
Abstract
Background and study aim
Carrying a pathogenic BRCA1/2 variant increases greatly young women’s risk of developing breast cancer (BC). This study aimed to provide the first genetic data on BC in Mauritania.
Methods
Using NGS based screening; we searched for BRCA1/2 variants in DNA samples from 137 patients diagnosed for hereditary BC.
Results
We identified 16 pathogenic or likely pathogenic (PV) variants carried by 38 patients. Two predominant BRCA1 PV variants were found: c.815_824dup and c.4986 + 6 T > C in 13 and 7 patients, respectively. Interestingly, three novels BRCA1/2 predicted pathogenic variants have also been detected. Notably, no specific distribution of BRCA1/2 variants was observed regarding triple negative breast cancer (TNBC) or patient gender status.
Conclusions
In this first genetic profiling of BC in Mauritania, we identified a substantial number of BRCA1/2 pathogenic variants. This finding could be important in the future diagnosis and prevention policy of hereditary BC in Mauritania.
Similar content being viewed by others

Background
Breast cancer (BC) is the second cause of death by cancer in women [1]. Screening for inherited variants in patients with BC has significantly increased over the past 30 years. About 20% of all BC cases described worldwide have a genetic origin with a large heterogeneity in the percentage of pathogenic variants (PVs) [2,3,4].
Somatic variants represent the most common cause of cancer while germline variants accounts for approximately 5% of BC. Although two-thirds of these variants were found in BRCA1/2, other genes such as ATM, PALB2, CHEK2, PTEN and TP53 have been also reported in hereditary BC, ovarian cancer (OC), and pancreatic cancer (PC) [5, 6]. Using NGS-based multi-gene panel testing, many cases with strong personal and/or family history of cancer were indeed found to be BRCA1/2-wild-type. For instance, a significant proportion of 15.1% of BC, OC, or PC germline pathogenic variants was observed in susceptibility genes other than BRCA1/A2 among bilateral BC patients and therefore would have been over looked [7, 8].
The major risk of developing BC due to these variants and the large benefit of their early detection have made genetic screening for hereditary BC recommended to patients with family history in many countries even though the implication of these PV variants in defining the cancer clinical and phenotypic features remained unanswered [9]. Due to its comprehensive genomic coverage and higher sensitivity, next generation sequencing (NGS) has become an essential tool in tumor profiling [10]. However, this technology remained poorly used in African countries mainly due to its elevated running cost [11,12,13].
In this study, using NGS methodology, we reported the first BRCA1/2 mutational profile of a BC patients’ cohort in Mauritania and assessed the relevance of detected variants to the carriers’ demographic and clinical characteristics.
Patients and methods
Patients
In this study, 137 Mauritanian BC patients (132 women and 5 men) whose demographic and clinical data were complete and available to participate to the genetic screening were selected if they met one of the following criteria:
-
Age below 45 years when diagnosed with BC
-
Bilateral BC
-
BC detected in two or more relatives (first or second degree) in the family
-
BC in men
-
Triple negative breast cancer (TNBC) diagnosed before the age of 60 years.
Demographic and clinical characteristics of recruited subjects included age at diagnosis, family history of the disease, cancer staging and histological grading. Immunohistochemical staining (IHC), when accessible, was carried out on patient tissue samples embedded in paraffin blocks. Grading (from T0 to T4) was performed according to the American joint committee on cancer/ /Union for international cancer control (AJCC /UICC) systems. Evaluation of cancer stage (from 0 to 4) used TNM staging. TNBC patients were referred to subjects with IHC slides showing no antibody staining or a tumor cells fluorescence less than 1% for, concomitantly, receptors of estrogen (ER), progesterone (PR) and hormone epidermal growth factor receptor 2 (HER-2).
BRCA1 and BRACA2 molecular screening
Extraction of DNA from peripheral blood collected in EDTA tubes was carried out using QIAampDNA Blood Midi Kit (Qiagen, Hilden, Germany). DNA concentration and quality were assessed by NanoDrop (Jenway) spectrophotometer and agarose electrophoresis, respectively. Screening of the BRCA1/2 variants was performed using the ONCO/Reveal™ BRCA1/BRCA2 panel (Pillar Biosciences, Natick, MA). This technique consisted of a robust NGS assay to sequence the entire coding regions of the BRCA1 and BRCA2 genes plus 20 bp of flanking introns. It is based on the use of proprietary Stem-Loop Inhibition-Mediated amplification (SLIMamp®), a tiled amplicon-based library prep chemistry for efficient single-tube target enrichment. Sequencing was then performed on MiSeq platform as recommended by the manufacturer (Illumina, Inc., San Diego, CA) at Colorado sequencing center (https://thesequencingcenter.com/).
Bioinformatics
Alignment of the sequence reads, in FastQ format, was referred to the reference human genome 19 (hg19). Variant annotations were carried out using databases including HapMap project (http://hapmap.ncbi.nlm.nih.gov/), 1000 Genome Project (http://www.1000genomes.org/), Exome Variant Server (EVS, http://evs.gs.washington.edu/EVS/), and Exome Aggregation Consortium (EXAC, http://exac.broadinstitute.org/).
DNA variants were considered as pathogenic if cited by ClinVar record or reported by in silico analysis with PolyPhen2, (http://genetics.bwh.harvard.edu/pph2/), Sorting Intolerant From Tolerant (SIFT, http://sift.jcvi.org/) or Mutation Taster (http://www.mutationtaster.org/).
Statistics
Demographic and clinic-pathologic characteristics of patients were statistically assessed by SPSS data analysis package version 23.0 software (Chicago, Ill).
Results
Demographic and pathological features of the cohort
The demographic and clinico-pathological characteristics of 137 BC patients (132 women and 5 men) were shown in Table 1. Women were aged between 26 and 76 years with an mean age of 45 years at time of diagnosis of BC. Family history (patients with first- or second-degree relatives with BC) was reported in 41(30%) BC patients and consanguinity observed in 63 (46%) patients.
Cells abnormality was diagnosed at poorly differentiated status (grade III) in 45 (32.8%) patients. BC was at stage III in 61 (44.5%) and metastasized (stage IV) in 32 (23.4%) patients.
Cancer stretching into breast surrounding tissues showed presence of invasive ductal carcinomas (IDC) and invasive lobular carcinomas (ILC) in 114 (83.2%) and 12 (8.8%) patients, respectively.
Among 96 patients with satisfactory immunohistochemical data, 45 (46.9%) were classified triple negative breast cancer (TNBC) as no antibody staining above cut off was detected simultaneously for ER, PR and HER-2 receptors.
The five men with BC recruited had an age between 42 and 95 years. They all had unilateral (left or right) advanced BC with IDC in 4 of them. Only one male was TNBC.
BRCA1 and BRCA2 sequence variants in the study population
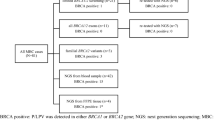
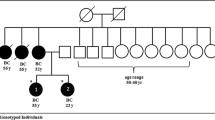
BRCA1/2 NGS-based screening of the cohort identified sixteen pathogenic (PV) or likely pathogenic variants (PLPVs) (8 in BRCA1 and 8 in BRCA2) observed in 38 patients (28.8% of total BC patients) (Table 2). PV/LPV variants were carried by 11 different families among 41 enrolled patients with family history (1st or 2nd degree affected) of BC recruited. Pedigrees of the most prevalent pathogenic variants were presented in Fig. 1. Among the PV/LPV variants, we had seven missense, four insertions, four deletions and one splice site variants. The two predominant variants were frameshift (c.815_824dup) in exon 10 and splice site variant (c.4986 + 6 T > C) in intron 15, found in 13 and 7 BC patients, respectively. Both changes were located in BRCA1 gene. Frameshift variants (c.7234_7235insG and c.6280_6286del) of BRCA2 gene were identified in 4 and 2 patients, respectively.

Examples of pedigrees with patients carrying variants in BRCA1 and BRCA2 genes in the Mauritanian population
Interestingly, we identified four variants never reported before (Tables 2 and 3): One (c.813_814insTAGCCATGTG) in BRCA1 and three (c.256del, c.2892_2893insC and c.10247A > G) in BRCA2. Three of these novel variants were predicted pathogenic (Table 2).
Twelve of the 42 TNBC patients carried pathogenic variants: 7 in BRCA1 and 5 in BRCA2. Nineteen non pathogenic variants (8 in BRCA1 and 11 in BRCA2) were detected in 96 BC patients (73.8%) (Table 3).
Carriers of all variants originated from different regions of the country (Fig. 2).

Geographic distribution of reported BRCA1/2 variants in Mauritania
Discussion
Although poor socioeconomic conditions and reduced access to adequate healthcare may have been determinant in BC development and outcome among our cohort, genetic origin was also likely giving the high rate of consanguinity (46%), the early average age of women (45 years) and advanced phase of BC at diagnosis observed in our cohort. Indeed, numerous data showed a higher incidence of common adult diseases such as hypertension, diabetes mellitus or cancer in patients from consanguineous marriages [14]. Reports also indicated that close-kin marriage continued to be preferential in North and sub-Saharan Africa with a prevalence of marriages between couples related as second cousins or closer exceeding 40% in countries including Mauritania [15, 16].
Gene predisposition has been suggested in the younger median age at BC diagnosis among Eastern Mediterranean women compared to women in Western European countries [17, 18]. An equivalent early age at BC detection (47 years) was also observed in Sub-Saharan and British black women while most Caucasian women had a BC onset at 67 years [19,20,21,22].
In this first study providing molecular data on BC in our country, among the 137 BC patients recruited at the only state cancer referring center, 38 (27.7%) carried a BRCA1/2 pathogenic or likely pathogenic variant. In neighboring North African region, the frequency of BRCA1/2 pathogenic variants in Moroccan population varied from 16.7 to 31.6% [23]. In Tunisia, 25% of hereditary BC patients carried a BRCA1/2 gene pathogenic variant [24]. The BRCA1/2 variant ratio we found (1:1.18) was also consistent with the fraction reported by these different studies which confirmed the significant contribution of BRCA1/2 germline variant in BC risk among North African populations [25, 26]. Although we did not perform functional validation of the BRCA1/2 deleterious variants identified in our cohort, most common PV/LPVs we found were already reported as associated with BC. Indeed (c.815_824dup p.Thr276Alafs*14) (found here in 34.2% of patients), was diagnosed in 15 of 27 (55.5%) patients of hereditary BC cases in neighboring Senegal with a founder effect in Afro-American patients [27]. This BRCA1 duplication leading to a premature stop codon was revealed as pathogenic variant by ClinVar here and in other studies [27, 28]. It was also submitted by HGSC-CL as pathogenic germline variant using clinical testing [29, 30]. The relatively high and concordant frequency in our cohort and in Senegal further highlights the pathogenic character of this variant in BC assessment and prevention in West African families [27, 31,32,33].
The second pathogenic variant (c.4986 + 6 T > C) in BRCA1 carried by seven of our BC patients has also been detected in multiple African patients with an early-onset of BC [34, 35]. This splicing site variant is located in highly conserved human genome region and seems to activate a cryptic splice donor site which alters the reading frame resulting in absent or truncated protein [36, 37]. It was proposed by ClinVar as pathogenic germline allele by numerous submitters [35, 38].
We also found that among the 12 TNBC patients carrying a pathogenic or likely pathogenic variant, 7 had a BRCA1 variant which substantiates data suggesting that patients with BRCA1 variants were more likely to have TNBC than those with BRCA2 variants [39, 40].
Due to the limited number of BC patients here, we could not conclude on the association of the pathogenic variant frequency and the BC phenotypic features (molecular tumor subtypes) as investigated elsewhere [40]. For instance, BRCA1-633delC was detected with relatively higher prevalence in patients with TNBC, whereas BRCA2-1466delT was found mainly in Luminal B tumors, but not in TNBC patient [40].
Our study also identified three novel predicted pathogenic BRCA1/2 variants never reported before. These variants could be specific to our population. Territorial prevalence has for instance reported among families with hereditary breast and ovarian cancer in families from southern Italy with a higher prevalence of PVs in Sicilian population [41].
One limitation of this single gene testing was that only BRCA1/2 was explored in this cohort while a NGS-based multi-gene panel testing could have revealed more potentially BC pathogenic variants. It was thus showed that 19 out of 53 positively tested bilateral BC patients harbored a germline PVs in a known (no-BRCA) BC susceptibility gene which clearly support the inclusion of multi-gene panel with high or intermediate penetrant BC genetic predisposition [7, 8]. Although highly pertinent, we could not currently perform such a study giving the high cost of this profiling.
Conclusions
Our study gave the first data on BRCA1/2 alterations likely underlying hereditary BC in Mauritania using a powerful NGS based screening.
A multi-gene panel testing of all BC patients followed by Sanger sequencing confirmation should avoid underestimation of affected patients and pave way to more cancer associated PV/LPV variants identification in hereditary tumor surveillance and targeted therapy choice.
Most of PV/LPVs variants identified in our cohort were previously reported in other African populations geographically remote and culturally apparently not related. This BC heterogeneous heredity confirmed the complex genetic structure of African populations shaped by successive voluntary or forced migrations, integrations and assimilations over many centuries.
Availability of data and materials
The datasets on unpublished pathogenic variants generated and/or analyzed during the current study are not publicly available as the study is still ongoing on other breast cancer genes and other cancers than breast cancer but are available from the corresponding author on reasonable request. The datasets of already published pathogenic variants study are available in [ClinVar] repository, via following WEB links-.
https://www-ncbi-nlm-nih-gov.proxy.insermbiblio.inist.fr/clinvar/docs/submit/
https://submit-ncbi-nlm-nih-gov.proxy.insermbiblio.inist.fr/clinvar/
Abbreviations
- BC:
-
Breast cancer
- BRCA:
-
Breast cancer gene
- PV:
-
Pathogenic variant
- LPV:
-
Likely pathogenic variant
- NGS:
-
Next generation sequencing
- TNBC:
-
Triple negative breast cancer
- OC:
-
Ovarian cancer
- PC:
-
Pancreatic cancer
- IHC:
-
Immunohistochemical staining
- IDC:
-
Invasive ductal carcinomas
- ILC:
-
Invasive lobular carcinomas
References
Globocan 2020: New Global Cancer Data | UICC. Accessed 22 Mar. 2022. https://www.uicc.org/news/globocan-2020-new-global-cancer-data
Epidemiology of breast cancer - ScienceDirect. Accessed 31 Jan. 2022. https://www.sciencedirect.com/science/article/abs/pii/S0755498219304105?via%3Dihub
Catana A, Apostu AP, Antemie RG. Multi gene panel testing for hereditary breast cancer - is it ready to be used? Med Pharm Rep. Published online July 4, 2019. Doi: https://doi.org/10.15386/mpr-1083
Turashvili G and Brogi E. Tumor Heterogeneity in Breast Cancer Front. Med., December 08, 2017| https://doi.org/10.3389/fmed.2017.00227.
Rosenthal ET, Evans B, Kidd J, et al. Increased identification of candidates for high-risk breast Cancer screening through expanded genetic testing. J Am Coll Radiol. 2017;14(4):561–8. https://doi.org/10.1016/j.jacr.2016.10.003.
Goidescu IG, Caracostea G, Eniu DT, Stamatian FV. Prevalence of deleterious mutations among patients with breast cancer referred for multigene panel testing in a Romanian population. Clujul Med 1957. 2018; 91(2):157–165. Doi: https://doi.org/10.15386/cjmed-894.
Bono M, Fanale D, Corsini LR, Brando C, Madonia G, et al. Impact of deleterious variants in other genes beyond BRCA1/2 detected in breast/ovarian and pancreatic cancer patients by NGS-based multi-gene panel testing: looking over the hedge. ESMO Open. 2021;6(4):100235. https://doi.org/10.1016/j.esmoop.2021.100235.
Fanale D, Incorvaia L, Filorizzo C, et al. Detection of germline mutations in a cohort of 139 patients with bilateral breast Cancer by multi-gene panel testing: impact of pathogenic variants in other genes beyond BRCA1/2. Cancers. 2020;12(9):E2415. https://doi.org/10.3390/cancers12092415.
Fanale D, Bono M, Calò V, et al. BRCA1/2 pathogenic variants in triple-negative versus luminal-like breast cancers: genotype-phenotype correlation in a cohort of 531 patients. Ther Adv. Med Oncol. 2020;12:1758835920975326. https://doi.org/10.1177/1758835920975326.
Kamps R, Brandão RD, van den Bosch BJ, et al. Next-generation sequencing in oncology: genetic diagnosis, risk prediction and Cancer classification. Int J Mol Sci. 2017;18(2):308. https://doi.org/10.3390/ijms18020308.
Vieira R, Biller G, Uemura G, Ruiz C, Curado M. Breast cancer screening in developing countries. Clinics. 2017; 72:244–253. doi:https://doi.org/10.6061/clinics/2017 (04)09,https://pubmed.ncbi.nlm.nih.gov/30130155/
Momenimovahed Z, Salehiniya H. Epidemiological characteristics of and risk factors for breast cancer in the world Breast Cancer (Dove Medical Press), 11, 151–164. https://doi.org/10.2147/BCTT.S176070.
Mouh FZ, Slaoui M, Razine R, El Mzibri M, Amrani M. Clinicopathological, treatment and event-free survival characteristics in a Moroccan population of triple-negative breast Cancer. Breast Cancer Basic Clin Res. 2020;14:117822342090642. https://doi.org/10.1177/1178223420906428.
Bhinder MA, Sadia H, Mahmood N, et al. Consanguinity: A blessing or menace at population level? Ann Hum Genet 2019;83(4):214–219. https://doi.org/10.1111/ahg.12308.
Anwar WA, Khyatti M, Hemminki K. Consanguinity and genetic diseases in North Africa and immigrants to Europe. Eur J Pub Health. 2014;24(suppl 1):57–63. https://doi.org/10.1093/eurpub/cku104.
Mohamed Brahim S, Zein E, Houmeida A, Tolba A. General Oncology Care in Mauritania. In: Al-Shamsi HO, Abu-Gheida IH, Iqbal F, Al-Awadhi A, eds. Cancer in the Arab World. Springer; 2022:149–161. doi:https://doi.org/10.1007/978-981-16-7945-2_10.
Zahedi R, Molavi Vardanjani H, Baneshi MR, et al. Incidence trend of breast Cancer in women of eastern Mediterranean region countries from 1998 to 2019: a systematic review and meta-analysis. BMC Womens Health 2020; 20(1):53. https://doi.org/10.1186/s12905-020-00903-z.
Dobson R. Black women have a higher risk of breast cancer than white women. BMJ. 2008;336(7636):116. https://doi.org/10.1136/bmj.39461.648750.DB.
Danforth DN. Disparities in breast cancer outcomes between Caucasian and African American women: a model for describing the relationship of biological and nonbiological factors. Breast Cancer Res BCR. 2013;15(3):208. https://doi.org/10.1186/bcr3429.
Fitzpatrick MB, Rendi MH, Kiviat NB, et al. Pathology of Senegalese breast cancers. Pan Afr Med J. 2019:34:67. https://doi.org/10.11604/pamj.2019.34.67.17993.
Pace LE, Shulman LN. Breast Cancer in sub-Saharan Africa: challenges and opportunities to reduce mortality. Oncologist. 2016;21(6):739–44. https://doi.org/10.1634/theoncologist.2015-0429.
Inequities in breast cancer treatment in sub-Saharan Africa: findings from a prospective multi-country observational study. Springermedizinde Accessed January 31, 2022. https://www.springermedizin.de/inequities-in-breast-cancer-treatment-in-sub-saharan-africa find/17064096.
Laraqui A, Uhrhammer N, Lahlou-Amine I, et al. Mutation screening of the BRCA1 gene in early onset and familial breast/ovarian Cancer in Moroccan population. Int J Med Sci. 2013;10(1):60–7. https://doi.org/10.7150/ijms.5014.
Boujemaa M, Hamdi Y, Mejri N, et al. Germline copy number variations in BRCA1/2 negative families: Role in the molecular etiology of hereditary breast cancer in Tunisia. Bandapalli OR, ed. PLOS ONE. 2021; 16(1):e0245362. https://doi.org/10.1371/journal.pone.0245362
El Ansari FZ, Jouali F, Marchoudi N, et al. Screening of BRCA1/2 genes mutations and copy number variations in patients with high risk for hereditary breast and ovarian cancer syndrome (HBOC). BMC Cancer. 2020;20(1):747. https://doi.org/10.1186/s12885-020-07250-0.
Laitman Y, Friebel TM, Yannoukakos D, et al. The spectrum of BRCA1 and BRCA2 pathogenic sequence variants in middle eastern, north African, and south European countries. Hum Mutat. 2019;40(11):e1–e23. https://doi.org/10.1002/humu.23842.
Ndiaye R, Diop JPD, Bourdon-Huguenin V, et al. Evidence for an ancient BRCA1 pathogenic variant in inherited breast cancer patients from Senegal. Npj. Genomic Med. 2020;5(1):1–6. https://doi.org/10.1038/s41525-020-0114-7.
Mefford HC, Baumbach L, Panguluri RC, et al. Evidence for a BRCA1 founder mutation in families of west African ancestry. Am J Hum Genet. 1999;65(2):575–8. https://doi.org/10.1086/302511.
Walsh T, Casadei S et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci. 2011;108(44):18032–7. https://doi.org/10.1073/pnas.1115052108.
Kour A, Sambyal V, Guleria K, et al. Screening of BRCA1 variants c.190T>C, 1307delT, g.5331G>a and c.2612C>T in breast cancer patients from North India. Genet Mol Biol. 2020;43(2):e20190014. https://doi.org/10.1590/1678-4685-gmb-2019-0014.
Biancolella M, Ouédraogo NLM, Zongo N, et al. Breast cancer in West Africa: molecular analysis of BRCA genes in early-onset breast cancer patients in Burkina Faso. Hum Genomics. 2021;15(1):65. https://doi.org/10.1186/s40246-021-00365-w.
Pal T, Bonner D, Cragun D et al. A high frequency of BRCA mutations in young black women with breast cancer residing in Florida. Cancer. 2015;121(23):4173–80. https://doi.org/10.1002/cncr.29645.
Rebbeck TR, Friebel TM, Friedman E, et al. Mutational spectrum in a worldwide study of 29,700 families with BRCA1 or BRCA2 mutations. Hum Mutat. 2018;39(5):593–620. https://doi.org/10.1002/humu.23406.
Chen X, Truong TTN, Weaver J, et al. Intronic alterations in BRCA1 and BRCA2: effect on mRNA splicing fidelity and expression. Hum Mutat. 2006;27(5):427–35. https://doi.org/10.1002/humu.20319.
Vreeswijk MPG, Kraan JN, van der Klift HM, et al. Intronic variants in BRCA1 and BRCA2 that affect RNA splicing can be reliably selected by splice-site prediction programs. Hum Mutat. 2009;30(1):107–14. https://doi.org/10.1002/humu.20811.
Salmi F, Maachi F, Tazzite A, et al. Next-generation sequencing of BRCA1 and BRCA2 genes in Moroccan prostate cancer patients with positive family history. PLoS One. 2021;16(7):e0254101. https://doi.org/10.1371/journal.pone.0254101.
Tian CQ, Darcy KM, Krivak TC, al. Assessment of the prognostic value of two common variants of BRCA1 and BRCA2 genes in ovarian Cancer patients treated with cisplatin and paclitaxel: a gynecologic oncology group study. Front. Oncol. 2013;3:206. https://doi.org/10.3389/fonc.2013.00206.
Rashid MU, Muhammad N, Amin A, Loya A, Hamann U. Contribution of BRCA1 large genomic rearrangements to early-onset and familial breast/ovarian cancer in Pakistan. Breast Cancer Res Treat. 2017;161(2):191–201. https://doi.org/10.1007/s10549-016-4044-0.
Chen H, Wu J, Zhang Z, et al. Association between BRCA status and triple-negative breast Cancer: a Meta-analysis. Front Pharmacol. 2018;9:909. https://doi.org/10.3389/fphar.2018.00909.
Incorvaia L, Fanale D, Bono M, et al. BRCA1/2 pathogenic variants in triple-negative versus luminal-like breast cancers: genotype-phenotype correlation in a cohort of 531 patients. Ther Adv. Med Oncol. 2020:12:1758835920975326. https://doi.org/10.1177/1758835920975326.
Incorvaia L, Fanale D, Badalamenti G, et al. Hereditary breast and ovarian Cancer in families from southern Italy (Sicily)-prevalence and geographic distribution of pathogenic variants in BRCA1/2 genes. Cancers. 2020;12(5):E1158. https://doi.org/10.3390/cancers12051158.
Acknowledgments
Not applicable.
Authors ‘contributions
SBR collected and organized patients files; SBR, CTH, MS and MVZ contributed to epidemiological and clinicopatholgical data analysis; AT examined Immunohistochemical slides (IHC) and contributed in paper conception; EZ, MK and AH contributed in paper conception; SBR, MK was the major contributor in writing the manuscript. All authors read and approved the final manuscript.
Funding
The authors declare that no funds, grants were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethics approval for this was granted by the Ethics Committee of the University of Nouakchott Al-Asriya, Mauritania and Informed consent was obtained from all patients of the study for participation. Research in this study has been performed in accordance with the Declaration of Helsinki and has been approved by an appropriate ethics committee. We confirm that all methods were performed in accordance with the relevant guidelines and regulations. Informed consent was obtained from all study participants.
Consent for publication
The informed consent of all patients was obtained for the data publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Brahim, S.M., Zein, E.E., Bonnet, C. et al. Screening of BRCA1/2 variants in Mauritanian breast cancer patients. BMC Cancer 22, 802 (2022). https://doi.org/10.1186/s12885-022-09903-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-022-09903-8