
Abstract
Background
As major regulators of DNA replication in eukaryotes, minichromosome maintenance (MCM) proteins play an important role in the initiation and extension of DNA replication. MCMs and their related genes may be new markers of cell proliferation activity, which is of great significance for the diagnosis and prognosis of cervical cancer.
Methods
To explore the role of MCMs and their related genes in cervical cancer, various bioinformatics methods were performed. First, the ONCOMINE and UALCAN databases were used to analyze the mRNA expression of different MCMs. The Human Protein Atlas database was used to analyze the protein expression of MCMs in normal and tumor tissues. The potential clinical value of MCMs was evaluated using the UALCAN, Kaplan-Meier plotter and cBioPortal databases. Then, the related genes and key coexpressed genes of MCMs were screened using GEPIA2 and cBioPortal analysis. For these genes, we used Metascape and the DAVID database to perform Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses, construct the related molecular interaction network, and obtain the key subnetworks and related hub genes. The Kaplan-Meier plotter database was used for survival analysis of cervical cancer patients to evaluate and predict the potential clinical value of the hub genes. Moreover, multiple gene comparisons of the expression of MCMs and related genes in different cancer types also showed the clinical significance of these potential targets.
Results
The mRNA and protein expression of MCMs increased in tumor tissue. Overexpression of MCM2/3/4/5/6/7/8/10 was found to be significantly associated with clinical cancer stage. Higher mRNA expression levels of MCM3/5/6/7/8 were found to be significantly associated with longer overall survival, and higher mRNA expression of MCM2/3/4/5/6/7/8 was associated with favorable OS. In addition, a high mutation rate of MCMs (71%) was observed. MCM2, MCM4, MCM8, MCM3 and MCM7 were the five genes with the most genetic alterations. In addition, the coexpressed genes and related genes of MCMs were successfully screened for enrichment analysis. These genes were significantly enriched in important pathways, such as the DNA replication, cell cycle, mismatch repair, spliceosome, and Fanconi anemia pathways. A protein-protein interaction network was successfully constructed, and a total of 13 hub genes (CDC45, ORC1, RPA1, CDT1, TARDBP, RBMX, SRSF3, SRSF1, RFC5, RFC2, MSH6, DTL, and MSH2) from 4 key subnetworks were obtained. These genes and MCM2/3/4/5/6/7/8 might have potential clinical value for the survival and prognosis of cervical cancer patients.
Conclusions
These findings promoted the understanding of the MCM protein family and clinically related molecular targets for cervical epithelial neoplasia and cervical cancer. Our results were helpful to evaluate the potential clinical value of MCMs and related genes in patients with cervical cancer.
Similar content being viewed by others

Background
The minichromosome maintenance (MCM) protein family is a group of proteins closely related to DNA replication and genome stability [1]. Highly conserved MCM complex proteins may have helicase activity and are essential for the initiation of DNA replication. MCM complex proteins contain ATPase domains, and energy is harnessed to affect DNA unwinding [2]. There are ten characterized homologous MCM genes. MCM2–7 form a replicative helicase complex [3], and MCM8 and MCM9 form a dimer involved in homologous recombination repair [4]. The ninth gene that encodes an MCM domain is named MCM domain-containing 2 (MCMDC2) [5]. MCM10 is a dynamic scaffold at eukaryotic replication forks [6].
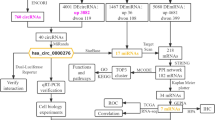
It has been reported that the expression of MCMs in the cell proliferation cycle is one of the important factors of DNA replication initiation and extension, and their positive expression is an important marker of cell proliferation [7]. Their expression levels are related to the proliferation and differentiation of tumor cells and can accurately reflect the proliferation activity of cells. MCMs have great reference value in the early diagnosis, classification and prognosis of clinical tumors [8, 9]. Therefore, it is necessary to strengthen the research on the basic theory and clinical application value of MCMs, including their mechanism of protein action, their expression characteristics and their related genes in cervical cancer tissue, as well as their value in clinical diagnosis and differential diagnosis. At present, there are few related studies in these areas. In this study, the roles of MCMs and related genes in cervical cancer were investigated by a variety of bioinformatics methods. By analyzing the mRNA and protein expression of MCM family members, their potential clinical value in cervical cancer was analyzed. The workflow of this study is shown in Fig. 1.

Flowchart of the integrated analysis
Methods
Expression of different MCMs in cervical cancer patients
To explore the distinct prognostic and potential therapeutic values of different MCM members in cervical cancer patients, the mRNA expression of different MCMs was analyzed by the ONCOMINE database [10] (www.oncomine. org) and UALCAN [11] (http://ualcan.path.uab.edu). After analyzing the mRNA expression, the protein expression of different MCMs in cervical cancer was explored by the Human Protein Atlas [12,13,14] (HPA, http://www.proteinatlas.org) database, and the results of immunohistochemistry from HPA showed the expression of MCMs in normal tissues and tumor tissues. The data used for analysis were from databases, and the expression results of different MCMs can provide a reference for evaluating their potential clinical value.
Potential clinical value of MCMs
After mRNA and protein expression analyses, the relationship between the mRNA expression of MCMs and the clinicopathological parameters of patients, such as individual cancer stages, was assessed by performing The Cancer Genome Atlas (TCGA) database analysis via UALCAN. The data used for analysis were from the database, and the analysis results showed the potential value of different MCMs in clinical pathology. Moreover, the survival of patients was analyzed, and specific MCMs related to better prognosis were identified.
Furthermore, the Kaplan-Meier plotter database [15, 16] (http://kmplot.com/analysis/) was used to analyze the prognostic value of the mRNA expression of different MCMs in cancer patients. The correlation between the mRNA expression of MCMs and the prognosis of patients with cervical cancer was explored and analysed. MCMs useful for predicting the survival of patients with cervical cancer were identified.
After mRNA expression of specific MCMs was found to be significantly associated with patient prognosis, genetic alterations in MCMs and their associations with overall survival (OS) and disease-free survival (DFS) of cervical squamous cell carcinoma (CESC) patients were analyzed by the cBioPortal database [17, 18] (www.cbioportal.org). Analysis of genetic alterations promoted the exploration and understanding of different MCMs in CESC, identified MCMs that are prone to alteration and provided information support for genetic alterations of MCMs in cervical cancer. In addition, key coexpressed genes of MCMs in CESC (TCGA, PanCancer Atlas) were screened and analyzed via a Venn diagram (http://bioinformatics.psb.ugent.be/webtools/Venn/).
Similar gene detection and enrichment analysis of related genes in CESC tumors
After the analysis of the ONCOMINE, UALCAN, HPA, Kaplan-Meier plotter and cBioPortal databases, the functions of the MCMs with potential value and their related genes in CESC tumors were further enriched and explored. Genes that had a similar expression pattern to MCMs in CESC tumors were analyzed by GEPIA2 [19] (http://gepia2.cancer-pku.cn/#similar). Then, the key coexpressed genes and related genes of MCMs in CESC tumors were analyzed by Metascape [20] (https://metascape.org/gp/index.html). The pathways and process enrichment of these genes were determined [21]. Database for Annotation, Visualization and Integrated Discovery (DAVID, https://david.ncifcrf.gov/) was also used to verify the biological processes, cellular components, molecular functions and KEGG pathways of these genes [22,23,24].
Protein-protein interaction (PPI) network construction and screening of hub genes
Next, the network of enriched terms and the PPI network were also analyzed by Metascape [25,26,27]. Then, key subnetworks and related hub genes were obtained, and the Kaplan-Meier plotter database was used for survival analysis of cervical cancer patients to evaluate and predict the potential clinical value of the hub genes [28]. Moreover, multiple gene comparisons of the expression of MCMs and related genes in different cancer types were performed to show the clinical significance of these potential targets by GEPIA2. In particular, MCMs and their related genes that are involved in the progression of cervical cancer might provide potential targets for the clinical prevention, treatment, and effective prognostication of cervical cancer.
Results
Expression of different MCMs in cervical cancer patients
As shown in Fig. 2 and Table 1, the mRNA expression levels of MCM2/3/4/5/6/7/8/9/10 in cervical cancer tissues and normal tissues were compared using the ONCOMINE database [29,30,31,32], and MCM2/3/4/5/6/7/8/10 expression was significantly increased in tumor tissues. Then, the mRNA expression of MCM2/3/4/5/6/7/8/9/10 was further analyzed by the UALCAN database. As shown in Fig. 3, the mRNA expression of MCM2/3/4/5/6/7/8/10 in tumor tissues was significantly higher than that in normal tissues (p < 0.05), while the expression of MCM8/9/10 in tumor tissues was lower than that of other MCMs (MCM2/3/4/5/6/7). However, the expression of MCM9 in tumor tissues was not significant. In addition to the above analysis, immunohistochemical information was obtained from the HPA database to analyze the protein expression of MCMs in normal and tumor tissues (Fig. 4). MCM2 and MCM5 were not detected in normal tissues, but their high expression was observed in tumor tissues. The expression of MCM6/9/10 was low in normal tissues, but medium and high protein expression was observed in tumor tissues. In addition, medium protein expression of MCM3/4/7 was observed in normal tissues, and high protein expression was observed in tumor tissues. Moreover, high expression of MCM2/3/5/7 was associated with good survival and prognosis (Fig. 5).

Transcriptional expression of MCMs in different types of cancer (ONCOMINE). Differences in transcriptional expression were compared by t-tests. The cutoff p-value, fold change, and other settings were as follows: p-value: 0.01, fold change: 1.5, gene rank: top 10%, data type: mRNA

mRNA expression of MCMs in CESC tissues and normal tissues (UALCAN). *p < 0.05, **p < 0.01, ***p < 0.001

Differential expression of MCMs in cervical cancer tissues and normal tissues (Human Protein Atlas). MCM8: No data in HPA database

Prognostic markers of cervical cancer (favorable) in the Human Protein Atlas database (p < 0.001)
Potential clinical value of MCMs and screening of coexpressed genes
As shown in Fig. 6, the results showed that the mRNA expression of MCMs was significantly correlated with individual cancer stages in the UALCAN database, and cancer patients (stage 1-stage 4) had higher mRNA expression of MCMs than normal controls. The effects of MCM2 and MCM5 expression are shown in Fig. 7. High expression of MCM2 and MCM5 had a significant impact on patients, and they might be better targets to promote the good survival and prognosis of patients.

Relationship between mRNA expression of MCMs and individual cancer stages in CESC, *p < 0.05, **p < 0.01, ***p < 0.001 (UALCAN)

Effect of MCM2 and MCM5 expression levels on CESC patient survival (UALCAN)
Furthermore, the prognostic value of the mRNA expression of MCMs was evaluated using the Kaplan-Meier plotter database (Fig. 8). The mRNA expression of MCM2/3/4/5/6/7/8 was significantly associated with CESC patient prognosis, and the results showed that higher mRNA expression of MCM2/3/4/5/6/7/8 was associated with favorable OS in CESC patients. Moreover, higher mRNA expression of MCM3/5/6/7/8 was significantly associated with longer OS, but the mRNA expression of MCM9, MCM10 and MCMDC2 had no significant effect on the prognosis of CESC patients (Fig. 9). These results indicated that MCM2/3/4/5/6/7/8 may serve as useful biomarkers for CESC patients.

Prognostic value of MCM2/3/4/5/6/7/8 mRNA expression in CESC patients (Kaplan-Meier plotter database)

Prognostic value of the mRNA expression of MCM9/10 and MCMDC2 in CESC patients (Kaplan-Meier plotter database)
In particular, genetic mutations in MCMs and their associations with OS and DFS were explored by the cBioPortal database. The cBioPortal analysis showed a high mutation rate (71%) of MCMs in CESC patients (TCGA, PanCancer Atlas). MCM2, MCM4, MCM8, MCM3 and MCM7 were the top five genes with the highest genetic alterations, and the mutation rates were 35, 20, 19, 15 and 14%, respectively (Fig. 10). Moreover, the results showed that genetic alterations in MCMs were not significantly associated with longer OS or DFS in CESC patients.

Analysis of MCMs in CESC patients (cBioPortal). A. Genetic mutations; B. Relationships between MCM gene mutations and OS and DFS in CESC patients (cBioPortal)
Furthermore, to deeply explore the potential clinical value of MCMs, their coexpressed genes were analyzed using the cBioPortal database. The coexpressed genes of MCM2/3/4/5/6/7/8 (top 25 correlated genes) were screened, and the results of intersection analysis are shown in Table 2. A total of 9 intersections and 16 key coexpressed genes (MCM5 [33], CHAF1B, MCM2, GINS2, MCM6, RFC5, FANCC, TIMELESS, CLSPN, BRIP1, RBL1, CDT1, RFC2, CDC45, ORC1, and TOP2A) were obtained for further study.
Detection and enrichment analysis of related genes
Next, the top 50 genes related to MCMs were identified by GEPIA2. The 57 selected genes (as shown in Table 3) and MCM7/8 were analyzed by Metascape. The pathway and process enrichment analysis results are shown in Fig. 11. The results showed that MCMs and related genes (59 total) were mainly enriched in DNA replication, DNA repair, the cell cycle, cell division and expression regulation. These genes were also analyzed by the DAVID database, and the Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment results (p-value< 0.05, false discovery rate < 0.05) are shown in Table 4, Table 5, Table 6 and Table 7. The MCM-related genes were significantly enriched in 21 biological processes, 5 cellular components, 14 molecular functions and 5 important KEGG pathways.

Enrichment results of the hub genes. A. Bar graph of the enriched terms (the top 20 clusters) and the top-ranked Gene Ontology biological processes; B. Network of the enriched terms colored by cluster ID. Nodes that share the same cluster ID are typically close to each other (Metascape)
PPI network construction and screening of hub genes
As shown in Fig. 12, the PPI network was successfully constructed. There were 4 Molecular Complex Detection (MCODE) components identified from the PPI network. MCODE 1 (MCM2/3/4/5/6/7/8, CLSPN, CDC45, ORC1, RPA1, and CDT1) played an important role in the activation of the prereplicative complex (R-HSA-68962), the activation of ATR in response to replication stress (R-HSA-176187) and DNA replication preinitiation (R-HSA-69002). MCODE 2 (TARDBP, RBMX, U2AF2, HNRNPM, SRSF3, and SRSF1) was important in the regulation of mRNA metabolic processes (GO:1903311) and spliceosomes (ko03040, hsa03040). MCODE 3 (USP1, RFC5, and RFC2) was important in the recognition of DNA damage by the PCNA-containing replication complex (R-HSA-110314), DNA damage response (GO:0042769) and PID Fanconi pathway (M1). MCODE 4 (MSH6, DTL, and MSH2) was important in the response to UV (GO:0009411), the response to light stimulus (GO:0009416) and DNA repair (R-HSA-73894). The survival analysis results of the hub genes from the 4 MCODE components are shown in Fig. 13 (the prognostic value of MCMs is shown in Fig. 8 and Fig. 9). The expression of HNRNPM, U2AF2, USP1 and CLSPN showed no correlation with the prognosis of CESC patients, while the high expression of the other 13 genes was significantly related to a better prognosis. Moreover, higher mRNA expression of RFC5, RFC2, DTL, RBMX, ORC1 and MSH2 was significantly associated with longer OS in CESC patients. These results indicated that the hub genes might play an important role in cervical cancer and provide potential molecular targets. In addition, GEPIA2 was used to generate an interactive heat map of the expression of MCMs and related genes in different cancer types (Fig. 14). The hub genes and MCM2/3/4/5/6/7/8 might have potential clinical value for the survival and prognosis of cervical cancer patients.

Protein-protein interaction network and MCODE components (Metascape)

Prognostic value of the mRNA expression of the hub genes related to MCMs in CESC patients (Kaplan-Meier plotter database)

Interactive heat map analysis of tissue-wise expression in different cancer types (GEPIA2)
Discussion
Cervical cancer is the most common cancer among women worldwide. High-risk human papillomavirus (HPV) infection causes high morbidity and mortality. Therefore, the development of cervical cancer vaccines and screening technology and the exploration of clinical targets with good application prospects are still important. MCMs are implicated in the development of multiple cancers, including cervical cancer. Thus, MCM proteins have emerged as exceptionally promising markers for cervical cancer screening and early diagnosis [34]. Mitali Das et al. [35] explored the role of MCM4/5/6/10 in cervical cancer and their correlation with the clinical parameters of cervical cancer, and further study indicated that cervical cancer cells may use excess MCMs as a backup for replicative stress [36]. V N Saritha et al. [37] showed that MCM2/5 expression was upregulated in low-grade lesions, high-grade lesions and malignancies to a greater extent than p16 and p63. Gurjeet Kaur et al. [38] evaluated MCM gene expression profiles and MCM2 protein in HPV-associated cervical carcinogenesis. There is growing evidence that MCMs may be used as biomarkers to predict the malignant potential of cervical lesions. However, as cervical cancer is a complex disease involving many molecular interactions and complex signaling pathways, reports of MCMs and their related genes in cervical cancer are few and insufficient, and more research is still needed for the prevention and treatment of cervical cancer. In this study, bioinformatics was used to mine expression data and perform subsequent comprehensive analyses, which were based on a large amount of public data of patients with cervical cancer, for the in-depth study of the related molecular mechanisms of MCMs and protein molecular interactions, the prediction of related biomarkers, and the exploration of factors related to the good survival and prognosis of patients.
The expression of different MCMs in cervical cancer patients obtained from professional databases (ONCOMINE, UALCAN and HPA) showed that the mRNA and protein expression of MCMs increased in tumor tissue. These findings promoted our understanding of the expression of different MCMs in cancer patients. In particular, to evaluate and predict the potential clinical value of MCMs, analyses of individual cancer stages, the survival of cancer patients and the genetic alterations of different MCMs were performed using the UALCAN, Kaplan-Meier plotter and cBioPortal databases. The multidatabase analysis revealed that MCMs had great potential clinical significance. MCM2/3/4/5/6/7/8 might be used as potential indicators for survival in patients with cervical cancer, which needs more research and verification. Moreover, a high mutation rate (71%) of MCMs was observed in cervical cancer patients. MCM2, MCM4, MCM8, MCM3 and MCM7 ranked as the top five genes with the highest number of genetic alterations, but genetic alterations in MCMs were not significantly associated with longer OS or DFS in CESC patients. According to these results, the intervention strategy of mutating MCM genes to achieve longer survival times in patients might not be effective. However, controlling the gene transcription and protein expression of MCMs might be an effective intervention method for CESC patients.
After the expression analysis and clinical value evaluation of MCMs, the coexpressed genes and related genes of MCMs were screened by cBioPortal and GEPIA2 analysis, and then the systematic enrichment analysis of related genes was performed by Metascape and the DAVID database to deepen the understanding of the role of MCMs and related genes in cervical cancer. A total of 59 genes were involved in the enrichment analysis. The enrichment results revealed that these genes played an important role in DNA replication, the cell cycle, DNA repair, the DNA damage response, the regulation of signal transduction by p53 class mediators and other important biological processes. Moreover, these genes were significantly enriched in some important cellular components, such as the nucleoplasm, nucleus, MCM complex, nuclear chromosome, telomeric region and nuclear speck, which were also involved in DNA binding, DNA helicase activity, protein binding, ATP binding, nucleoside binding, DNA replication origin binding and RNA binding. We found that MCMs and their related genes were significantly enriched in some important pathways, such as the DNA replication (RFC5, MCM7, RFC2, PRIM1, RPA1, MCM3, MCM4, MCM5, MCM6, and MCM2), cell cycle (RBL1, CDC45, MCM7, ORC1, MCM3, MCM4, MCM5, MCM6, and MCM2), mismatch repair (RFC5, MSH6, MSH2, RFC2, and RPA1), spliceosome (HNRNPM, U2AF2, SRSF1, HNRNPU, SRSF3, and RBMX) and Fanconi anemia (BRIP1, RPA1, USP1, and FANCC) pathways. Further study of these pathways can deepen the understanding of the molecular mechanisms related to the occurrence and development of cervical cancer.
In the current study, genes coexpressed with MCMs and their related genes were successfully screened for PPI network construction. A total of 4 key subnetworks were screened. Survival analysis showed that 13 hub genes (CDC45, ORC1, RPA1, CDT1, TARDBP, RBMX, SRSF3, SRSF1, RFC5, RFC2, MSH6, DTL, and MSH2) from the key subnetworks might have potential clinical value. These genes and MCM2/3/4/5/6/7/8 were significantly related to the survival and prognosis of cervical cancer patients. Thus, our results might provide bioinformatics support for MCMs and their related genes in the prevention and clinical treatment of cervical cancer.
However, this study had three limitations. First, the data analyzed in this study were from public databases, and the analysis results might be affected by the quantity and quality of the data. Second, we did not evaluate the potential therapeutic or diagnostic effects of MCMs in detail. Finally, we did not explore the potential mechanisms of MCMs and hub genes in cervical cancer in detail, and the effect on prognosis requires follow-up data. Therefore, further research is needed to verify our findings and to explore the clinical application of MCMs and their related genes for the treatment of cervical cancer.
Conclusion
In conclusion, this study used a variety of bioinformatics methods to explore the transcriptional expression of MCMs as potential indicators of survival in patients with cervical cancer, obtained target genes with potential application value, and deepened the understanding of the influence of MCMs and their related genes in cervical cancer. These genes can be used to diagnose the progression of the disease before it leads to cancer. Moreover, our findings promoted the understanding of the MCM protein family and clinically related molecular targets for cervical epithelial neoplasia and cervical cancer, which provided new insight into the biological functions of MCMs in cervical cancer.
Availability of data and materials
The data used for analysis were all from online public databases and could be retrieved. Based on these data, we conducted further analysis and research. The datasets generated and analyzed during the current study are available in the ONCOMINE database, UALCAN database, Human Protein Atlas database, Kaplan-Meier plotter database, and GEPIA2 database repository. Preliminary enrichment analysis, molecular interaction network construction and hub gene screening were performed via the Metascape website, and the DAVID database was used for further verification. The websites of the databases are as follows: [https://www.oncmine.org], [http://ualcan.path.uab.edu/], [https://www.proteinatlas.org/], [http://kmplot.com/analysis], [https://metascape.org/gp/index.html], [https://david.ncifcrf.gov/], and [http://gepia2.cancer-pku.cn].
Abbreviations
- MCM:
-
minichromosome maintenance
- MCMDC2:
-
MCM domain-containing 2
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- PPI:
-
protein-protein interaction
- CESC:
-
cervical squamous cell carcinoma
- HPA:
-
Human Protein Atlas
- DAVID:
-
Database for Annotation, Visualization and Integrated Discovery
- TCGA:
-
The Cancer Genome Atlas
- OS:
-
overall survival
- DFS:
-
disease-free survival
- MCODE:
-
Molecular Complex Detection
- HPV:
-
Human papillomavirus
- CESCE:
-
cervical squamous cell carcinoma epithelia
- HG-CIN:
-
high-grade cervical squamous intraepithelial neoplasia epithelia
References
Forsburg SL. Eukaryotic MCM proteins: beyond replication initiation. Microbiol Mol Biol Rev. 2004;68(1):109–31. https://doi.org/10.1128/MMBR.68.1.109-131.2004.
Miller JM, Arachea BT, Epling LB, Enemark EJ. Analysis of the crystal structure of an active MCM hexamer. Elife. 2014;3:e03433. https://doi.org/10.7554/eLife.03433.
Bell SD, Botchan MR. The minichromosome maintenance replicative helicase. Cold Spring Harb Perspect Biol. 2013;5(11):a012807. https://doi.org/10.1101/cshperspect.a012807.
Griffin WC, Trakselis MA. The MCM8/9 complex: a recent recruit to the roster of helicases involved in genome maintenance. DNA Repair (Amst). 2019;76:1–10. https://doi.org/10.1016/j.dnarep.2019.02.003.
Finsterbusch F, Ravindranathan R, Dereli I, Stanzione M, Tränkner D, Tóth A. Alignment of homologous chromosomes and effective repair of programmed DNA double-Strand breaks during mouse meiosis require the Minichromosome maintenance domain containing 2 (MCMDC2) protein. PLoS Genet. 2016;12(10):e1006393. https://doi.org/10.1371/journal.pgen.1006393.
Baxley RM, Bielinsky AK: Mcm10: A Dynamic Scaffold at Eukaryotic Replication Forks. Genes (Basel). 2017;8(2):73.
Malinowski DP. Molecular diagnostic assays for cervical neoplasia: emerging markers for the detection of high-grade cervical disease. Biotechniques. 2005;Suppl:17–23.
Zheng J. Diagnostic value of MCM2 immunocytochemical staining in cervical lesions and its relationship with HPV infection. Int J Clin Exp Pathol. 2015;8(1):875–80.
Wang D, Li Q, Li Y, Wang H. The role of MCM5 expression in cervical cancer: correlation with progression and prognosis. Biomed Pharmacother. 2018;98:165–72. https://doi.org/10.1016/j.biopha.2017.12.006.
Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, et al. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6(1):1–6. https://doi.org/10.1016/S1476-5586(04)80047-2.
Chandrashekar DS, Bashel B, Balasubramanya SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi B, et al. UALCAN: a portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia. 2017;19(8):649–58. https://doi.org/10.1016/j.neo.2017.05.002.
Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347(6220):1260419.
Thul PJ, Åkesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H, Alm T, Asplund A, Björk L, Breckels LM, et al. A subcellular map of the human proteome. Science. 2017;356(6340):eaal3321.
Uhlen M, Zhang C, Lee S, Sjöstedt E, Fagerberg L, Bidkhori G, Benfeitas R, Arif M, Liu Z, Edfors F et, al. A pathology atlas of the human cancer transcriptome. Science. 2017;357(6352):eaan2507.
Nagy Á, Lánczky A, Menyhárt O, Győrffy B. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci Rep. 2018;8(1):9227. https://doi.org/10.1038/s41598-018-27521-y.
Nagy Á, Munkácsy G, Győrffy B. Pancancer survival analysis of cancer hallmark genes. Sci Rep. 2021;11(1):6047.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–4. https://doi.org/10.1158/2159-8290.CD-12-0095.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. https://doi.org/10.1126/scisignal.2004088.
Tang Z, Kang B, Li C, Chen T, Zhang Z. GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019;47(W1):W556–w560. https://doi.org/10.1093/nar/gkz430.
Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10(1):1523. https://doi.org/10.1038/s41467-019-09234-6.
Hochberg Y, Benjamini Y. More powerful procedures for multiple significance testing. Stat Med. 1990;9(7):811–8. https://doi.org/10.1002/sim.4780090710.
Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, et al. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4(5):P3. https://doi.org/10.1186/gb-2003-4-9-r60.
Huang DW, Sherman BT, Tan Q, Kir J, Liu D, Bryant D, et al. DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007;35(Web Server issue):W169–75.
Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. https://doi.org/10.1093/nar/28.1.27.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504. https://doi.org/10.1101/gr.1239303.
Stark C, Breitkreutz BJ, Reguly T, Boucher L, Breitkreutz A, Tyers M. BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 2006;34(Database issue):D535–9. https://doi.org/10.1093/nar/gkj109.
Li T, Wernersson R, Hansen RB, Horn H, Mercer J, Slodkowicz G, et al. A scored human protein-protein interaction network to catalyze genomic interpretation. Nat Methods. 2017;14(1):61–4. https://doi.org/10.1038/nmeth.4083.
Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics. 2003;4(1):2. https://doi.org/10.1186/1471-2105-4-2.
Scotto L, Narayan G, Nandula SV, Arias-Pulido H, Subramaniyam S, Schneider A, et al. Identification of copy number gain and overexpressed genes on chromosome arm 20q by an integrative genomic approach in cervical cancer: potential role in progression. Genes Chromosomes Cancer. 2008;47(9):755–65. https://doi.org/10.1002/gcc.20577.
Zhai Y, Kuick R, Nan B, Ota I, Weiss SJ, Trimble CL, et al. Gene expression analysis of preinvasive and invasive cervical squamous cell carcinomas identifies HOXC10 as a key mediator of invasion. Cancer Res. 2007;67(21):10163–72. https://doi.org/10.1158/0008-5472.CAN-07-2056.
Pyeon D, Newton MA, Lambert PF, den Boon JA, Sengupta S, Marsit CJ, et al. Fundamental differences in cell cycle deregulation in human papillomavirus-positive and human papillomavirus-negative head/neck and cervical cancers. Cancer Res. 2007;67(10):4605–19. https://doi.org/10.1158/0008-5472.CAN-06-3619.
Biewenga P, Buist MR, Moerland PD, Ver Loren Van Themaat E, Van Kampen AH, Ten Kate FJ, et al. Gene expression in early stage cervical cancer. Gynecol Oncol. 2008;108(3):520–6. https://doi.org/10.1016/j.ygyno.2007.11.024.
Nowinska K, Ciesielska U, Piotrowska A, Jablonska K, Partynska A, Paprocka M, et al. MCM5 expression is associated with the grade of malignancy and Ki-67 antigen in LSCC. Anticancer Res. 2019;39(5):2325–35. https://doi.org/10.21873/anticanres.13349.
Laskey R. The Croonian lecture 2001 hunting the antisocial cancer cell: MCM proteins and their exploitation. Philos Trans R Soc Lond Ser B Biol Sci. 2005;360(1458):1119–32. https://doi.org/10.1098/rstb.2005.1656.
Das M, Prasad SB, Yadav SS, Govardhan HB, Pandey LK, Singh S, et al. Over expression of minichromosome maintenance genes is clinically correlated to cervical carcinogenesis. PLoS One. 2013;8(7):e69607. https://doi.org/10.1371/journal.pone.0069607.
Das M, Prasad SB, Yadav SS, Modi A, Singh S, Pradhan S, et al. HPV-type-specific response of cervical cancer cells to cisplatin after silencing replication licensing factor MCM4. Tumour Biol. 2015;36(12):9987–94. https://doi.org/10.1007/s13277-015-3782-7.
Saritha VN, Veena VS, Jagathnath Krishna KM, Somanathan T, Sujathan K. Significance of DNA replication licensing proteins (MCM2, MCM5 and CDC6), p16 and p63 as markers of premalignant lesions of the uterine cervix: its usefulness to predict malignant potential. Asian Pac J Cancer Prev. 2018;19(1):141–8. https://doi.org/10.22034/APJCP.2018.19.1.141.
Kaur G, Balasubramaniam SD, Lee YJ, Balakrishnan V, Oon CE. Minichromosome maintenance complex (MCM) genes profiling and MCM2 protein expression in cervical Cancer development. Asian Pac J Cancer Prev. 2019;20(10):3043–9. https://doi.org/10.31557/APJCP.2019.20.10.3043.
Acknowledgments
We thank the company leaders for their support and the editors and reviewers for their time spent handling and reviewing this manuscript. We also acknowledge the ONCOMINE database, UALCAN database, Human Protein Atlas database, Kaplan-Meier plotter database, cBioPortal databases, DAVID database, KEGG database and GEPIA2 database for providing public data and Metascape and the Venn diagram tool for providing analytical tools.
Funding
Not applicable. The funding source played no role in the design of the study, the collection, analysis, and interpretation of the data, or the writing of the manuscript.
Author information
Authors and Affiliations
Contributions
Conceived and designed the study: BW and SX. Analyzed the data: BW. Interpreted/analyzed the data and results: BW. Wrote the paper: BW. All authors reviewed and approved the final manuscript.
Authors’ information
Corresponding author and first author: Baojie Wu (June 1987), male, senior engineer of the Pilot Department, Master’s degree in biochemistry and molecular biology, mainly engaged in optimization of the purification process, amplification of the purification process, and bioinformatics data mining. Email: w3522021987@163.com, zrwbj@walvax.com. Second author: Shuyi Xi (February 1978), male, department head, mainly engaged in management of the Pilot Department. Email: xishuyi@walvax.com. The authors are committed to the study of HPV vaccines.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that there are no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wu, B., Xi, S. Bioinformatics analysis of the transcriptional expression of minichromosome maintenance proteins as potential indicators of survival in patients with cervical cancer. BMC Cancer 21, 928 (2021). https://doi.org/10.1186/s12885-021-08674-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-021-08674-y