi I. Adipokines, Myokines, and Cardiokines: the role of Nutritional Interventions. Int J Mol Sci. 2020;21(21).
Tanaka M, Ishizaka Y, Ishiyama Y, Kato J, Kida O, Kitamura K, et al. Exercise-induced secretion of brain natriuretic peptide in essential hypertension and normal subjects. Hypertens Res. 1995;18(2):159–66.
Saha S, Singh PK, Roy P, Kakar SS. Cardiac Cachexia: unaddressed aspect in Cancer Patients. Cells. 2022;11(6).
Valentova M, Anker SD, von Haehling S. Cardiac cachexia revisited: the role of wasting in heart failure. Heart Fail Clin. 2020;16(1):61–9.
Wu YS, Zhu B, Luo AL, Yang L, Yang C. The role of Cardiokines in Heart Diseases: beneficial or detrimental? Biomed Res Int. 2018;2018:8207058.
Belloum Y, Rannou-Bekono F, FAvIER FB. Cancer-induced cardiac cachexia: pathogenesis and impact of physical activity. Oncol Rep. 2017;37(5):2543–52.
Okoshi MP, Capalbo RV, Romeiro FG, Okoshi K. Cardiac cachexia: perspectives for prevention and treatment. Arquivos brasileiros de cardiologia. 2016;108:74–80.
Stefan N, Häring H-U. The role of hepatokines in metabolism. Nat Reviews Endocrinol. 2013;9(3):144–52.
Jensen-Cody SO, Potthoff MJ. Hepatokines and metabolism: deciphering communication from the liver. Mol Metabolism. 2021;44:101138.
Qiu W, Kuo CY, Tian Y, Su GH. Dual roles of the activin signaling pathway in pancreatic Cancer. Biomedicines. 2021;9(7).
Neto NIP, Boldarine VT, Hachul ACL, Oyama LM, Lima J, Fernandez ES, et al. Association between ANGPTL-4 and the proinflammatory process in cancer cachexia patients. Oncotarget. 2019;10(60):6444–55.
Cao Z, Zhao K, Jose I, Hoogenraad NJ, Osellame LD. Biomarkers for Cancer Cachexia: a Mini Review. Int J Mol Sci. 2021;22(9):4501.
Choi K, Jang HY, Ahn JM, Hwang SH, Chung JW, Choi YS, et al. The association of the serum levels of myostatin, follistatin, and interleukin-6 with sarcopenia, and their impacts on survival in patients with hepatocellular carcinoma. Clin Mol Hepatol. 2020;26(4):492–505.
Penna F, Bonetto A, Muscaritoli M, Costamagna D, Minero VG, Bonelli G, et al. Muscle atrophy in experimental cancer cachexia: is the IGF-1 signaling pathway involved? Int J Cancer. 2010;127(7):1706–17.
Stefan N, Sun Q, Fritsche A, Machann J, Schick F, Gerst F, et al. Impact of the adipokine adiponectin and the hepatokine fetuin-A on the development of type 2 diabetes: prospective cohort-and cross-sectional phenotyping studies. PLoS ONE. 2014;9(3):e92238.
Kucukoglu O, Sowa J-P, Mazzolini GD, Syn W-K, Canbay A. Hepatokines and adipokines in NASH-related hepatocellular carcinoma. J Hepatol. 2021;74(2):442–57.
Starling S. Metformin-induced hepatokine reduces appetite. Nat Reviews Endocrinol. 2020;16(3):131.
Kim JS, Galvão DA, Newton RU, Gray E, Taaffe DR. Exercise-induced myokines and their effect on prostate cancer. Nat Reviews Urol. 2021;18(9):519–42.
Park SY, Hwang BO, Song NY. The role of myokines in cancer: crosstalk between skeletal muscle and tumor. BMB Rep. 2023;56(7):365–73.
Huang Q, Wu M, Wu X, Zhang Y, Xia Y. Muscle-to-tumor crosstalk: the effect of exercise-induced myokine on cancer progression. Biochim et Biophys acta Reviews cancer. 2022;1877(5):188761.
de Castro GS, Correia-Lima J, Simoes E, Orsso CE, Xiao J, Gama LR, et al. Myokines in treatment-naïve patients with cancer-associated cachexia. Clin Nutr. 2021;40(4):2443–55.
Gholamali M, Nourshahi M, Hedayati M. The Effect of Acute endurance Exercise on plasma myostastin in Healthy Elderly Men. Iran J Ageing. 2015;10(1):82–91.
Trobec K, von Haehling S, Anker SD, Lainscak M. Growth hormone, insulin-like growth factor 1, and insulin signaling—a pharmacological target in body wasting and cachexia. J cachexia Sarcopenia Muscle. 2011;2:191–200.
Pugh JK, Faulkner SH, Jackson AP, King JA, Nimmo MA. Acute molecular responses to concurrent resistance and high-intensity interval exercise in untrained skeletal muscle. Physiological Rep. 2015;3(4).
Hittel DS, Axelson M, Sarna N, Shearer J, Huffman KM, Kraus WE. Myostatin decreases with aerobic exercise and associates with insulin resistance. Med Sci Sports Exerc. 2010;42(11):2023–9.
Han D-S, Hsiao M-Y, Wang T-G, Chen S-Y, Yang W-S. Association of serum myokines and aerobic exercise training in patients with spinal cord injury: an observational study. BMC Neurol. 2016;16(1):142.
Shabani R, Izaddoust F. Effects of aerobic training, resistance training, or both on circulating irisin and myostatin in untrained women. Acta Gymnica. 2018;48(2):47–55.
He Z, Tian Y, Valenzuela PL, Huang C, Zhao J, Hong P et al. Myokine/Adipokine response to Aerobic Exercise: is it just a matter of Exercise load? Front Physiol. 2019;10.
Kabak B, Belviranli M, Okudan N. Irisin and myostatin responses to acute high-intensity interval exercise in humans. Horm Mol Biol Clin Investig. 2018;35(3).
Dalbo VJ, Roberts MD, Sunderland KL, Poole CN, Stout JR, Beck TW et al. Acute loading and aging effects on myostatin pathway biomarkers in human skeletal muscle after three sequential bouts of resistance exercise. The journals of gerontology Series A, Biological sciences and medical sciences. 2011;66(8):855–65.
Fernandez-Gonzalo R, Lundberg TR, Tesch PA. Acute molecular responses in untrained and trained muscle subjected to aerobic and resistance exercise training versus resistance training alone. Acta Physiol. 2013;209(4):283–94.
MacKenzie MG, Hamilton DL, Pepin M, Patton A, Baar K. Inhibition of myostatin signaling through notch activation following acute resistance exercise. PLoS ONE. 2013;8(7):e68743.
Shabkhiz F, Khalafi M, Rosenkranz S, Karimi P, Moghadami K. Resistance training attenuates circulating FGF-21 and myostatin and improves insulin resistance in elderly men with and without type 2 diabetes mellitus: a randomised controlled clinical trial. Eur J Sport Sci. 2021;21(4):636–45.
Aminian F, Birjandi SC. Low-intensity blood Flow Restriction Training does not modulate myostatin concentration in Elderly Females. Med Lab J. 2020;14(5):30–4.
Laurentino GC, Ugrinowitsch C, Roschel H, Aoki MS, Soares AG, Neves M Jr, et al. Strength training with blood flow restriction diminishes myostatin gene expression. Med Sci Sports Exerc. 2012;44(3):406–12.
Aminian F, hejazi M, Birjandi SC. Low-intensity blood Flow Restriction Training does not modulate myostatin concentration in Elderly Females. Med Lab J. 2020;14(5):30–4.
Bagheri R, Rashidlamir A, Motevalli MS, Elliott BT, Mehrabani J, Wong A. Effects of upper-body, lower-body, or combined resistance training on the ratio of follistatin and myostatin in middle-aged men. Eur J Appl Physiol. 2019;119:1921–31.
Farzanegi P, Zamani M, Khalili A, Dehghani H, Fotohi R, Ghanbarpour M, et al. Effects of upper-and lower-extremity resistance training on serum vascular endothelial growth factor, myostatin, endostatin and follistatin levels in sedentary male students. Sci Sports. 2021;36(2):139. e1-. e6.
Kazemipour N, Faramarzi M, Banitalebi E. Effect of elastic-band resistance training on myostatin, follistatin levels in elderly women with osteosarcopenic obesity. Metabolism and Exercise. 2019;9(2):117–36.
Kon M, Tanimura Y, Yoshizato H. Effects of acute endurance exercise on follistatin-like 1 and apelin in the circulation and metabolic organs in rats. Arch Physiol Biochem. 2022;128(5):1254–8.
Kon M, Ebi Y, Nakagaki K. Effects of acute sprint interval exercise on follistatin-like 1 and apelin secretions. Arch Physiol Biochem. 2021;127(3):223–7.
Cosio PL, Crespo-Posadas M, Velarde-Sotres Á, Pelaez M. Effect of chronic resistance training on circulating irisin: systematic review and meta-analysis of randomized controlled trials. Int J Environ Res Public Health. 2021;18(5):2476.
Kim H-J, Lee H-J, So B, Son JS, Yoon D, Song W. Effect of aerobic training and resistance training on circulating irisin level and their association with change of body composition in overweight/obese adults: a pilot study. Physiol Res. 2016;65(2):271.
Haghighi AH, Hajinia M, Askari R, Abbasian S, Goldfied G. Effect of high-intensity interval training and high-intensity resistance training on Irisin and fibroblast growth factor 21 in men with overweight and obesity. Can J Physiol Pharmacol. 2022;100(9):937–44.
Anastasilakis AD, Polyzos SA, Saridakis ZG, Kynigopoulos G, Skouvaklidou EC, Molyvas D, et al. Circulating irisin in healthy, young individuals: day-night rhythm, effects of food intake and exercise, and associations with gender, physical activity, diet, and body composition. J Clin Endocrinol Metabolism. 2014;99(9):3247–55.
Daskalopoulou SS, Cooke AB, Gomez Y-H, Mutter AF, Filippaios A, Mesfum ET, et al. Plasma irisin levels progressively increase in response to increasing exercise workloads in young, healthy, active subjects. Eur J Endocrinol. 2014;171(3):343–52.
Huh JY, Siopi A, Mougios V, Park KH, Mantzoros CS. Irisin in response to exercise in humans with and without metabolic syndrome. J Clin Endocrinol Metabolism. 2015;100(3):E453–E7.
Rioux BV, Brunt KR, Eadie AL, Bouchard DR, Fox J, Sénéchal M. Impact of acute circuit training on irisin in younger and older overweight adults. Applied physiology, nutrition, and metabolism = physiologie appliquee, nutrition et metabolisme. 2021;46(10):1248–56.
Reisi J, Rajabi H, Ghaedi K, Marandi S-M, Dehkhoda M-R. Effect of Acute Resistance Training on plasma irisin protein level and expression of muscle FNDC5 and adipose tissue UCP1 genes in male rats. J Isfahan Med School. 2013;31(256).
Fox J, Rioux B, Goulet E, Johanssen N, Swift D, Bouchard D, et al. Effect of an acute exercise bout on immediate post-exercise irisin concentration in adults: a meta‐analysis. Scand J Med Sci Sports. 2018;28(1):16–28.
Fischer CP. Interleukin-6 in acute exercise and training: what is the biological relevance. Exerc Immunol rev. 2006;12(6–33):41.
Islam H, Townsend LK, McKie GL, Medeiros PJ, Gurd BJ, Hazell TJ. Potential involvement of lactate and interleukin-6 in the appetite-regulatory hormonal response to an acute exercise bout. J Appl Physiol. 2017;123(3):614–23.
Lee EC, Watson G, Casa D, Armstrong LE, Kraemer W, Vingren JL, et al. Interleukin-6 responses to water immersion therapy after acute exercise heat stress: a pilot investigation. J Athl Train. 2012;47(6):655–63.
Raines C, Frosig T, Escobar KA, Cotter JA, Schick EE. Acute resistance exercise at varying volume loads does not enhance plasma interleukin-6. Int J Kinesiol Sports Sci. 2020;8(1):37–42.
Tajra V, Tibana RA, Vieira DCL, de Farias DL, Teixeira TG, Funghetto SS, et al. Identification of high responders for interleukin-6 and creatine kinase following acute eccentric resistance exercise in elderly obese women. J Sci Med Sport. 2014;17(6):662–6.
Trenerry MK, Della Gatta PA, Larsen AE, Garnham AP, Cameron-Smith D. Impact of resistance exercise training on interleukin‐6 and JAK/STAT in young men. Muscle Nerve. 2011;43(3):385–92.
Nash D, Hughes MG, Butcher L, Aicheler R, Smith P, Cullen T, et al. IL-6 signaling in acute exercise and chronic training: potential consequences for health and athletic performance. Scand J Med Sci Sports. 2023;33(1):4–19.
MacDonald C, Wojtaszewski JF, Pedersen BK, Kiens B, Richter EA. Interleukin-6 release from human skeletal muscle during exercise: relation to AMPK activity. J Appl Physiol. 2003;95(6):2273–7.
Dreyer HC, Fujita S, Cadenas JG, Chinkes DL, Volpi E, Rasmussen BB. Resistance exercise increases AMPK activity and reduces 4E-BP1 phosphorylation and protein synthesis in human skeletal muscle. J Physiol. 2006;576(2):613–24.
Nitzsche N, Schulze R, Weigand F, Hummer N, Schulz H. Comparison of an acute resistance training on the lactate concentration with and without blood flow restriction at different loads. Dtsch Z Sportmed. 2018;69(11):337–43.
Kim KH, Kim SH, Min Y-K, Yang H-M, Lee J-B, Lee M-S. Acute exercise induces FGF21 expression in mice and in healthy humans. PLoS ONE. 2013;8(5):e63517.
Tanimura Y, Aoi W, Takanami Y, Kawai Y, Mizushima K, Naito Y, et al. Acute exercise increases fibroblast growth factor 21 in metabolic organs and circulation. Physiological Rep. 2016;4(12):e12828.
Pedersen BK, Febbraio MA. Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nat Reviews Endocrinol. 2012;8(8):457–65.
Konopka AR, Wolff CA, Suer MK, Harber MP. Relationship between intermuscular adipose tissue infiltration and myostatin before and after aerobic exercise training. Am J Physiology-Regulatory Integr Comp Physiol. 2018;315(3):R461–R8.
LeBrasseur NK, Schelhorn TM, Bernardo BL, Cosgrove PG, Loria PM, Brown TA. Myostatin inhibition enhances the effects of exercise on performance and metabolic outcomes in aged mice. Journals of Gerontology Series A: Biomedical Sciences and Medical Sciences. 2009;64(9):940–8.
Karimi R, Fakhrpour R, Zarneshan A. Effect of resistance training with milk protein concentrate (MPC) supplementation on serum levels of Follistatin and myostatin and muscle hypertrophy in Beginner Bodybuilders. J Appl Health Stud Sport Physiol. 2022;9(1):151–63.
Braun T, Gautel M. Transcriptional mechanisms regulating skeletal muscle differentiation, growth and homeostasis. Nat Rev Mol Cell Biol. 2011;12(6):349–61.
Ko IG, Jeong JW, Kim YH, Jee YS, Kim SE, Kim SH, et al. Aerobic exercise affects myostatin expression in aged rat skeletal muscles: a possibility of antiaging effects of aerobic exercise related with pelvic floor muscle and urethral rhabdosphincter. Int Neurourol J. 2014;18(2):77–85.
Ryan AS, Li G, Blumenthal JB, Ortmeyer HK. Aerobic exercise + weight loss decreases skeletal muscle myostatin expression and improves insulin sensitivity in older adults. Obesity. 2013;21(7):1350–6.
Shahrokhian S, Habibi AH, Shakerian S. The effect of nonlinear resistance training on serum myostatin and insulin resistance in women with breast cancer. Afr J Diabetes Med. 2022;30(1).
Bagheri R, Rashidlamir A, Motevalli MS, Elliott BT, Mehrabani J, Wong A. Effects of upper-body, lower-body, or combined resistance training on the ratio of follistatin and myostatin in middle-aged men. Eur J Appl Physiol. 2019;119(9):1921–31.
Willoughby DS. Effects of heavy resistance training on myostatin mRNA and protein expression. Med Sci Sports Exerc. 2004;36(4):574–82.
Bagheri R, Rashidlamir A, Attarzadeh Hosseini SR. Effect of resistance training with blood flow restriction on follistatin to myostatin ratio, body composition and anaerobic power of trained-volleyball players. Med Lab J. 2018;12(6):28–33.
Asadpour SM, Daryanoosh F, Salesi M, Nemati J. Effect of eight weeks of resistance training on myostatin and folistatin proteins content in gastrocnemius muscle tissue of Elderly rats. SSU_Journals. 2020;28(10):3134–43.
Sharp M, Lowery RP, Shields K, Ormes J, McCleary SA, Rauch J, et al. The effects of a myostatin inhibitor on lean body mass, strength, and power in resistance trained males. J Int Soc Sports Nutr. 2014;11(1):1.
Buford TW, Cooke MB, Willoughby DS. Resistance exercise-induced changes of inflammatory gene expression within human skeletal muscle. Eur J Appl Physiol. 2009;107(4):463–71.
Della Gatta PA, Garnham AP, Peake JM, Cameron-Smith D. Effect of exercise training on skeletal muscle cytokine expression in the elderly. Brain Behav Immun. 2014;39:80–6.
Quiles JM, Klemp A, Dolan C, Maharaj A, Huang C-J, Khamoui AV, et al. Impact of resistance training program configuration on the circulating brain-derived neurotrophic factor response. Appl Physiol Nutr Metab. 2020;45(6):667–74.
Bugera EM, Duhamel TA, Peeler JD, Cornish SM. The systemic myokine response of decorin, interleukin-6 (IL-6) and interleukin-15 (IL-15) to an acute bout of blood flow restricted exercise. Eur J Appl Physiol. 2018;118(12):2679–86.
Kanzleiter T, Rath M, Görgens SW, Jensen J, Tangen DS, Kolnes AJ, et al. The myokine decorin is regulated by contraction and involved in muscle hypertrophy. Biochem Biophys Res Commun. 2014;450(2):1089–94.
Hofmann M, Schober-Halper B, Oesen S, Franzke B, Tschan H, Bachl N, et al. Effects of elastic band resistance training and nutritional supplementation on muscle quality and circulating muscle growth and degradation factors of institutionalized elderly women: the Vienna active ageing study (VAAS). Eur J Appl Physiol. 2016;116(5):885–97.
Negaresh R, Ranjbar R, Habibi A, Mokhtarzade M, Fokin A, Gharibvand M. The effect of resistance training on quadriceps muscle volume and some growth factors in elderly and young men. УспеÑ и геронтологии. 2017;30(6).
Ellefsen S, Vikmoen O, Slettaløkken G, Whist JE, Nygård H, Hollan I, et al. Irisin and FNDC5: effects of 12-week strength training, and relations to muscle phenotype and body mass composition in untrained women. Eur J Appl Physiol. 2014;114(9):1875–88.
Figueiredo C, Antunes BM, Giacon TR, Vanderlei LC, Campos EZ, Peres FP, et al. Influence of acute and chronic high-intensity intermittent aerobic plus strength exercise on BDNF, lipid and autonomic parameters. J Sports Sci Med. 2019;18(2):359.
Attarzadeh Hosseini SR, Moeinnia N, Motahari Rad M. The effect of two intensities resistance training on muscle growth regulatory myokines in sedentary young women. Obes Med. 2017;5.
Bagheri R, Moghadam BH, Church DD, Tinsley GM, Eskandari M, Moghadam BH, et al. The effects of concurrent training order on body composition and serum concentrations of follistatin, myostatin and GDF11 in sarcopenic elderly men. Exp Gerontol. 2020;133:110869.
Scharhag-Rosenberger F, Meyer T, Wegmann M, Ruppenthal S, Kaestner L, Morsch A, et al. Irisin does not mediate resistance training-induced alterations in resting metabolic rate. Med Sci Sports Exerc. 2014;46(9):1736–43.
Hecksteden A, Wegmann M, Steffen A, Kraushaar J, Morsch A, Ruppenthal S, et al. Irisin and exercise training in humans–results from a randomized controlled training trial. BMC Med. 2013;11(1):1–8.
Kim H-j, So B, Choi M, Kang D, Song W. Resistance exercise training increases the expression of irisin concomitant with improvement of muscle function in aging mice and humans. Exp Gerontol. 2015;70:11–7.
Zhao J, Su Z, Qu C, Dong Y. Effects of 12 weeks resistance training on serum irisin in older male adults. Front Physiol. 2017;8:171.
Manabe Y, Takagi M, Nakamura-Yamada M, Goto-Inoue N, Taoka M, Isobe T, et al. Redox proteins are constitutively secreted by skeletal muscle. J Physiological Sci. 2014;64(6):401–9.
Rahimi M, Nazarali P, Alizadeh R. Pilates and TRX training methods can improve insulin resistance in overweight women by increasing an exercise-hormone. Irisin J Diabetes Metabolic Disorders. 2021;20(2):1455–60.
Timmons JA, Baar K, Davidsen PK, Atherton PJ. Is irisin a human exercise gene? Nature. 2012;488(7413):E9–E10.
Olarescu NC, Ueland T, Godang K, Lindberg-Larsen R, Jørgensen J, Bollerslev J. Inflammatory adipokines contribute to insulin resistance in active acromegaly and respond differently to different treatment modalities. Eur J Endocrinol. 2014;170(1):39–48.
Rahmatollahi M, Valizade-Orang A, Bahram ME, Jafarnezhad A. Effect of 12 weeks TRX training on Irisin and Chemerin Profile in male older adults. The Scientific Journal of Rehabilitation Medicine; 2021.
Chaliki K, Delgado D, Dong D, Moreno M, Lee J, McCulloch P, et al. editors. Acute Effects of a Single Session of Hot Yoga on Caloric Expenditure, Range of Motion, and Metabolism. International Journal of Exercise Science: Conference Proceedings; 2019.
Cahn BR, Goodman MS, Peterson CT, Maturi R, Mills PJ. Yoga, meditation and mind-body health: increased BDNF, cortisol awakening response, and altered inflammatory marker expression after a 3-month yoga and meditation retreat. Front Hum Neurosci. 2017;11:315.
Yadav R, Yadav RK, Khadgawat R, Pandey RM. Comparative efficacy of a 12 week yoga-based lifestyle intervention and dietary intervention on adipokines, inflammation, and oxidative stress in adults with metabolic syndrome: a randomized controlled trial. Translational Behav Med. 2019;9(4):594–604.
Supriya R, Yu AP, Lee PH, Lai CW, Cheng KK, Yau SY, et al. Yoga training modulates adipokines in adults with high-normal blood pressure and metabolic syndrome. Scand J Med Sci Sports. 2018;28(3):1130–8.
Yadav R, Yadav RK, Khadgawat R, Mehta N. OS 28 – 06 beneficial effects of a 12-week yoga-based lifestyle intervention on cardio-metabolic risk factors and adipokines in subjects with pre-hypertension or hypertension. J Hypertens. 2016;34:e252.
Kapilevich LV, Kironenko T, Zakharova A, Kabachkova A, Orlov S. Level of interleukins IL-6 and IL-15 in blood plasma of mice after forced swimming test. Bull Exp Biol Med. 2017;163(1):10–3.
Peng C-C, Chen K-C, Hsieh C-L, Peng RY. Swimming exercise prevents fibrogenesis in chronic kidney disease by inhibiting the myofibroblast transdifferentiation. PLoS ONE. 2012;7(6):e37388.
Nakamura M, Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Reviews Cardiol. 2018;15(7):387–407.
Mohr M, Nordsborg NB, Lindenskov A, Steinholm H, Nielsen HP, Mortensen J, et al. High-intensity intermittent swimming improves cardiovascular health status for women with mild hypertension. Biomed Res Int. 2014;2014:728289.
Zahraei H, Mogharnasi M, Afzalpour ME, Fanaei H. The effect of 8 weeks of continuous and high intensity interval swimming on chemerin levels in liver and visceral fat tissues and insulin resistance in male rats with metabolic syndrome. J Sport Exerc Physiol. 2022;15(1):33–44.
Cabrera A, Pineda W, del Pilar Correa Valencia N, Gutierrez M. Body mass conversion and improved insulin response in colombian Paso horses subjected to a swimming training program. Comp Exerc Physiol. 2022;18(3):211–8.
Hansen J, Brandt C, Nielsen AR, Hojman P, Whitham M, Febbraio MA, et al. Exercise induces a marked increase in plasma follistatin: evidence that follistatin is a Contraction-Induced Hepatokine. Endocrinology. 2011;152(1):164–71.
Dobashi S, Nakamura A, Saito K, Ando D, Koyama K. Acute swimming exercise, but not exposure to moderate hypoxic conditions reduces circulating selenoprotein P levels in short-term, high-fat diet-fed rats. J Phys Fit Sports Med. 2019;8(4):181–4.
Nazari M, Kordi M, Minasian V, Saffar Kohneh Quchan AH. Ameliorating effect of 6-week swimming exercise on mice with experimental autoimmune encephalomyelitis (EAE) by reducing fetuin-A and increasing AMPK & NAD+ levels in liver tissue. Iran J Basic Med Sci. 2022;25(8):1016–20.
Silva R, Bueno P, Avó L, Nonaka K, Selistre-Araújo H, Leal A. Effect of physical training on liver expression of activin A and follistatin in a nonalcoholic fatty liver disease model in rats. Braz J Med Biol Res. 2014;47:746–52.
Hutchinson KA, Mohammad S, Garneau L, McInnis K, Aguer C, Adamo KB. Examination of the myokine response in pregnant and non-pregnant women following an acute bout of moderate-intensity walking. Front Physiol. 2019;10:1188.
da Silva Lage VK, de Paula FA, Lima LP, Santos JNV, Dos Santos JM, Viegas ÂA et al. Plasma levels of myokines and inflammatory markers are related with functional and respiratory performance in older adults with COPD and sarcopenia. Exp Gerontol. 2022:111834.
Gmiat A, Mieszkowski J, Prusik K, Kortas J, Kochanowicz A, Radulska A, et al. Changes in pro-inflammatory markers and leucine concentrations in response to nordic walking training combined with vitamin D supplementation in elderly women. Biogerontology. 2017;18(4):535–48.
Wahl P, Hein M, Achtzehn S, Bloch W, Mester J. Acute effects of superimposed electromyostimulation during cycling on myokines and markers of muscle damage. J Musculoskel Neuronal Interact. 2015;15(1):53.
Arabzadeh E, Samadian Z, Tofighi A, Tolouei Azar J. Alteration of follistatin-like 1, neuron-derived neurotrophic factor, and vascular endothelial growth factor in diabetic cardiac muscle after moderate-intensity aerobic exercise with insulin. Sport Sci Health. 2020;16(3):491–9.
Llanos AA, Krok JL, Peng J, Pennell ML, Vitolins MZ, Degraffinreid CR, et al. Effects of a walking intervention using mobile technology and interactive voice response on serum adipokines among postmenopausal women at increased breast cancer risk. Horm Cancer. 2014;5(2):98–103.
Neumayr G, Engler C, Lunger L, Lechleitner P. Effects of a one-week vacation with various activity programs on metabolism and adipokines. Int J Sports Med. 2021;42(08):703–7.
Tota Ł, Pilch W, Piotrowska A, Pałka T, Pilch P. The effect of 12-week-long nordic walking exercise on body composition, changes in lipid and carbohydrate metabolism indices, concentration of selected adipokines and calcidiol in healthy middle-aged women. Cent Eur J Sport Sci Med. 2017;20:69–80.
Seo D-I, So W-Y, Sung DJ. Changes in insulin resistance and adipokines in obese women following a 12-week programme of combined exercise training. South Afr J Res Sport Phys Educ Recreation. 2016;38(1):39–147.
Stefanyk LE, Dyck DJ. The interaction between adipokines, diet and exercise on muscle insulin sensitivity. Curr Opin Clin Nutr Metabolic Care. 2010;13(3):255–9.
Babaei P, Hosseini R. Exercise training modulates adipokines dysregulations in metabolic syndrome. Sports Med Health Sci. 2022.
Gonzalez-Gil AM, Elizondo-Montemayor L. The role of exercise in the interplay between myokines, hepatokines, osteokines, adipokines, and modulation of inflammation for energy substrate redistribution and fat mass loss: a review. Nutrients. 2020;12(6):1899.
Sargeant JA. Exercise and insulin sensitivity: interaction with intrahepatic triglyceride and hepatokines. Loughborough University; 2018.
Garneau L, Parsons SA, Smith SR, Mulvihill EE, Sparks LM, Aguer C. Plasma myokine concentrations after acute exercise in non-obese and obese sedentary women. Front Physiol. 2020;11:18.
Catoire M, Mensink M, Kalkhoven E, Schrauwen P, Kersten S. Identification of human exercise-induced myokines using secretome analysis. Physiol Genom. 2014;46(7):256–67.
Chikamoto K, Misu H, Takayama H, Kikuchi A, Ishii K-a, Lan F, et al. Rapid response of the steatosis-sensing hepatokine LECT2 during diet-induced weight cycling in mice. Biochem Biophys Res Commun. 2016;478(3):1310–6.
Takata N, Ishii K-a, Takayama H, Nagashimada M, Kamoshita K, Tanaka T, et al. LECT2 as a hepatokine links liver steatosis to inflammation via activating tissue macrophages in NASH. Sci Rep. 2021;11(1):1–10.
Vosadi E, Ravasi AA, Soori R, Mazaheri Z, Shabkhiz F. The effect of 4 weeks of endurance exercise on the expression of the muscle myonectin levels and insulin resistance in the adult rat. Pathobiology Res. 2016;19(2):89–97.
Pourranjbar M, Arabnejad N, Naderipour K, Rafie F. Effects of aerobic exercises on serum levels of myonectin and insulin resistance in obese and overweight women. J Med Life. 2018;11(4):381.
Gullu E, Gullu A, Düzova H, Ozgor B, Kilinç E, Akçinar F. The relationship between serum leptin and VO2max levels in pre-puberty swimmer girls: effect of acute exercise. Prog Nutr. 2020;22(1):177–84.
Kraemer R, Johnson L, Haltom R, Kraemer G, Hebert E, Gimpel T et al. Serum leptin concentrations in response to acute exercise in postmenopausal women with and without hormone replacement therapy. Proceedings of the Society for Experimental Biology and Medicine. 1999;221(3):171-7.
Elias A, Pandian M, Wang L, Suarez E, James N, Wilson A. Leptin and IGF-I levels in unconditioned male volunteers after short-term exercise. Psychoneuroendocrinology. 2000;25(5):453–61.
Van Aggel-Leijssen D, Van Baak M, Tenenbaum R, Campfield L, Saris W. Regulation of average 24 h human plasma leptin level; the influence of exercise and physiological changes in energy balance. Int J Obes. 1999;23(2):151–8.
Mota GRd, Orsatti FL, Delbin MA, Zanesco A. Resistance exercise improves metabolic parameters and changes adipocyte-derived leptin: a comparison between genders in untrained adults. Motriz: Revista de Educação Física. 2016;22:217–22.
Sari İ, Habipoğlu S, Seydel G, Erşan S, Güntürk İ. The effect of acute step-aerobic exercise on adiponectin and leptin levels in premenopausal women. J Sports Med Phys Fit. 2020;61(5):725–31.
Baltaci AK, Vurucu N, Uzun A, Mogulkoc R, Kilic M. The effect of acute swimming exercise on plasma leptin in rats. Bratisl Lek Listy. 2012;113(10):592–4.
Leal-Cerro A, Garcia-Luna PP, Astorga R, Parejo J, Peino R, Dieguez C, et al. Serum leptin levels in male marathon athletes before and after the marathon run. J Clin Endocrinol Metabolism. 1998;83(7):2376–9.
Dündar A. Effect of Acute Handball Training on Irisin, Leptin and some biochemical parameters for Adolescence Handball Players. Univers J Educational Res. 2019;7(2):318–22.
Jamurtas AZ, Theocharis V, Koukoulis G, Stakias N, Fatouros IG, Kouretas D, et al. The effects of acute exercise on serum adiponectin and resistin levels and their relation to insulin sensitivity in overweight males. Eur J Appl Physiol. 2006;97(1):122–6.
Kraemer RR, Aboudehen KS, Carruth AK, Durand RJ, Acevedo EO, Hebert EP, et al. Adiponectin responses to continuous and progressively intense intermittent exercise. Med Sci Sports Exerc. 2003;35(8):1320–5.
Jürimäe J, Hofmann P, Jürimäe T, Mäestu J, Purge P, Wonisch M, et al. Plasma adiponectin response to sculling exercise at individual anaerobic threshold in college level male rowers. Int J Sports Med. 2006;27(04):272–7.
Racil G, Ben Ounis O, Hammouda O, Kallel A, Zouhal H, Chamari K, et al. Effects of high vs. moderate exercise intensity during interval training on lipids and adiponectin levels in obese young females. Eur J Appl Physiol. 2013;113:2531–40.
Ferguson MA, White LJ, McCoy S, Kim H-W, Petty T, Wilsey J. Plasma adiponectin response to acute exercise in healthy subjects. Eur J Appl Physiol. 2004;91:324–9.
Simpson KA, Singh MAF. Effects of exercise on adiponectin: a systematic review. Obesity. 2008;16(2):241–56.
Numao S, Katayama Y, Hayashi Y, Matsuo T, Tanaka K. Influence of acute aerobic exercise on adiponectin oligomer concentrations in middle-aged abdominally obese men. Metabolism. 2011;60(2):186–94.
Numao S, Suzuki M, Matsuo T, Nomata Y, Nakata Y, Tanaka K. Effects of acute aerobic exercise on high-molecular-weight adiponectin. Med Sci Sports Exerc. 2008;40(7):1271–6.
Lim K-I, Suk M-H, Shin Y-A. Effects of acute aerobic exercise on circulating adiponectin and inflammatory makers in obese middle-aged women. Korean J Health Promotion. 2012:203–10.
Lloyd JW, Evans KA, Zerfass KM, Holmstrup ME, Kanaley JA, Keslacy S. Effect of an acute bout of aerobic exercise on chemerin levels in obese adults. Diabetes & Metabolic Syndrome: Clinical Research & Reviews. 2016;10(1):37–42.
Riyahi Malayeri S, Hoseini M. The effect of acute exercise on vaspin and chemerin levels in obese men. J Basic Res Med Sci. 2021;8(1):58–66.
Malayeri SR, Hoseini M. The effect of acute exercise on vaspin and chemerin levels in obese men. 2021.
Fathi R, Mosayebi Z, Nazarali P, Aslani S. Effect of resistance training on plasma levels of chemerin and insulin in two groups of healthy and insulin resistance male rats. Res Med. 2015;38(4):207–13.
Soori R, Rezaeian N, Khosravi N, Ahmadizad S, Taleghani H, Jourkesh M, et al. Effects of water-based endurance training, resistance training, and combined water and resistance training programs on visfatin and ICAM-1 levels in sedentary obese women. Sci Sports. 2017;32(3):144–51.
Kargarfard M, Shariat A, Shaw I, Haddadi P, Shaw BS. Effects of resistance and aerobic exercise training or education associated with a dietetic program on visfatin concentrations and body composition in overweight and obese women. Asian J Sports Med. 2017;8(4).
Vatani DS, Faraji H, Rahimi R, Ahmadizad S. Acute effect of exercise type on serum visfatin in healthy men. Med Sport. 2011;65:75–83.
Haus J, Solomon T, Marchetti C, O’Leary V, Brooks L, Gonzalez F, et al. Decreased visfatin after exercise training correlates with improved glucose tolerance. Medicine + Science in Sports + Exercise. 2009;41(6):1255.
Rezaei Nasab H, Ranjbar R, Habibi AH, Shakerian S. Comparison of acute aerobic exercise in different intensities on plasma visfatin concentration in type 2 diabetic males. Iran J Diabetes Metabolism. 2015;14(2):133–40.
Mardalizade H, Soori R, Akbarnejad A. The study of the acute effect of BODY PUMP and High Intensity Circuit Training on serum levels of Omentin-1 and insulin resistance in overweight women. Sport Physiol Manage Investigations. 2022;14(3).
BASHIRI J, Rahbaran A, Gholami F, Ahmadizad S, Moradi NIKOOKHESLATS. A. The effect of acute exercise on serum vaspin level and its relation to insulin sensitivity in overweight elderly men. 2014.
Cnop M, Havel PJ, Utzschneider K, Carr D, Sinha M, Boyko E, et al. Relationship of adiponectin to body fat distribution, insulin sensitivity and plasma lipoproteins: evidence for independent roles of age and sex. Diabetologia. 2003;46(4):459–69.
Bouassida A, Chamari K, Zaouali M, Feki Y, Zbidi A, Tabka Z. Review on leptin and adiponectin responses and adaptations to acute and chronic exercise. Br J Sports Med. 2010;44(9):620–30.
Aly FA, Alghadir AH, Gabr SA. Adiponectin response to supervised aerobic training in type II diabetic patients. Asian Biomed. 2014;8(5):597–602.
Dehghani K, Mogharnasi M. Effects of ten weeks of aerobic interval training and four weeks detraining on plasma adiponectin level in male student non-athletes. Zahedan J Res Med Sci. 2015;17(10):1–7.
Busch AJ, Overend TJ, Schachter CL. Fibromyalgia treatment: the role of exercise and physical activity. Int J Clin Rheumtol. 2009;4(3):343–80.
You T, Nicklas BJ. Effects of exercise on adipokines and the metabolic syndrome. Curr Diab Rep. 2008;8(1):7–11.
Olean-Oliveira T, Figueiredo C, de Poli RAB, Lopes VHF, Jimenez-Maldonado A, Lira FS, et al. Menstrual cycle impacts adipokine and lipoprotein responses to acute high-intensity intermittent exercise bout. Eur J Appl Physiol. 2022;122(1):103–12.
He Z, Tian Y, Valenzuela PL, Huang C, Zhao J, Hong P, et al. Myokine/adipokine response to aerobic exercise: is it just a matter of exercise load? Front Physiol. 2019;10:691.
Ennequin G, Sirvent P, Whitham M. Role of exercise-induced hepatokines in metabolic disorders. Am J Physiology-Endocrinology Metabolism. 2019;317(1):E11–E24.
Weigert C, Hoene M, Plomgaard P. Hepatokines—a novel group of exercise factors. Pflügers Archiv-European Journal of Physiology. 2019;471(3):383–96.
de Piano A, de Mello MT, Sanches PdL, da Silva PL, Campos RM, Carnier J, et al. Long-term effects of aerobic plus resistance training on the adipokines and neuropeptides in nonalcoholic fatty liver disease obese adolescents. Eur J Gastroenterol Hepatol. 2012;24(11):1313–24.
Ward LJ, Nilsson S, Hammar M, Lindh-Åstrand L, Berin E, Lindblom H, et al. Resistance training decreases plasma levels of adipokines in postmenopausal women. Sci Rep. 2020;10(1):1–9.
Saeidi A, Jabbour G, Ahmadian M, Abbassi-Daloii A, Malekian F, Hackney AC, et al. Independent and combined effects of antioxidant supplementation and circuit resistance training on selected adipokines in postmenopausal women. Front Physiol. 2019;10:484.
Akbarpour M, Ozgoli G, Aryamanesh Z, Mojab F, Majd HA, Karami M et al. XML the Effect of Resistance Training on serum levels of Adipokine and inflammatory markers of Cardiovascular Disease in obese men. 2013.
Coqueiro AY, Raizel R, Bonvini A, Godois AdM, Hypólito TM, Pereira JRR, et al. Effects of glutamine and alanine supplementation on adiposity, plasma lipid profile, and adipokines of rats submitted to resistance training. J Diet Supplements. 2019;16(6):676–88.
Keihanian A, Arazi H, Kargarfard M. Effects of aerobic versus resistance training on serum fetuin-A, fetuin-B, and fibroblast growth factor-21 levels in male diabetic patients. Physiol Int. 2019;106(1):70–80.
Fedewa MV, Hathaway ED, Ward-Ritacco CL, Williams TD, Dobbs WC. The Effect of Chronic Exercise Training on Leptin: a systematic review and Meta-analysis of Randomized controlled trials. Sports Med. 2018;48(6):1437–50.
Fatouros IG, Tournis S, Leontsini D, Jamurtas AZ, Sxina M, Thomakos P, et al. Leptin and adiponectin responses in overweight inactive Elderly following Resistance Training and Detraining are Intensity Related. J Clin Endocrinol Metabolism. 2005;90(11):5970–7.
Shalitin S, Ashkenazi-Hoffnung L, Yackobovitch-Gavan M, Nagelberg N, Karni Y, Hershkovitz E, et al. Effects of a twelve-week randomized intervention of exercise and/or diet on weight loss and weight maintenance, and other metabolic parameters in obese preadolescent children. Hormone Res Paediatrics. 2009;72(5):287–301.
Lambert CP, Sullivan DH, Evans WJ. Effects of testosterone replacement and/or resistance training on interleukin-6, tumor necrosis factor alpha, and leptin in elderly men ingesting megestrol acetate: a randomized controlled trial. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences. 2003;58(2):M165–M70.
Lucotti P, Monti LD, Setola E, Galluccio E, Gatti R, Bosi E, et al. Aerobic and resistance training effects compared to aerobic training alone in obese type 2 diabetic patients on diet treatment. Diabetes Res Clin Pract. 2011;94(3):395–403.
Kraemer RR, Chu H, Castracane VD. Leptin and exercise. Experimental Biology and Medicine. 2002;227(9):701–8.
Bouassida A, Zalleg D, Bouassida S, Zaouali M, Feki Y, Zbidi A, et al. Leptin, its implication in physical exercise and training: a short review. J Sports Sci Med. 2006;5(2):172.
Zarei F, Shadmehri S, Daryanoosh F, Sherafati Moghadam M, Mahmoodi MT. The effect of eight weeks of high-intensity interval training (HIIT) on the serum levels of chemerin, omentin-1 and apelin on overweight female Sprague-Dawley rats. SSU_Journals. 2018;26(6):473–82.
Zarei M, Beheshti Nasr SMB, Hamedinia M, Taheri Chadorneshin H, Askari Majdabadi H. Effects of 12 weeks of combined aerobic-resistance exercise training on levels of chemerin, omentin and insulin resistance in men with type 2 diabetes. Koomesh. 2020;22(1):155–63.
Neuparth MJ, Proença JB, Santos-Silva A, Coimbra S. The positive effect of Moderate walking Exercise on chemerin levels in portuguese patients with type 2 diabetes Mellitus. J Investig Med. 2014;62(2):350–3.
Saremi A, Shavandi N, Parastesh M, Daneshmand H. Twelve-week aerobic training decreases chemerin level and improves cardiometabolic risk factors in overweight and obese men. Asian J Sports Med. 2010;1(3):151.
Malin S, Navaneethan S, Mulya A, Huang H, Kirwan JP. Exercise-induced lowering of chemerin is associated with reduced cardiometabolic risk and glucose-stimulated insulin secretion in older adults. J Nutr Health Aging. 2014;18(6):608–15.
Aghapour A, Farzanegi P. Effect of six-week aerobic exercise on chemerin and resistin concentration in hypertensive postmenopausal women. Electron Physician. 2013;5(1):623.
Faramarzi M, Banitalebi E, Nori S, Farzin S, Taghavian Z. Effects of rhythmic aerobic exercise plus core stability training on serum omentin, chemerin and vaspin levels and insulin resistance of overweight women. J Sports Med Phys Fitness. 2016;56(4):476–82.
Costa LR, Castro CAd, Marine DA, Fabrizzi F, Furino VdO, Malavazi I, et al. High-intensity interval training does not change vaspin and omentin and does not reduce visceral adipose tissue in obese rats. Front Physiol. 2021;12:564862.
Asadi V, Azizbeigi K, Khosravi N, Hagh Nazari N. Effect of Exercise Training on Omentin-1 and Vaspin: comparison of continuous endurance, Circuit Resistance, and high intensity interval Trainings in obese young men. Sci J Rehabilitation Med. 2019;8(4):103–12.
Parry TL, Hayward R. Exercise protects against Cancer-induced Cardiac Cachexia. Med Sci Sports Exerc. 2018;50(6):1169–76.
Okoshi MP, Capalbo RV, Romeiro FG, Okoshi K. Cardiac Cachexia: perspectives for Prevention and Treatment. Arquivos brasileiros de cardiologia. 2017;108(1):74–80.
Antunes J, Ferreira RM, Moreira-Gonçalves D. Exercise training as therapy for cancer-induced cardiac cachexia. Trends Mol Med. 2018;24(8):709–27.
Engelmann MD, Niemann L, Kanstrup I-L, Skagen K, Godtfredsen J. Natriuretic peptide response to dynamic exercise in patients with atrial fibrillation. Int J Cardiol. 2005;105(1):31–9.
Xi Y, Gong D-W, Tian Z. FSTL1 as a potential mediator of Exercise-Induced Cardioprotection in Post-Myocardial Infarction rats. Sci Rep. 2016;6(1):32424.
Kaminski K, Oledzka E, Bialobrzewska K, Kozuch M, Musial W, Winnicka M. The effects of moderate physical exercise on cardiac hypertrophy in interleukin 6 deficient mice. Adv Med Sci. 2007;52:164–8.
McGinnis GR, Ballmann C, Peters B, Nanayakkara G, Roberts M, Amin R, et al. Interleukin-6 mediates exercise preconditioning against myocardial ischemia reperfusion injury. Am J Physiol Heart Circ Physiol. 2015;308(11):H1423–H33.
Pedersen BK. Anti-inflammatory effects of exercise: role in diabetes and cardiovascular disease. Eur J Clin Invest. 2017;47(8):600–11.
Kersten S, Lichtenstein L, Steenbergen E, Mudde K, Hendriks HF, Hesselink MK, et al. Caloric restriction and exercise increase plasma ANGPTL4 levels in humans via elevated free fatty acids. Arterioscler Thromb Vasc Biol. 2009;29(6):969–74.
Ingerslev B, Hansen JS, Hoffmann C, Clemmesen JO, Secher NH, Scheler M, et al. Angiopoietin-like protein 4 is an exercise-induced hepatokine in humans, regulated by glucagon and cAMP. Mol Metabolism. 2017;6(10):1286–95.
Hansen JS, Rutti S, Arous C, Clemmesen JO, Secher NH, Drescher A, et al. Circulating follistatin is liver-derived and regulated by the glucagon-to-insulin ratio. J Clin Endocrinol Metabolism. 2016;101(2):550–60.
Xiong Y, Chen Y, Liu Y, Zhang B. Moderate-intensity continuous training improves FGF21 and KLB expression in obese mice. Biochem (Moscow). 2020;85(8):938–46.
Willis SA, Sargeant JA, Thackray AE, Yates T, Stensel DJ, Aithal GP, et al. Effect of exercise intensity on circulating hepatokine concentrations in healthy men. Appl Physiol Nutr Metab. 2019;44(10):1065–72.
Catoire M, Alex S, Paraskevopulos N, Mattijssen F, Evers-van Gogh I, Schaart G, et al. Fatty acid-inducible ANGPTL4 governs lipid metabolic response to exercise. Proc Natl Acad Sci. 2014;111(11):E1043–E52.
Cullberg K, Christiansen T, Paulsen S, Bruun J, Pedersen S, Richelsen B. Effect of weight loss and exercise on angiogenic factors in the circulation and in adipose tissue in obese subjects. Obesity. 2013;21(3):454–60.
Webster JM, Kempen L, Hardy RS, Langen RCJ. Inflammation and skeletal muscle wasting during Cachexia. Front Physiol. 2020;11:597675.
Hoffman-Goetz L, Pedersen BK. Exercise and the immune system: a model of the stress response? Immunol Today. 1994;15(8):382–7.
Agha-Alinejad H, Ahmadi Hekmatikar AH, Ruhee RT, Shamsi MM, Rahmati M, Khoramipour K, et al. A guide to different intensities of Exercise, Vaccination, and Sports Nutrition in the course of preparing Elite athletes for the management of Upper Respiratory Infections during the COVID-19 pandemic: a narrative review. Int J Environ Res Public Health. 2022;19(3):1888.
Carney EF. Walking reduces inflammation in predialysis CKD. Nat Rev Nephrol. 2014;10(6):300.
Lavín-Pérez AM, Collado-Mateo D, Abbasi S, Ferreira-Júnior JB, Hekmatikar AHA. Effects of exercise on immune cells with tumor-specific activity in breast cancer patients and survivors: a systematic review and meta-analysis. Support Care Cancer. 2023;31(9):507.
Acknowledgements
Thanks to all the authors who contributed to this article.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
Idea, Conceptualization, Project Administration and editing, A.A.H and A.P.; writing—original draft preparation, A.A.H, A.P and A.N; Designing graphic shapes, A.A.H.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Not applicable.
Consent for publication
NA.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ahmadi Hekmatikar, A., Nelson, A. & Petersen, A. Highlighting the idea of exerkines in the management of cancer patients with cachexia: novel insights and a critical review. BMC Cancer 23, 889 (2023). https://doi.org/10.1186/s12885-023-11391-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-023-11391-3